2.2. Femtosecond Transient Absorption Monitors Electronic State Population Dynamics
To delineate the electron movements and charge transfer of GFP-S205V mutant in the excited state and also to help select the Raman pump tuning range, we first performed femtosecond transient absorption (fs-TA) spectroscopy on this protein mutant [
43]. The 2D contour plot of the TA spectra from 420 to 650 nm is shown in
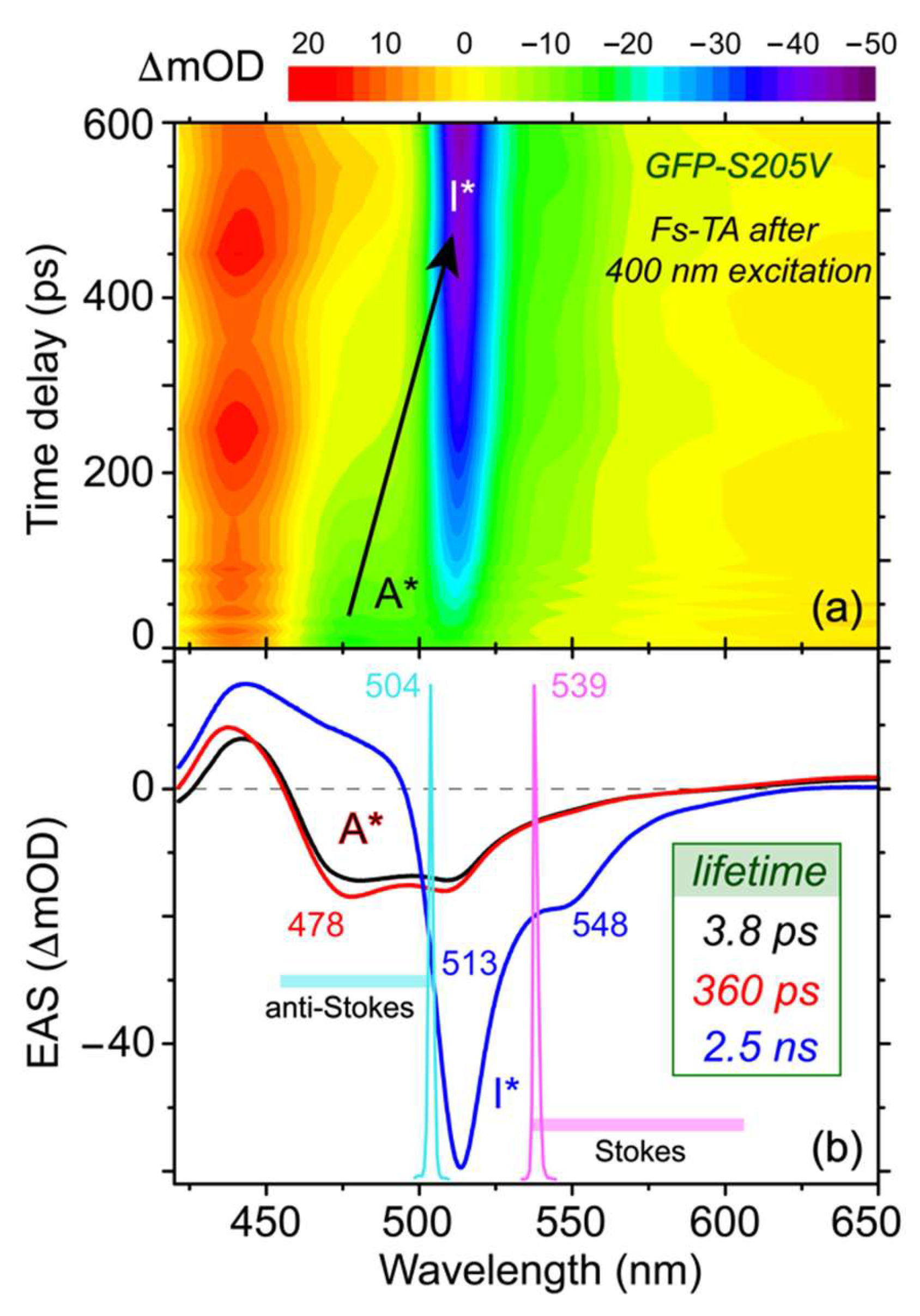
Figure 2a. The spectra mainly consist of three regions: an excited state absorption (ESA) band below 460 nm, two stimulated emission (SE) bands (centered at ~478 and 513 nm) from 460 to 620 nm, and a broad ESA band above 620 nm. The SE band at 478 nm and the ESA band above 620 nm are both attributed to the reactant A* species [
4,
11], while the SE band at 513 nm is mainly from the product I* species. With progression of the photochemical reaction as the chromophore phenolic proton gets transferred in the protein interior, the SE band at 478 nm and ESA band above 620 nm gradually decay away, while the SE band at 513 nm gains intensity, indicating a two-state A*→I* transition after photoexcitation.
To uncover the molecular evolution during a photochemical reaction where the broad electronic features commonly overlap with each other, we performed global analysis using the Glotaran software with a sequential model and resolved three main stages [
44]. The kinetic model for transient molecular species and the match between experimental and fitted traces across the spectral detection window at four representative time delay points are shown in
Figure S1 (see the
Supplementary Materials). Previous studies on related proteins have shown that a purely concerted model does not fit the TA spectra well, and some preparation stages typically exist before the main ESPT reaction step [
21,
26]. The resultant evolution-associated spectra (EAS) are plotted in
Figure 2b with a root-mean-square (RMS) value of 1.318 × 10
−3. Moreover, a four-component sequential kinetic model in global analysis leads to a ~13 fs initial component that lacks physical meaning. The resultant RMS is 1.309 × 10
−3, which represents a mere ~0.7% improvement from the aforementioned three-component fit, indicating that the three-component model is sufficient in analyzing the fs-TA data in this work. The earliest EAS shows a 478 nm emission peak and a 442 nm ESA band, which are associated with the A* species that promptly emerge upon actinic photoexcitation. The small emission peak at 512 nm is likely from the fluorescence background (because it is present to a certain extent at negative time delay points) or some ultrafast ESPT channel via those pre-existing, largely optimal H-bonding chains [
29,
32,
45]. The initial EAS then evolves to the second EAS with a time constant of 3.8 ps, during which the intensity of the A* SE band near 478 nm increases, indicating the necessity of a preparation stage before the ESPT process [
4,
21,
26,
46]. The ESA band below 450 nm also blue shifts, suggesting that the A* S
1 state is stabilized on this time scale. Judging from the TA profile of A* species before the main ESPT step, the initial two EAS can be attributed to a well-defined A* state with a notable slope out of the Franck–Condon (FC) region, wherein the chromophore remains protonated. Reminiscent of a previously proposed stepwise-concerted-hybrid ESPT reaction mechanism in a GFP-S65T/S205V double mutant [
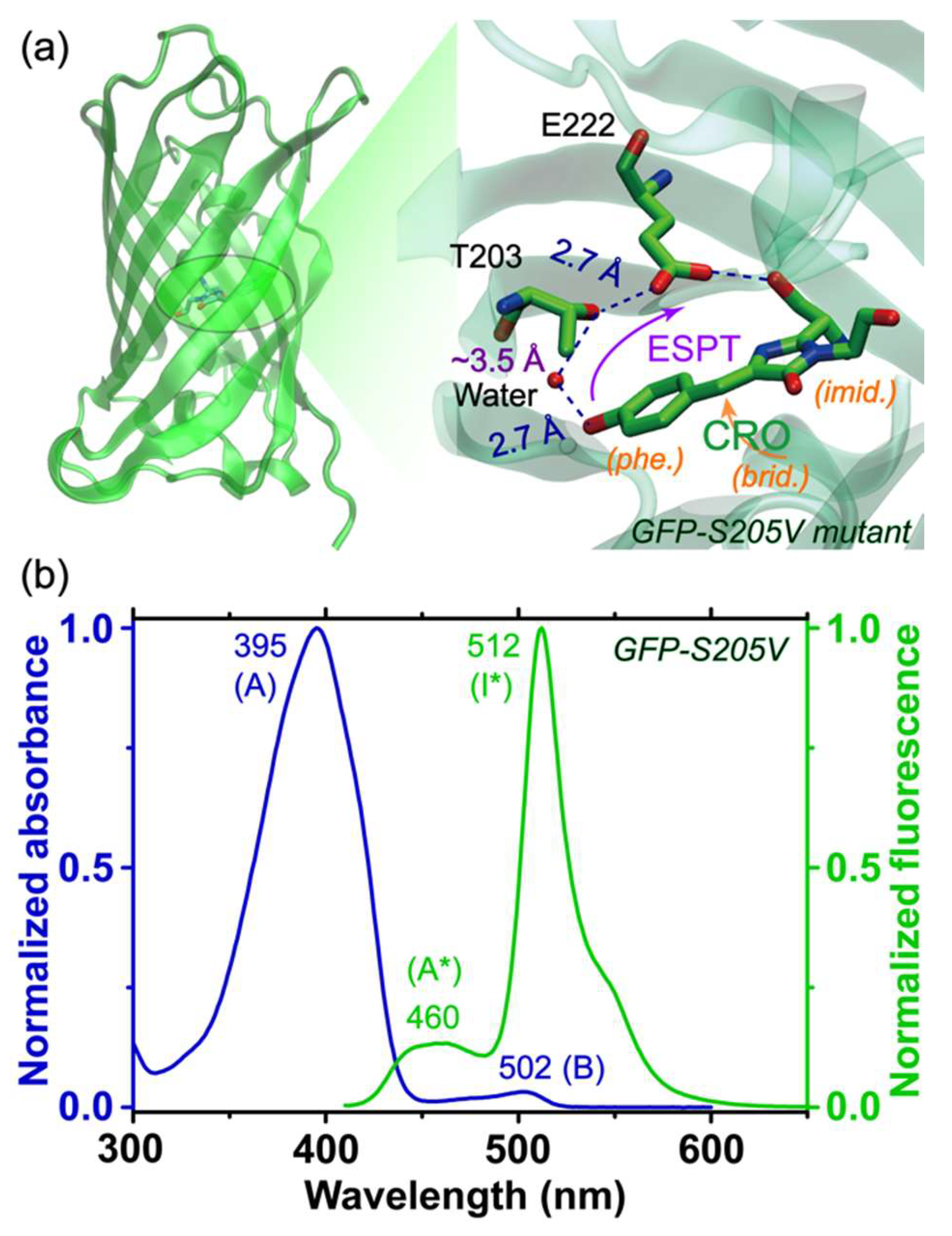
26], the initial ~3.8 ps process likely involves some small-scale proton motions in close proximity to the chromophore (e.g., CRO···water, see
Figure 1a) and a charge transfer (CT) step [
26,
32,
47] in the partially deprotonated chromophore, which presumably set up the stage for the subsequent main ESPT step on the hundreds of ps time scale (see below).
The progression of the second EAS manifests the most drastic change with disappearance of the 478 nm SE band (A*) and growth of the 513 nm SE band (I*), corroborated by the steady-state fluorescence peak positions at ~460 and 512 nm (see
Figure 1b). The retrieved time constant of this electronic population transition is 360 ps. Since the green fluorescence quantum yield of the GFP-S205V mutant is 0.78 [
38], which is very close to wtGFP (0.80) that undergoes efficient ESPT on the 5–10 ps time scale [
4,
35,
37], this 360 ps time constant can be mainly attributed to a lengthened ESPT process on ultrafast time scales, which does not negatively impact the overall fluorescence outcome in the fully equilibrated state. The third EAS then diminishes with a 2.5 ns time constant, largely in accord with the kinetic parameter of 0.31 ns
−1 previously retrieved by fitting the time-resolved fluorescence data for both A* (at 450 nm) and I* (at 510 nm) after 388 nm excitation [
38]. This component reflects a typical fluorescence process that depletes the excited state population [
4,
35,
38].
For comparison, we performed target analysis with A* evolving into two excited state species in parallel, namely A*’ and I*, which decay away (i.e., back to the ground state). The same time constants of 3.8 ps, 360 ps, and 2.5 ns (like those in
Figure 2b) were obtained which correspond to the lifetime of A*, A*’, and I*, respectively. Since the ESPT reaction (A*→I*) in this GFP-S205V mutant has been reported to be on the hundreds of ps time scale [
38,
40], the 360 ps lifetime should be associated with a deprotonation process of the chromophore (i.e., not just a relaxation within the protonated state), rendering this parallel model inconsistent with the main photochemical reaction pathway.
Based on the broad yet clearly assigned excited state electronic features of this protein mutant, three Raman pump wavelengths were carefully chosen in this study. First, an 800 nm Raman pump (not shown in
Figure 2b that displays a spectral region up to 650 nm) is selected, which is pre-resonant with the A* broad ESA band above 620 nm. This Raman pump is suitable to track the A* electronic population dynamics, and has proven to be highly effective in studying GFPs and the GFP-based Ca
2+-biosensors [
4,
11,
21,
24]. The other reason to choose this wavelength is that an 800 nm Raman pump is readily achievable in an optical setup from a commercially available laser amplifier system with an 800 nm fundamental pulse output (see
Section 3.2 below). However, this wavelength is not suitable to track the I* species because they have no ESA or SE bands in this region (e.g., see the blue trace in
Figure 2b). The second Raman pump we selected is at 539 nm (magenta spike in
Figure 2b), which can achieve pre- or quasi-resonance (but not on resonance) with the broad I* SE band at 513 nm, capable of enhancing the I* features, as demonstrated before for similar protein systems [
11,
23]. In both cases, the Stokes FSRS was performed while the Raman probe wavelength (ca. 820–955 nm or 545–605 nm) is longer than the Raman pump (i.e., 801 nm or 539 nm). The third Raman pump we picked is at 504 nm (cyan spike in
Figure 2b), with the Raman probe now located on the blue side (455–500 nm, i.e., anti-Stokes FSRS) [
31,
32,
48]. Under this condition, the Raman pump is pre-resonant with the A* SE band at 478 nm, while the probe better overlaps with this SE band [
32] without being absorbed by the A ground state species near 400 nm. Therefore, strong Raman signal of the transient A* species with sufficiently high signal-to-noise ratio (SNR) can be achieved on ultrafast time scales.
2.3. Ground State Raman Spectra with Different Raman Pumps Provide Insightful Comparisons
To benchmark the chromophore vibrational modes, the ground state (GS) FSRS spectra of the GFP-S205V mutant are plotted in
Figure 3, which make use of the Raman pump–probe pair without the photoexcitation pulse. Most of the Raman peaks appear at similar locations regardless of the Raman pump wavelengths, validating their association with the photosensitive chromophore. However, the mode intensity increases dramatically as we tune the Raman pump closer to resonance conditions with ground state absorption of the protein chromophore (see
Figure 1b). As seen for the ~1570 cm
−1 mode, it is significantly enhanced as the Raman pump changes from 801 to 504 nm, approaching the B (A) absorption peak at 502 (395) nm. Besides the increased intensity, an important benefit of performing FSRS using a Raman pump not at 800 nm is the higher sensitivity to detect low-frequency motions. Since the laser fundamental output was used to generate the supercontinuum white light as the near-IR Raman probe, the strong incident 800 nm light cannot be completely filtered or eliminated from the resultant weak white light (see
Section 3.2 below for details). As a result, the 800 nm residual in Raman probe can interfere with the 800 nm Raman pump and contaminate the low-frequency vibrational region (e.g., 802–820 nm, which corresponds to vibrational modes from ~30–300 cm
−1) [
4,
21]. Moreover, the lowest frequency we observed for a fluorescent protein sample is ~400 cm
−1 [
22], which is likely due to the scattering issues intrinsic to protein size. For comparison, a smaller organic chromophore, like a photoacid in solution, allows the observation of low-frequency modes below 300 cm
−1, such as the ~180 cm
−1 H-bond stretching mode for pyranine in water [
12]. In a nice contrast, tuning the Raman pump to the visible region allows the use of broadband Raman probe photons away from 800 nm which enables a much better observation of the low-frequency motions below ~600 cm
−1 [
10,
12]. The direct observation of multiple vibrational modes across a broad spectral window (typically over 1500 cm
−1) is a notable advantage of FSRS over other ultrafast spectroscopic techniques. To observe even lower frequency modes (e.g., <300 cm
−1) for protein samples in solution, which may report slower protein motions or collective (likely intermolecular in nature) vibrational motions that are coupled to the ultrafast processes such as ESPT reaction, other advanced spectroscopic techniques such as the femtosecond time-domain Raman spectroscopy [
25] and terahertz absorption spectroscopy [
49,
50] can be implemented.
When comparing the GS peaks collected by the Stokes FSRS (with 539 nm Raman pump and 545–605 nm Raman probe) and the anti-Stokes FSRS (with 504 nm Raman pump and 455–500 nm Raman probe), it becomes clear that some of the vibrational motions exhibit frequency red shifts. For example, the 852 cm
−1 mode shows an intensity increase without frequency change as the Raman pump is tuned from 801 to 539 nm in the Stokes FSRS, however, the mode not only gains further intensity but also shifts its frequency to 839 cm
−1 in the anti-Stokes FSRS with a bluer Raman pump at 504 nm. Similarly, two lower frequency modes at 486 and 550 cm
−1 also exhibit stronger intensity and notable red shifts. This observation substantiates the improved resonance Raman enhancement when both the Raman pump and probe wavelengths better match an electronic absorption band [
51,
52] and, in this particular case, the deprotonated chromophore Raman signal gets enhanced due to its absorption peak at ~502 nm (
Figure 1b). Such a Raman pump-dependent FSRS peak center frequency in association with different molecular subpopulations of an inhomogeneous sample has been demonstrated [
10,
13,
53]. Moreover, some excited state vibrational coherences (typically broader peaks) may be generated when the Raman pump effectively overlaps with the absorption band of a certain species [
31,
54]. Further comparisons to the excited state FSRS can be found below in
Section 2.5,
Section 2.6 and
Section 2.7, and these intriguing mode frequency red shifts in
Figure 3, associated with the enhancement of deprotonated chromophore species, can corroborate the transient Raman mode assignment during the ESPT reaction (see below). The essence of performing time-resolved FSRS is to capture transient Raman modes in the excited state before the system returns to the electronic ground state [
7,
10], so the resonance conditions of the Raman pump–probe pair need to be carefully selected to avoid generating significant ground and excited state species simultaneously (which could lead to spectral overlap and dispersive line shapes [
31,
52]). Fortunately, pre-resonance conditions have been experimentally validated to track excited state vibrational dynamics during a photochemical reaction, such as ESPT in solution [
12,
32] or protein environment [
8,
11,
13,
23,
26], also showing apparent vibrational frequency shifts from the ground to excited state (e.g., S
0→S
1) of the chromophore.
2.4. Stokes FSRS with an 801 nm Raman Pump Benchmarks A* Vibrational Dynamics
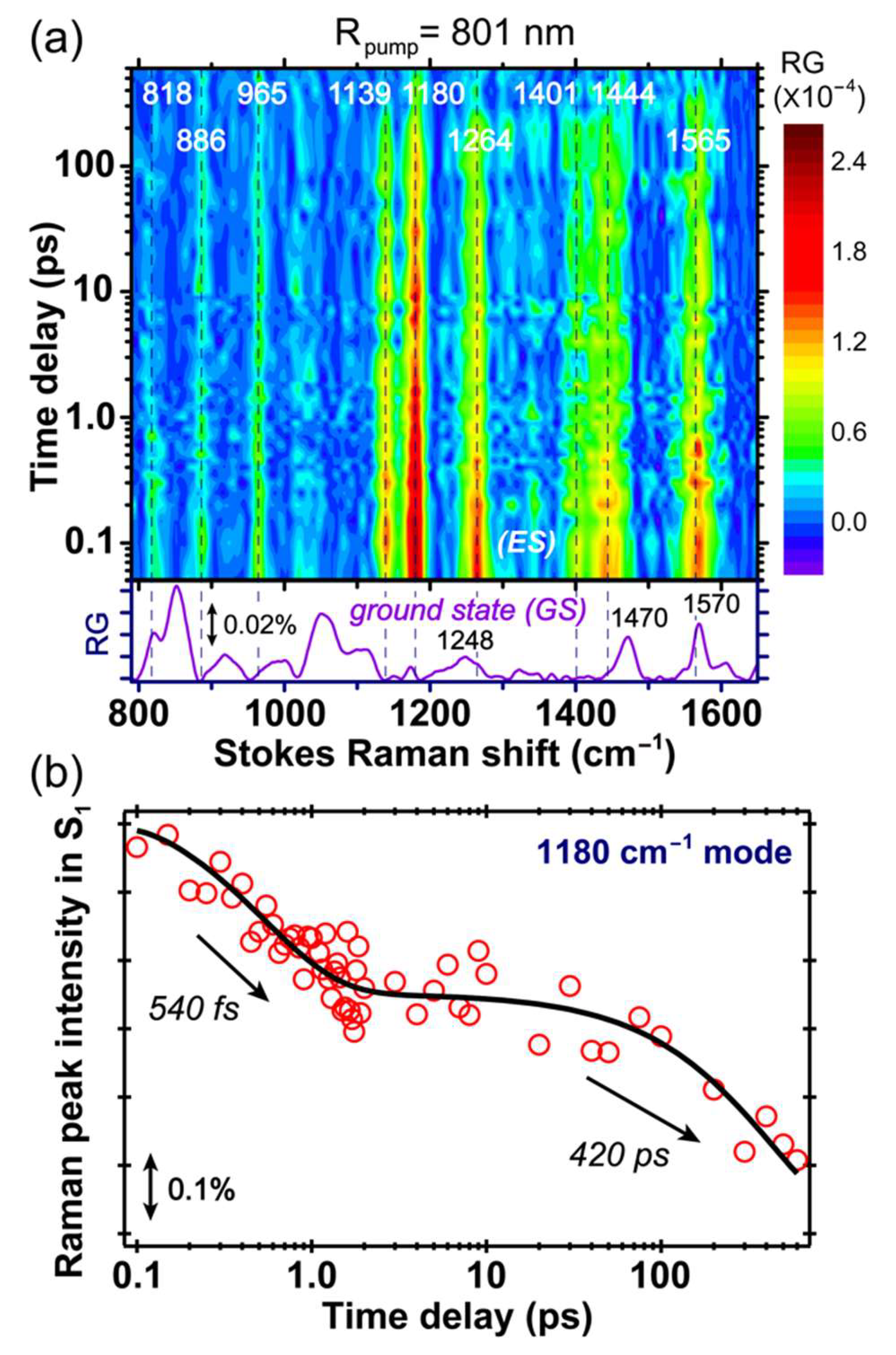
The excited state FSRS spectra of GFP-S205V were first collected with an 801 nm Raman pump at the Stokes side. The 2D contour plot from ca. 800 to 1650 cm
−1 and up to 600 ps after photoexcitation is presented in
Figure 4a. Note that the ground state spectrum is subtracted from all the experimentally collected excited state spectrum, so the resultant difference spectrum at each time delay point after photoexcitation corresponds to the “pure” excited state species [
4,
7]. Further verification of the excited state mode dominance in our data analysis comes from the observed dynamics and distinct frequencies/line shapes in a non-equilibrium state (versus the electronic ground state), and the broad peak width that does not significantly vary starting from photoexcitation time zero on the relevant time scale for the photochemical reaction under study (without a significant internal conversion pathway back to a hot ground state) [
4,
8,
10,
12]. All the transient modes reach their maximal intensities within the cross-correlation time (~140 fs) between the fs actinic pump and Raman probe, followed by decay on the hundreds of ps time scale. Based on their intensity dynamics, these motions are attributed to the reactant species A* with a protonated chromophore in S
1, because the modes from the product I* species should mainly rise on the hundreds of ps time scale as the ESPT reaction occurs inside the protein [
11,
21].
In
Figure 4b, we plotted the intensity dynamics of the A* 1180 cm
−1 mode. This mode consists of the chromophore phenol ring-H and bridge C–H rocking motion, and is a sensitive probe to track the ESPT process based on our former studies [
11,
13,
21,
24]. The mode intensity plot on a linear scale, from negative time points to 10 ps after photoexcitation to the A* S
1 state, is shown in
Figure S2 in the
Supplementary Materials, and the signal rise around time zero within the cross-correlation time (~140 fs) is clearly visible. A semilogarithmic plot across a much wider detection time window (~600 ps) is displayed in
Figure 4b to better represent the excited state signal decay, and its vibrational intensity dynamics can be fit with a biexponential decay with ~540 fs and 420 ps time constants. The first time constant is likely due to the photoexcited chromophore moving out of the FC region, along with adjacent solvent molecular motions, and since a rapid change of the energy gap between electronic states (e.g., S
1–S
2, S
1–S
0) is expected, the resonance condition for the transient A* species changes along with the electric polarizability of the chromophore in its dynamic, rapidly fluctuating local environment. The second time constant is similar to the 360 ps retrieved from global analysis of the fs-TA data in
Figure 2b, therefore confirming the time scale for ESPT reaction. It is notable that the Raman mode coherences, as shown in
Figure 4,
Figure 5,
Figure 6 and
Figure 7 (see below), are generated by the ps-Raman-pump and fs-Raman-probe pair, at well-defined time delay points after actinic photoexcitation [
6,
8,
10], so these vibrational coherences effectively act as an array of “probing” modes, to capture the conformational snapshots at those particular time points and their associated transient electronic state population. In this context, the similarity between time constants of 360 and 420 ps in
Figure 2b and
Figure 4b supports the 1180 cm
−1 mode assignment to the A* state, while the larger time constant from time-resolved FSRS may arise from (1) some spectral overlap with the neighboring vibrational modes, (2) spectral overlap in TA that leads to some uncertainties in retrieving time constants from global analysis, and (3) resonance Raman condition change for the A* modes during the A*→I* transition [
31,
52].
Since the initial energy relaxation has occurred within ~2 ps as shown in
Figure 4b (i.e., the early-time inflection point on the A* marker band intensity dynamics fitting curve), this clear separation of two characteristic time constants reveals an activation energy barrier for ESPT as the A*→I* transition becomes the rate-limiting step for A* Raman mode decay, as well as the I* mode rise. The latter process represents a more direct link because ESPT is the only channel generating I* species, whereas it is one of the pathways depleting A* species.
Notably, a comparison between the FSRS spectra of the GFP-S205V mutant and wtGFP with the same 800 nm Raman pump [
4] shows that all the excited state vibrational mode frequencies are largely conserved, as well as the instantaneous mode frequency shift around photoexcitation time zero (see the vertical dashed lines in
Figure 4a) due to electron redistribution upon 400 nm electronic (π→π*) excitation of the A species. These spectral changes upon photoexcitation are deeply rooted in the photoacid nature of the protein chromophore, and the rapid protonation state change in the excited state [
4,
12,
21] as the overall electron density migrates from the phenolic ring to the imidazolinone ring (i.e., charge transfer that occurs on ultrafast time scale). However, the dynamics of Raman marker bands in the GFP-S205V mutant are much longer than wtGFP, which match the ESPT time constant obtained by other ultrafast spectroscopic techniques [
38,
40]. This result not only benchmarks the characteristic A* modes of an embedded SYG chromophore, but also proves the sensitivity and reliability of using FSRS to track an array of vibrational motions and monitor a photochemical reaction such as ESPT.
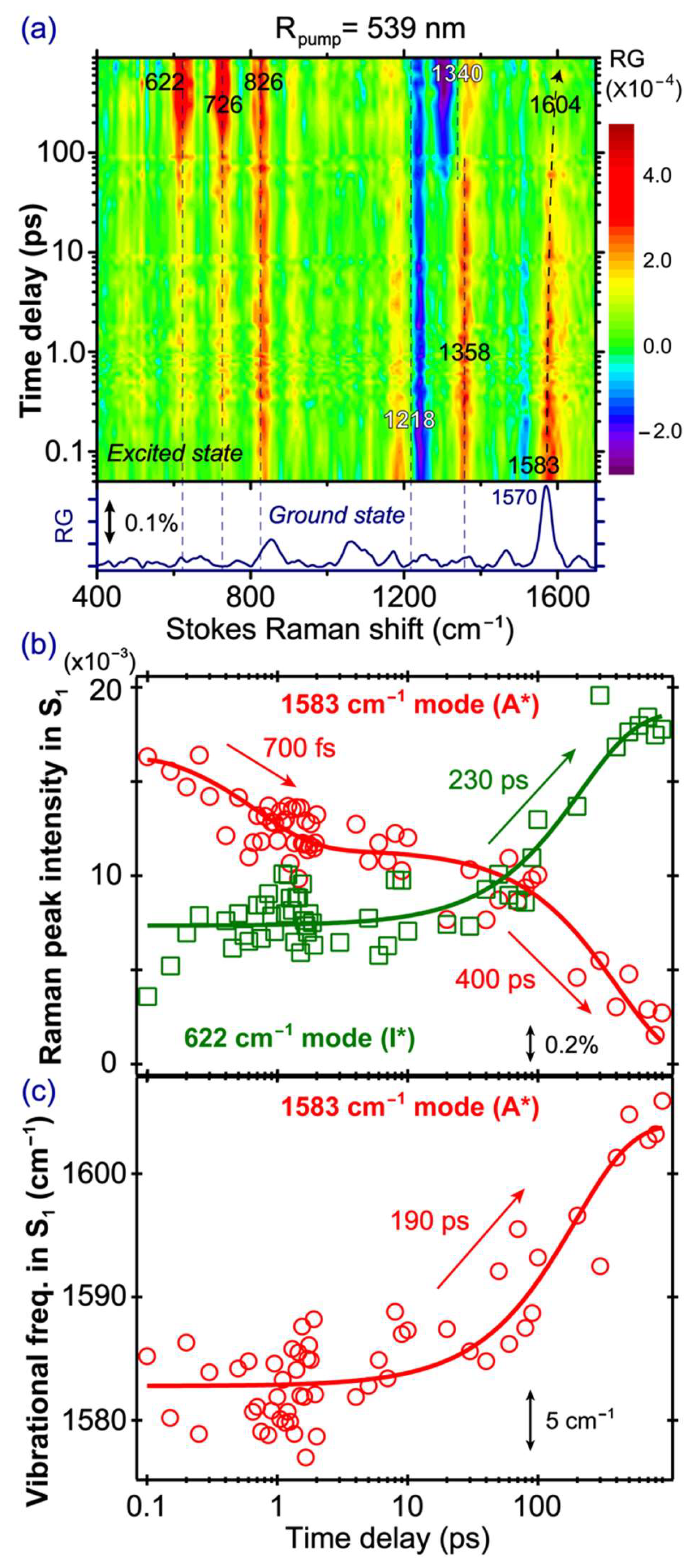
2.5. Stokes FSRS with a 539 nm Raman Pump Tracks I* Vibrational Dynamics
To unravel the characteristic I* vibrational dynamics of the SYG chromophore, we tuned our Raman pump at 539 nm to be pre-resonant with the SE band of the emerging I* species (
Figure 2b), and
Figure 5a presents the time-resolved 2D contour plot of the FSRS spectra of the GFP-S205V mutant. In contrast to the abovementioned FSRS spectra with an 800 nm Raman pump, major differences upon tuning the Raman pump to 539 nm are that some pronounced modes (622, 726, and 826 cm
−1) reach their maximal intensities after several hundreds of ps, not at time zero. This phenomenon infers the I* nature of these modes, and confirms that tuning the Raman pump toward particular electronic bands can effectively enhance and detect the vibrational features of transient molecular species. In addition to the I* modes with largely absorptive line shapes (e.g., positive peaks that have a gaussian profile for these chromophore modes inside a protein pocket), we also observe a dispersive line shape at ~1218 cm
−1 (the negative portion peaks at ~1241 cm
−1) at early times when the chromophore is mainly in the A* state. This dispersive line shape diminishes with time, while a new dispersive feature appears at ~1340 cm
−1. This rather unusual observation indicates that the dispersive line shape still allows the dynamic tracking of a photochemical reaction. The 1241 cm
−1 mode could be strongly coupled with the A* electronic transition to a nearby state (e.g., S
1→S
0), while the 1340 cm
−1 mode is coupled with an I* electronic transition (e.g., S
1→S
0). Such a mode-dependent line shape change due to vibronic transitions in Stokes FSRS has been reported for photoexcited chromophores in solution [
18,
52], especially on or near resonance conditions. We conjecture an intricate dependence of the A* Raman peak line shape on the resonance conditions with respect to overlapping electronic bands at early times (
Figure 2b). Since the evolving A* SE band and ESA band likely overlap in the spectral region of the Raman pump and probe we used in this case (i.e., 539 nm and ~545–605 nm), we observed clear dispersive line shapes at certain Raman marker bands which undergo vibronic transitions in a mode-specific manner during the FSRS signal generation [
52,
55].
With this tunable FSRS dataset, the aforementioned Raman mode frequency red shift in
Figure 3 can be further rationalized. In the low-frequency region, the ground state 852 cm
−1 mode appears at 839 cm
−1 when the Raman pump was tuned from 539 to 504 nm, indicating that when the deprotonated chromophore (B species in the ground state) gets further enhanced, a lower frequency is observed. The related I* species, when accumulated in time, exhibit the mode at 826 cm
−1 (see
Figure 5a). Similarly for the high-frequency mode at 1364 and 1333 cm
−1 with the 539 and 504 nm Raman pump, respectively, the nearby I* mode is at ~1340 cm
−1 (
Figure 5a). In essence, the deprotonated chromophore exhibits these characteristic lower frequencies of Raman modes due to change of the conjugation structure, as well as the overall energy stabilization achieved by a stronger H-bonding network surrounding the photoacidic chromophore [
12,
56]. The general consideration to enhance the I* species is thus to use a Raman pump in the bluer wavelength region, but away from the SE peak.
In
Figure 5b, the 1583 cm
−1 A* mode decay and the 622 cm
−1 I* mode rise are overlaid and correlated in one graph (see the mode assignments in
Table 1 below). The 1583 cm
−1 mode is mainly a C=N stretch mixed with some C=C and C=O stretching motions in the excited state [
4,
40]. It appears at a higher frequency than the 1565 cm
−1 mode observed in the 800 nm Raman pump FSRS (
Figure 4a), suggesting that different Raman pumps can enhance different A* species [
13]. We consider that different resonance conditions achieved by the Raman pump pulse cannot directly lead to the observed Raman mode frequency variation because the vibrational energy gaps within the same electronic state remain the same, regardless of the Raman pump wavelength being used [
13]. In particular, the 539 nm Raman pump likely enhances a slightly more deprotonated A* population, because in those chromophores with different H-bonding configurations from the mainly protonated A* population, more electrons are distributed across the phenol and imidazolinone rings, and can cause the frequencies of certain mixed C=N, C=C, and C=O stretching motions to blue shift [
13,
23]. For example, a higher frequency of the C=N stretching motion is expected when charge transfer occurs from the phenolate end to the imidazolinone end in a deprotonated SYG chromophore, which is characteristic of a photoacid that undergoes ESPT in solution or protein matrix [
26,
32]. The 1583 cm
−1 mode decays biexponentially with the 700 fs and 400 ps time constants. The similarity of the time constants between this A* mode and the 1180 cm
−1 A* mode in conventional FSRS (800 nm Raman pump, see
Figure 4b) indicates that although the two Raman pumps allow the probing of different A* subpopulations, their intensity decay dynamics remain largely unchanged, which we attribute to the large O···O distance between the bridging water and T203 (i.e., ~3.5 Å; the three subpopulations from MD simulations show the dynamic distance distribution peaks at ~2.75, 4.0, and 4.5 Å) in the ESPT chain that is the rate-determining step for ESPT (see
Figure 1a) [
38,
39]. This represents the reaction “bottleneck” as infrequent proton transfer occurs between these key players in the ESPT chain. By contrast, the distance observed from crystal structures of wtGFP between the chromophore hydroxyl oxygen atom and the adjacent conserved water molecule is ~2.6 Å [
57,
58,
59] which was supported by classical MD simulations and multiconfigurational electronic structure calculations [
47], also matching that same distance in the GFP-S205V mutant at ~2.7 Å [
38] (see
Figure 1a).
Notably, the 622 cm
−1 I* mode exhibits a faster rise with a ~230 ps time constant, when compared to the A* mode decay (~400 ps,
Figure 5b). A closer examination of the rise dynamics of this I* mode shows that its intensity promptly rises upon photoexcitation, reaching a plateau from ca. 300 fs–10 ps, and then rises again on the hundreds of ps time scale. This observation could stem from a small A* subpopulation that undergoes proton dissociation rapidly upon photoexcitation, consistent with a pre-existing largely optimal H-bonding chain (see above,
Figure 2b) as well as multiple conformations of key players, such as T203 and bridging water in the chain [
29,
32,
39,
45]. The mode intensity of the early-time-generated I* species accounts for ~39% of the overall I* intensity, which could slightly decrease the main ESPT time constant by fitting the later intensity rise of the mode. However, the clear trend of a smaller time constant for I* rise (~230 ps) than that for A* decay (~400 ps) is reasonable, because besides ESPT, there are other A* energy dissipation pathways that occur on longer time scales (e.g., non-radiative ring twisting, fluorescence) [
11,
21,
46] or from some trapped A* species undergoing vibrational cooling, which effectively compete with the proton transfer kinetics. The ESPT time constant in the GFP-S205V mutant is therefore better reflected by the 230 ps time constant. To corroborate this new experimental observation, we performed a quantitative estimate of the proton transfer rate for S205V in water on the basis of a temperature-dependent study of the decay of A* blue fluorescence, which is slowed down upon lowering the temperature from 318 to 132 K [
38,
61]. Using the reported ESPT activation energy of
≈ 18.2 kJ/mol and an exponential prefactor
≈ 6.7 × 10
12 s
−1 (corresponding to an intrinsic proton transfer rate of ~150 fs [
62], which also matches a barrierless reaction rate constant approximately as the frequency factor
≈ 6.1 × 10
12 s
−1), we used the Arrhenius equation
at room temperature (
K), and derived the ESPT reaction rate constant
s
−1, which corresponds to a time constant of ~250 ps. This is clearly consistent with the 230 ps time constant we retrieved from the I* vibrational marker band intensity rise in the excited state FSRS experiment.
Furthermore, the 1583 cm
−1 mode exhibits a notable frequency blue shift (1583→1604 cm
−1) with time (
Figure 5c), likely due to the vibrational cooling process in A*. This contrasts with the previously observed mode frequency red shift (1585→1565 cm
−1) in wtGFP [
4] and blue shift (1570→1584 cm
−1) in the calcium biosensor GEM-GECO1-P377R mutant [
23] with an 800 nm Raman pump, both closely tracking the ESPT reaction of the largely protonated SYG chromophore. Given the sudden influx of photoexcitation energy, the chromophore vibrational cooling through high-frequency motions can be an effective means for energy dissipation [
24,
63,
64]. Similarly, in a small organic photoacid HPTS, its 1530 cm
−1 mode (ring C=C stretching and phenolic COH rocking motion) from its excited photoacid state (PA*) also shows a frequency blue shift, and the shift magnitude increases with more energetic excitation photons [
12,
64]. Because of the spectral overlap in this high-frequency region, the mode frequency red shift due to photochemistry (associated with the A* species undergoing ESPT reaction to I*) and mode frequency blue shift due to photophysics (associated with the A* species undergoing vibrational cooling within A*) could mix, and exhibit some cancellation effect. In the current work with a different resonance condition achieved by the 539 nm Raman pump, due to the observed frequency blue shift (
Figure 5c) and intensity decay (
Figure 5b) of the 1583 cm
−1 marker band, this mode is dominantly an A* mode, and the observed time constant of ~190 ps can thus be attributed to vibrational cooling in parallel with ESPT. Interestingly, we previously reported a vibrational cooling time constant of ~70 ps based on the 1265 cm
−1 A* marker band for an embedded SYG chromophore in a calcium biosensor GEX-GECO1 using an 800 nm Raman pump [
24]. The significant lengthening of this time constant (70→190 ps) could be a result of the GFP-S205V mutant having a well-protected chromophore at the center of the GFP β-barrel, while the Ca
2+-free GEX-GECO1 has an opening on the circularly permuted GFP, so the lack of flexible water molecules and a more rigid environment in the GFP-S205V mutant results in a longer time for vibrational cooling and energy relaxation into the surroundings [
31,
65]. Moreover, the H-bonding network differs in the chromophore pocket of these two proteins. Larger interatomic distances between key players in the H-bonding chain (see
Figure 1a), which significantly lengthen the ESPT time constant in the GFP-S205V mutant when compared to the GEX-GECO1 protein biosensor, could also be responsible for a lengthened vibrational cooling time constant for the A* species that remains in the same electronic state.
2.6. Anti-Stokes FSRS with 504 nm Raman Pump Provides Further Insights into ESPT and Other Pathways
To gain more information about the transient A* species by taking advantage of the A* SE band (see
Figure 2b), we performed the anti-Stokes FSRS with a 504 nm Raman pump that locates to the red side of the main A* SE peak, while also to the blue side of the main I* SE peak. The wavelength selection adopts a balanced approach to track A* modes at early times and I* modes at later times, while avoiding an overlap with the bluer ground state bleaching band of A* species. This setup represents a rare case that anti-Stokes FSRS [
10,
20,
48] has been applied to study proteins, and the choice of Raman pump wavelength is important in the information content being retrieved from such an experiment. The time-frequency 2D contour plot is presented in
Figure 6, and most peaks are negative due to the FSRS signal generation pathways when the bluer Raman probe better matches the A* SE band than the redder Raman pump [
32,
66]. The 1139 and 1247 cm
−1 modes are attributed to the A* species because they display the monotonic decay behavior. The 1566 cm
−1 mode shows a very strong dispersive line shape at early time (see
Figure S3a for the raw experimental spectrum at 100 fs time delay and the spline baseline drawn), which gradually decays with time, so it can also be attributed to an A* mode. The line shape eventually changes to a negative peak after ~300 ps (see
Figure S3b for the raw experimental spectrum at 600 ps, and the spline baseline drawn), which implies that when the A* population decreases via ESPT and other relaxation pathways, there is likely an electronic feature change of the A* species, so the resonance condition worsens with the dispersive line shape diminished [
52,
66]. This is because the pertinent hot luminescence terms mainly responsible for such mode-dependent dispersive line shapes require the resonance or quasi-resonance condition to be favorable and maintained (i.e., the Raman pump and/or probe wavelength matching the transient electronic peak position associated with each mode). If the initially dominant dispersive line shape at ~1566 cm
−1 is due to the overlap between the Raman pump and probe pair and the A* SE band around 478 nm (e.g., for observation of the 1566 cm
−1 mode with the Raman pump at 504 nm, the corresponding Raman probe is at 467 nm), then, as the ESPT reaction progresses on the hundreds of ps time scale, the transient electronic band shifts to the I* SE band around 513 nm (
Figure 2) that is farther away from the Raman probe. As a result, the reduction of hot luminescence terms leads to the apparent diminishment of dispersive line shapes for this mode (see
Figure 6a and
Figure S3b).
Notably, besides the prominent A* modes above 1100 cm
−1 (including the one at 1324 cm
−1 before ~50 ps), several I* modes are enhanced and clearly observed at later time points. We consider that although the Raman probe overlaps with the A* SE band at early times, the 504 nm Raman pump is still within the I* SE band. At later times, as ESPT reaction proceeds and generates a significant I* population, the Raman probe better overlaps with the I* ESA band that “takes over” the A* SE band below 500 nm (see
Figure 2b). All these factors favor the improved resonance conditions for the photoproduct state and, consequently, increase the stimulated Raman signal intensity of the nascent I* species (mainly negative peaks, and one mode with dispersive line shape at ~1295 cm
−1 in
Figure 6a). Interestingly, the dispersive mode frequency largely matches the energy gap between the I* SE main peak at 513 nm and a vibronic transition shoulder at 548 nm (
Figure 2b), corresponding to an excited state vibrational mode at ~1250 cm
−1 that could be used as a sensitive vibrational probe for ESPT in the protein matrix.
In
Figure 6b, the intensity dynamics of the 1247 cm
−1 A* mode and the 830 cm
−1 I* mode are displayed as representative vibrational probes for the ESPT reaction inside the GFP-S205V protein pocket based on the anti-Stokes FSRS measurement. The evolution of the 1247 cm
−1 mode shows a dramatic difference from the dynamics of other A* modes observed in Stokes FSRS with the 801 nm (
Figure 4) or 539 nm (
Figure 5) Raman pump. This mode has a small but distinct rise phase, with a 5 ps time constant, which largely matches the transition time of ~4 ps from the first to second EAS in global analysis of the fs-TA spectra (
Figure 2b). It is noteworthy that this A* marker band (assigned to the chromophore C–O stretching motion) appears at ~1264 cm
−1 in Stokes FSRS with an 801 nm Raman pump (
Figure 4a) and ~1218 cm
−1 at the “zero crossing point” of a dispersive line shape [
52] in Stokes FSRS with a 539 nm Raman pump (
Figure 5a), and similar modes were observed for the SYG chromophore inside wtGFP [
4] and the abovementioned GFP-derived calcium ion biosensor GEM-GECO1 [
21]. Therefore, this key result corroborates the occurrence of an initial phase of protonated chromophore conformational motions which helps the A* species to evolve into a more stable state, likely better solvated by the surrounding water molecules and protein residues, leading to an increased A* SE intensity as well as a blue shift of the ESA band (
Figure 2b). Notably, in a time-resolved fluorescence up-conversion experiment on GFP-S205V monitoring the A* emission at 470 nm [
40], a ~9 ps component was observed in the A* fluorescence decay with 28% weight, preceded by a ~200 fs component that has 50% weight (solvation dynamics or vibrational relaxation could contribute) and followed by a ~250 ps component with 22% weight (consistent with the A*→I* ESPT time constant). Since the 9 ps step does not exhibit KIE and hence does not involve proton transfer, this temporal component could be related to the ~4 ps component recovered from our global analysis of the fs-TA data (
Figure 2b), as well as the ~5 ps component retrieved from kinetic analysis of the transient Raman mode intensity (
Figure 6b). The time-resolved fluorescence time constant is slightly longer than that from the time-resolved electronic dynamics, due to the stimulated nature of our fs-TA measurement [
43,
67].
Furthermore, the main reason we observed this distinct ~5 ps step prior to the main ESPT reaction is the unique resonance condition achieved by the 504 nm Raman pump and a bluer Raman probe, wherein the Raman probe essentially overlaps with the A* SE peak region. For example, the Raman probe generating the 1247 cm
−1 mode is at 474 nm, which is very close to the A* SE peak at 478 nm. Our previous study on a similar resonance condition with the Raman pump pre-resonance but Raman probe on-resonance with respect to an electronic absorption band in the ground state for a fluorescent dye rhodamine 6G in methanol, also showed a non-dispersive mode line shape [
66], which constitutes a useful strategy to track the high-frequency Raman modes of transient species with improved SNR while maintaining a Raman gain or loss line shape on the anti-Stokes side.
From the least-squares fit of the intensity dynamics plot in
Figure 6b, the ~1247 cm
−1 A* mode decays with an 82 ps time constant, much faster than the ~400 ps ESPT time constant. The nearby ~1139 cm
−1 mode exhibits a similar dynamic plot with these time constants. A possible explanation is that this tens of ps process is related to the protein chromophore relaxation from A* to A. Interestingly, an earlier study of the same protein mutant using time-resolved IR spectroscopy and species-associated global analysis reported a ~38 ps process that was attributed to an A*→A nonradiative transition (e.g., intramolecular motions of the chromophore that promotes its internal conversion back to S
0) [
40]. Four reasons may explain why this peculiar process is observed in the anti-Stokes FSRS with a 504 nm Raman pump. First, the Raman probe overlaps with the A* SE band, and can sensitively track the structural dynamics of all the A* subpopulations, regardless of their ESPT capabilities. Second, this Raman mode exhibits a dispersive line shape in Stokes FSRS with a 539 nm Raman pump, indicative of its strong coupling to the A* downward transition (i.e., S
1→S
0). Third, at late time delay points, the positive component of the emerging I* dispersive peak (center at ~1295 cm
−1 in
Figure 6a) overlaps with the negative peak at ~1247 cm
−1, resulting in an apparent intensity loss. Fourth, even if the pertinent A*→A transition is actually a vibrational relaxation process within A*, and the Raman intensity decay is a result of significant loss of resonance enhancement condition and electric polarizability, the conformational inhomogeneity of the chromophore still holds (i.e., some undergo ESPT to I*, while some undergo vibrational cooling within A*) [
23,
24,
40]. An interesting correlation can be made to the previously observed vibrational cooling time constant of ~70 ps based on an adjacent 1265 cm
−1 A* marker band for the SYG chromophore of a calcium biosensor GEX-GECO1 using Stokes FSRS with an 800 nm Raman pump [
24]. We conjecture that this mainly C–O stretching motion could be more sensitive to the A* decay dynamics along the slope of the A* potential energy surface (PES), and once the ESPT proceeds from the lower portion of the A* state, a higher frequency mode (than the 1247 cm
−1 mode, see
Figure 6a and
Figure S3b) appears, which can be analyzed to accurately retrieve the I* rise dynamics [
4,
11,
21,
23]. Moreover, the difference between this 82 ps time constant (1247 cm
−1 mode decay, anti-Stokes FSRS with 504 nm Raman pump in
Figure 6b) and 190 ps time constant (1583 cm
−1 mode frequency blue shift, Stokes FSRS with 539 nm Raman pump in
Figure 5c) stems from the mode-dependent nature of the trapped A* relaxation dynamics on a multidimensional excited state PES [
12,
52,
55,
64], as well as different dynamic resonance Raman conditions [
31,
52]. In particular, because the chromophore electronic absorption band typically red shifts upon gaining more charge transfer character in reaching a deprotonated state [
32,
68,
69], the anti-Stokes FSRS resonance condition with a 504 nm Raman pump and a bluer Raman probe of ~455–500 nm highlights the more protonated A* species at early times, so the gradually diminishing A* SE band within ~200 ps (
Figure 2) could contribute to a faster decay time constant of an A* Raman mode. By contrast, the Stokes FSRS with a 539 nm Raman pump and a redder Raman probe of ~545–605 nm, allows better tracking of more deprotonated A* species at later time points, with a more stagnant TA profile for the wavelength region above ~510 nm (
Figure 2b).
On the photoproduct side, the 830 cm
−1 mode reflects a relatively pure I* intensity rise (a negative peak becoming more negative) with a ~370 ps time constant. Although it has an initial small intensity, the early-time intensity is only ~13.5% of the overall intensity (
Figure 6b). This could be reflective of an initial ESPT component along a pre-existing, largely optimal H-bonding chain, that leads to some I* population within ~1 ps (see
Figure 2). While the anti-Stokes FSRS with a 504 nm Raman pump mainly enhances the A* species at early times, it also tracks the I* rise dynamics as the I* population accumulates at later times so overall there could be some interference or convolution effect. In comparison, the I* mode intensity rise, as reflected by the 622 cm
−1 mode in Stokes FSRS with a 539 nm Raman pump, exhibits a larger initial intensity (~39% of the overall I* mode intensity, see above) and a smaller time constant of ~230 ps (
Figure 5b), which indicates that the 539 nm Raman pump with redder Raman probe photons better enhance the deprotonated chromophore species, including those generated on the fs to few ps time scales [
26,
35] before the main ESPT step. In other words, due to a “cleaner” pre-resonance condition with the I* SE band only, the Stokes FSRS with a 539 nm Raman pump is a more reliable way to monitor the I* intensity rise on ultrafast time scales.
2.7. Different Raman Pumps Generate Characteristic Raman Line Shapes and Track Transient Species
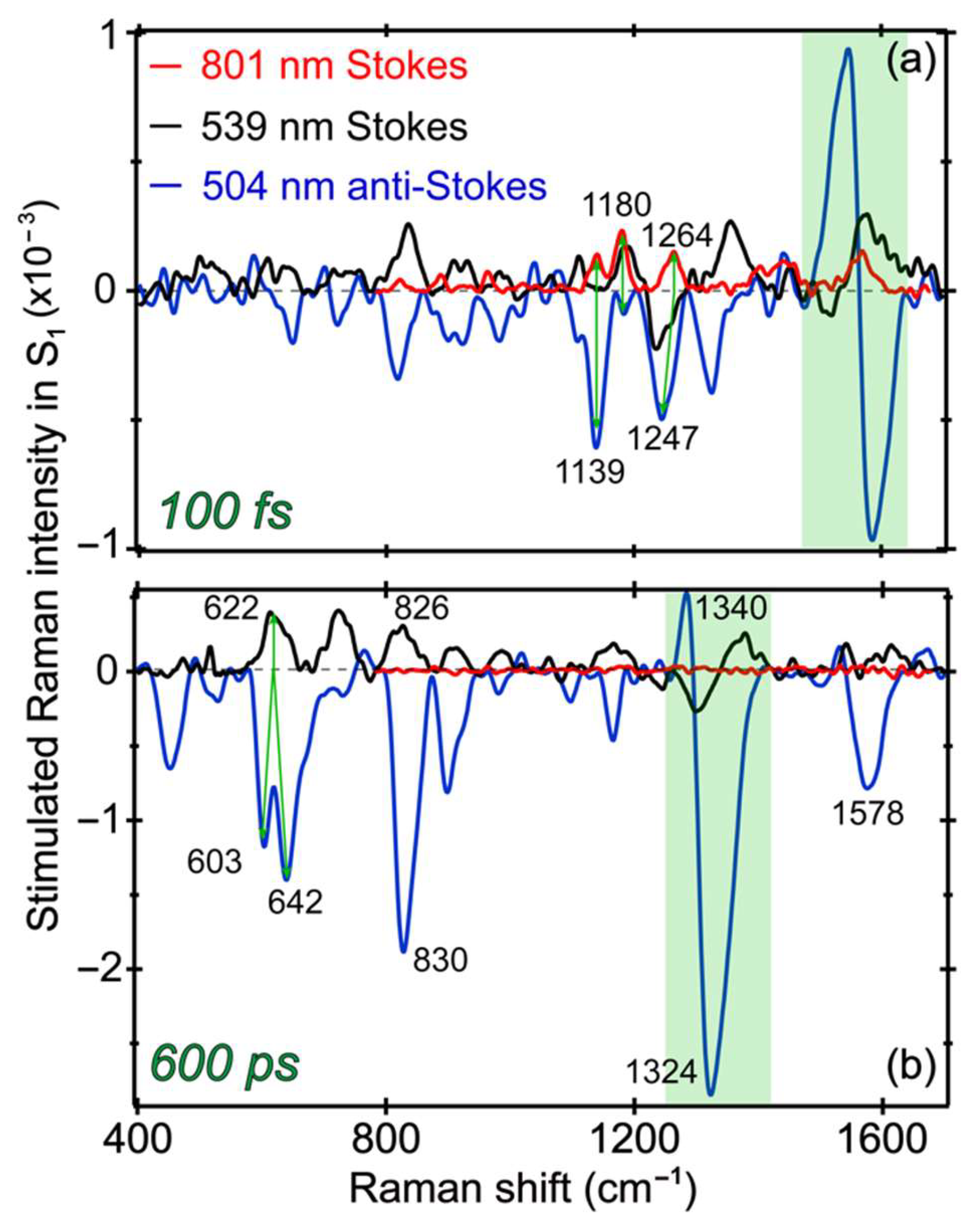
In this comprehensive work, a systematic comparison between the transient Raman features along an ultrafast ESPT reaction inside a protein pocket is made possible by tuning the Raman pump to three strategic wavelength locations (801, 539, and 504 nm) from the near-IR to the visible region. The versatile selection of the Raman probe from a supercontinuum white light, across the near-UV to near-IR range, enables the FSRS to be readily collected on both the Stokes and anti-Stokes sides. A wealth of information becomes available as the photoreactant (A*) converts via an ESPT reaction into photoproduct (I*) species with intrinsic conformational inhomogeneity, so a portion of the A* species undergoes energy dissipation via other pathways (generally faster than the ESPT reaction inside the GFP-S205V mutant) as reflected by detailed analysis of the electronic and vibrational dynamics and associated time constants in a non-equilibrium state. During these dynamic processes, charge transfer likely occurs in concert with proton motions from the chromophore phenolic hydroxyl end to an internal water molecule and further down the H-bonding chain for ESPT (see
Figure 1a). Depending upon the degree and stage of chromophore deprotonation, charge transfer mainly facilitates the dipole change for system–bath interactions as well as energy stabilization of the excited state prior to the main radiative pathway (i.e., fluorescence in this case). To further visualize the photochemical reaction inside GFP-S205V and highlight the sensitivity of tunable FSRS technique [
8,
10],
Figure 7 directly contrasts the excited state Raman modes at two representative time delay points with three distinct Raman pump wavelengths. Besides characteristic line shapes, these vibrational mode frequencies clearly differ from their ground state counterparts, as shown in
Figure 3, confirming their excited state nature and the rich information content we can glean by analyzing their temporal dynamics across the experimental time window (
Figure 4,
Figure 5 and
Figure 6) and correlating with the fs-TA results (
Figure 2).
At very early time (100 fs) in
Figure 7a, the Raman modes belong to A* species, since they are the major population immediately following photoexcitation. With the tuning of the Raman pump–probe pair, most of the modes remain at similar positions, while some modes exhibit clear frequency shifts. For example, in FSRS with an 801 nm Raman pump, a peak doublet at 1139 and 1180 cm
−1 is observed [
4,
24]. However, the 1180 cm
−1 mode is prominent (as a positive peak, Raman gain) in FSRS with a 539 nm Raman pump, whereas the 1139 cm
−1 mode is dominant (as a negative peak, Raman loss) in the anti-Stokes FSRS with a 504 nm Raman pump. We note that a much smaller negative peak at 1180 cm
−1 also exists in the anti-Stokes FSRS spectrum. Since the 539 nm (pre-resonant with the I* SE band) and the 504 nm (pre-resonant with the A* SE band at early times) Raman pump focus on different species, this result indicates that the 1139 cm
−1 mode is more related to the protonated chromophore, whereas the 1180 cm
−1 mode has more contribution from a slightly deprotonated chromophore [
13]. A related observation is the small peak intensity buildup at ~1170 cm
−1 (i.e., higher frequency than the ~1139 cm
−1 A* mode) past 100 ps in
Figure 6a. Notably, complications could arise in the Stokes FSRS when the resonance conditions of the Raman pump become favorable to induce vibronic transitions involving certain Raman marker bands, and for the deprotonated chromophore of GFP-S205V, the ~1180 cm
−1 mode merges into a broad dispersive line shape with the adjacent ~1240 cm
−1 mode (see
Figure 7a and
Figure S3a for the raw experimental data with spectral baselines). For this reason, we cannot easily track its intensity dynamics as those reported for the related GFP derivatives [
13,
23,
26].
At a much later time (600 ps) in
Figure 7b, the Raman modes should be mostly coming from I* species, because the ESPT reaction has a main time constant of ~360 ps (
Figure 2b). FSRS with an 800 nm Raman pump can hardly resolve any I* features [
4,
11] because the I* species has no ESA or SE band in this near-IR region. In FSRS with a 539 or 504 nm Raman pump, the I* Raman modes are clearly observed, and most of peaks appear at similar locations with an opposite sign corresponding to the Raman gain and loss in Stokes and anti-Stokes FSRS, respectively [
10,
31,
52]. Moreover, the 1330 cm
−1 mode displays a dispersive line shape in both FSRS measurements with a 539 or 504 nm Raman pump, confirming that this particular vibrational motion is strongly coupled with the electronic transitions from the I* state. Given the phase difference between the dispersive line shapes in this spectral region, the resonance conditions are different with the 539 nm Raman pump in Stokes FSRS (overlap with the I* SE band red shoulder) versus the 504 nm Raman pump in anti-Stokes FSRS (overlap with the I* SE band blue shoulder mixed with the I* ESA band red shoulder). Notably, the 1330 cm
−1 mode consists of the chromophore bridge C–H rocking, phenolic ring-H rocking, and imidazolinone ring in-plane deformation (see
Table 1), which is a very delocalized mode. As shown in
Figure 5,
Figure 6 and
Figure 7, this particular mode rises at later times that correlate well with the ESPT time constant, and the mode dispersive line shape becomes more prominent as I* species accumulate (e.g., could be related to vibronic coupling). Therefore, this mode could effectively facilitate the I* species to interact with surrounding water molecules and protein residues to stabilize the nascent I* species on an ultrafast time scale, in accord with the high sensitivity of bridge-H motions to the protonation state change of the chromophore [
4,
13,
21]. In essence, a significant involvement with improved electric polarizabilities of certain nuclear motions (not every mode, as shown in
Figure 5 and
Figure 6) along the CT and ESPT reaction coordinate on the excited state PES, pinpoints the effective dissipation pathway(s) of the photoexcitation energy via intra- and intermolecular interactions. Though at this point, we could not gain deeper insights due to the lack of knowledge about the exact time scale for intramolecular (e.g., within the chromophore) and intermolecular (e.g., between the chromophore and surrounding residues) motions in a protein pocket, it is plausible that primary structural motions on ultrafast time scales could have a notable effect on molecular events occurring on much longer timescales from large-scale proton transfer [
4,
12] and drug-enzyme binding [
70] to other functions in chemical and biological systems [
71].
The other interesting observation is that the 622 cm
−1 mode in FSRS with a 539 nm Raman pump splits into two modes (603 and 642 cm
−1) in the anti-Stokes FSRS with a 504 nm Raman pump, indicative of conformational inhomogeneity of the nascent I* species, which are enhanced by a different electronic resonance condition achieved by the 504 nm Raman pump (because of its overlap with both the ESA and SE bands of I* at later times). Nevertheless, performing a series of tunable FSRS experiments with dynamic resonance conditions still provides an effective way to validate the excited state peak positions, as well as gain additional structural dynamics insights into the vibronic coupling, vibrational relaxation, and distinctive subpopulations of a transient species before and after a photochemical reaction (see
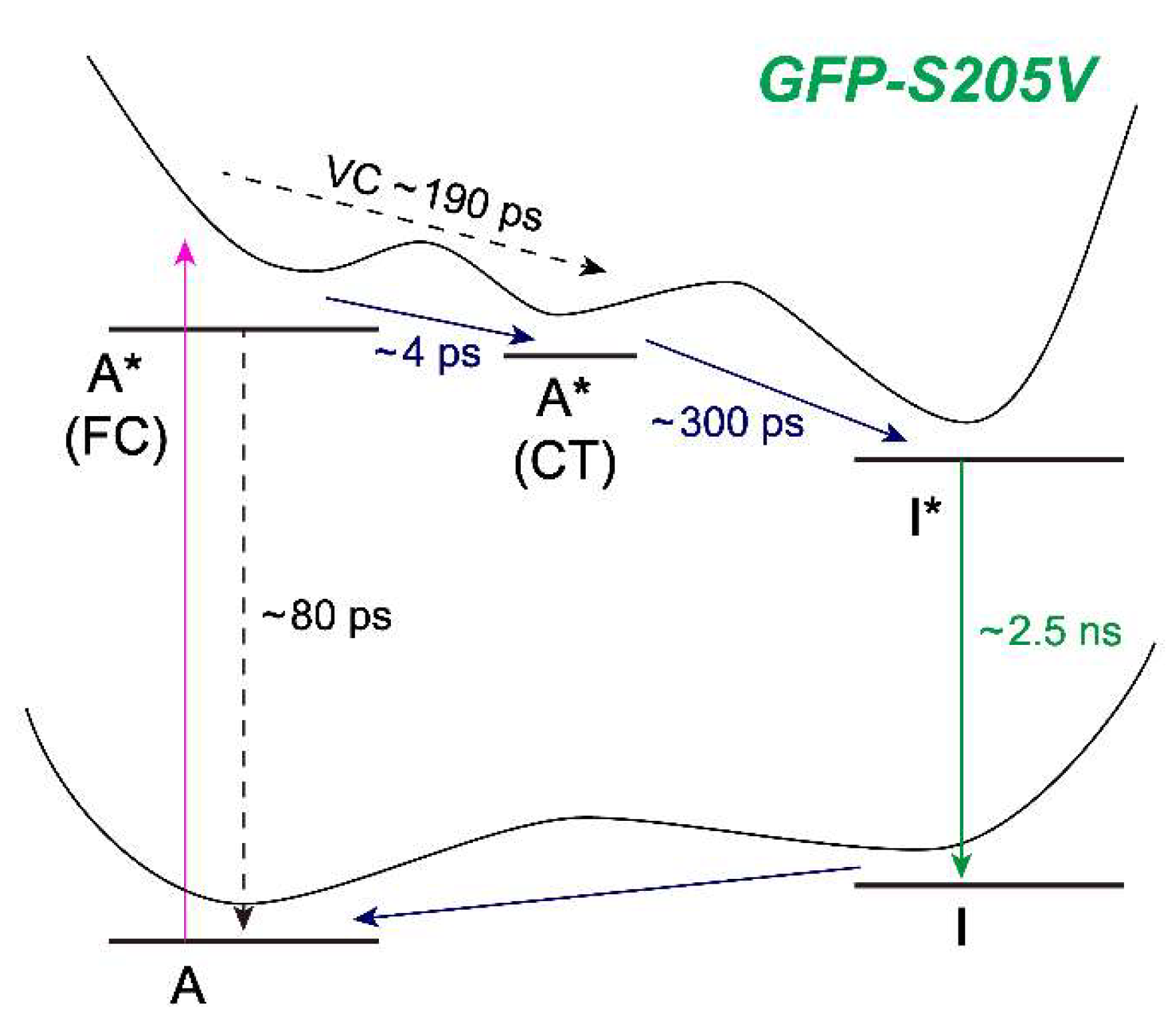
Figure 8 for the photocycle kinetic scheme of GFP-S205V).
When comparing the raw experimental FSRS data with different Raman pumps (see
Figure S3 in the
Supplementary Materials), the conventional FSRS with an 800 nm Raman pump shows all positive Raman peaks and the flattest baseline due to a pre-resonance condition with a weak A* ESA band. Tunable FSRS with a 539 or 504 nm Raman pump, which achieves pre-resonance with the strong 513 nm I* SE band, the 478 nm A* SE band, or the ~445 nm I* ESA band (
Figure 2b), all exhibit highly sloping baselines due to the Raman-pump-induced change to the TA profile [
12,
72], and the resultant dispersive line shapes of certain excited state Raman modes [
52]. The drawing of smooth and largely featureless spectral baselines (see
Figure S3 for example) allows subtraction of the TA-like electronic background, so the much narrower vibrational features (i.e., Raman modes) can be obtained with high fidelity [
8,
10,
12]. Notably, the low-frequency modes and the characteristic I* modes can only be clearly resolved in the tunable FSRS with a blue Raman pump. Consequently, a combination of various Raman pumps can be used to cross-examine the excited state Raman bands throughout a photoinduced process on crucial molecular time scales, to gain a more complete understanding of a complicated system like the GFP-S205V mutant in aqueous solution.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}