Aflatoxin B1–Formamidopyrimidine DNA Adducts: Relationships between Structures, Free Energies, and Melting Temperatures



Abstract
:
1. Introduction
2. Results
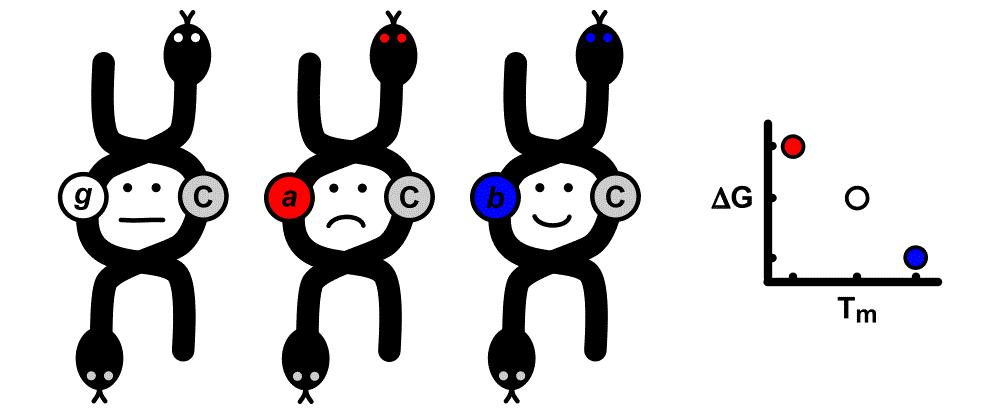
2.1. Sizes
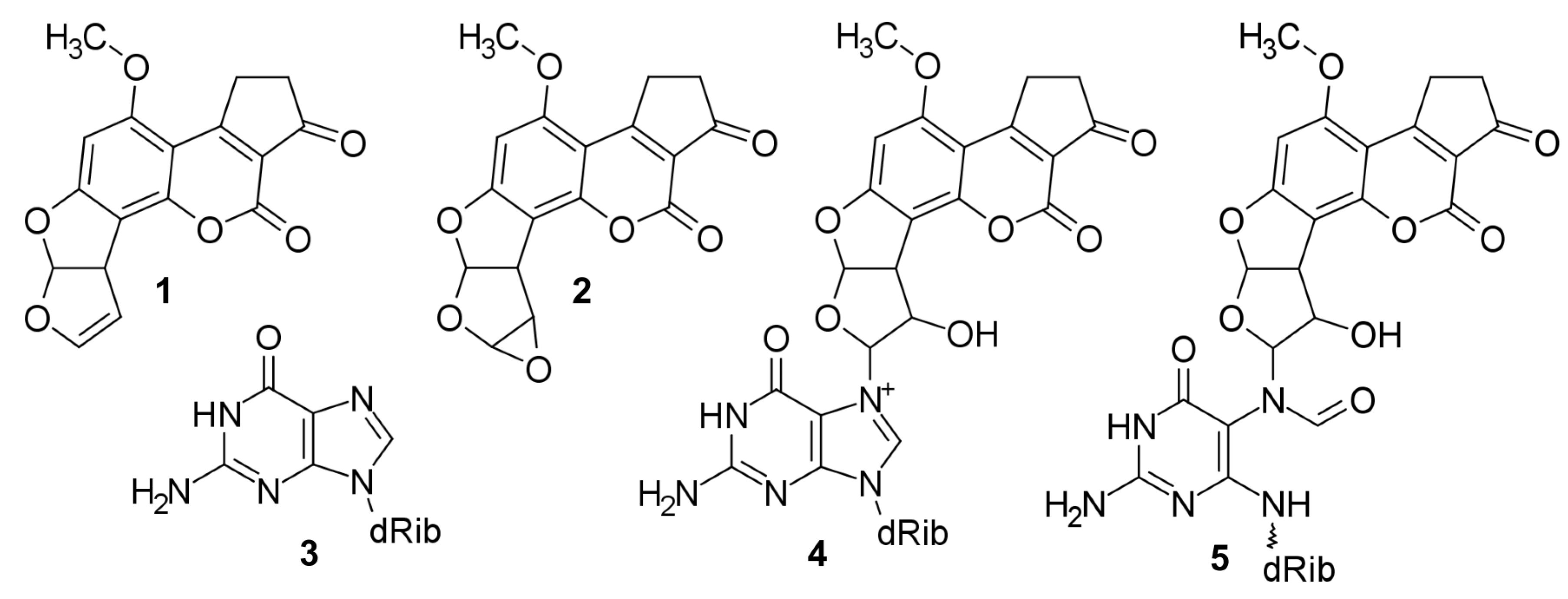
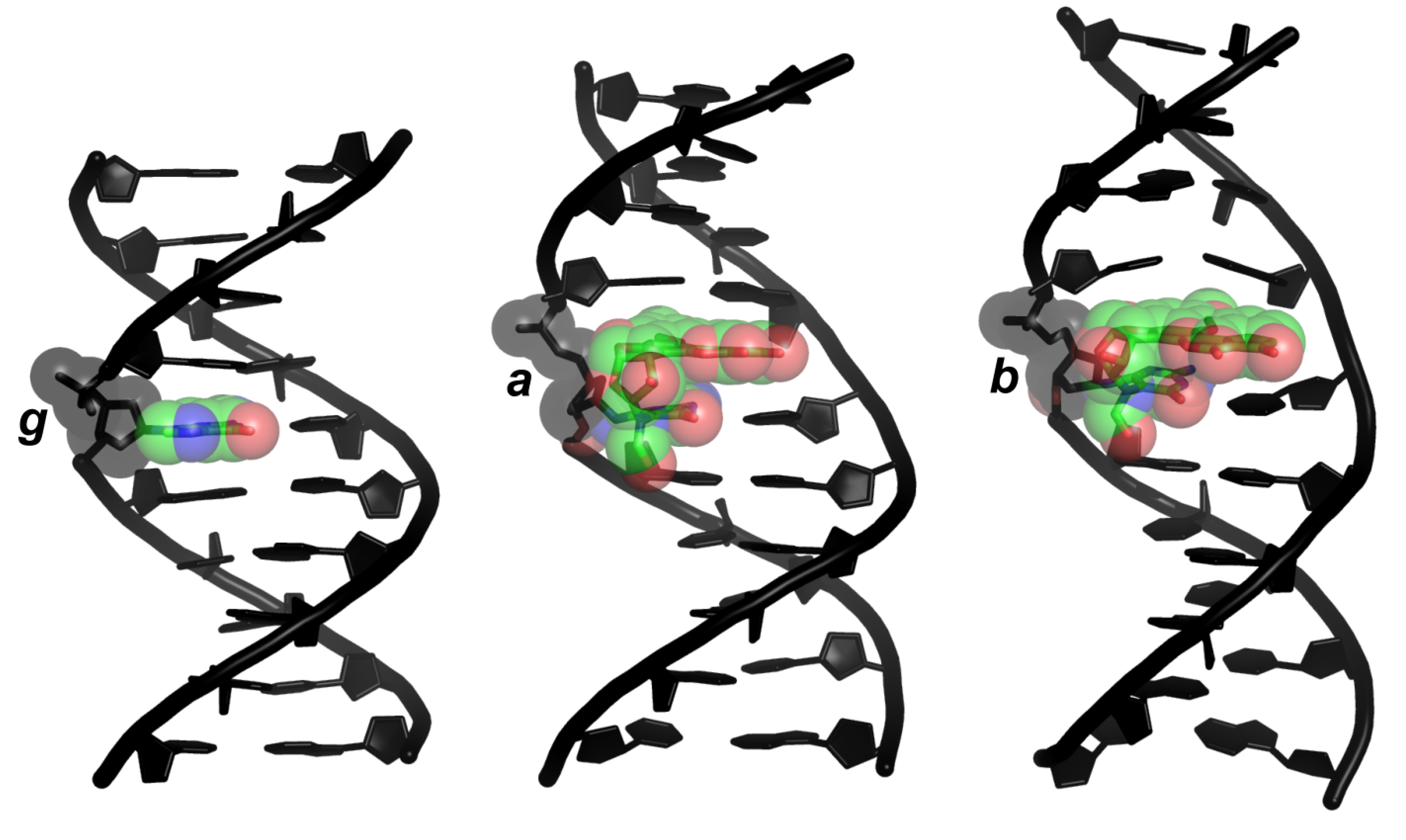
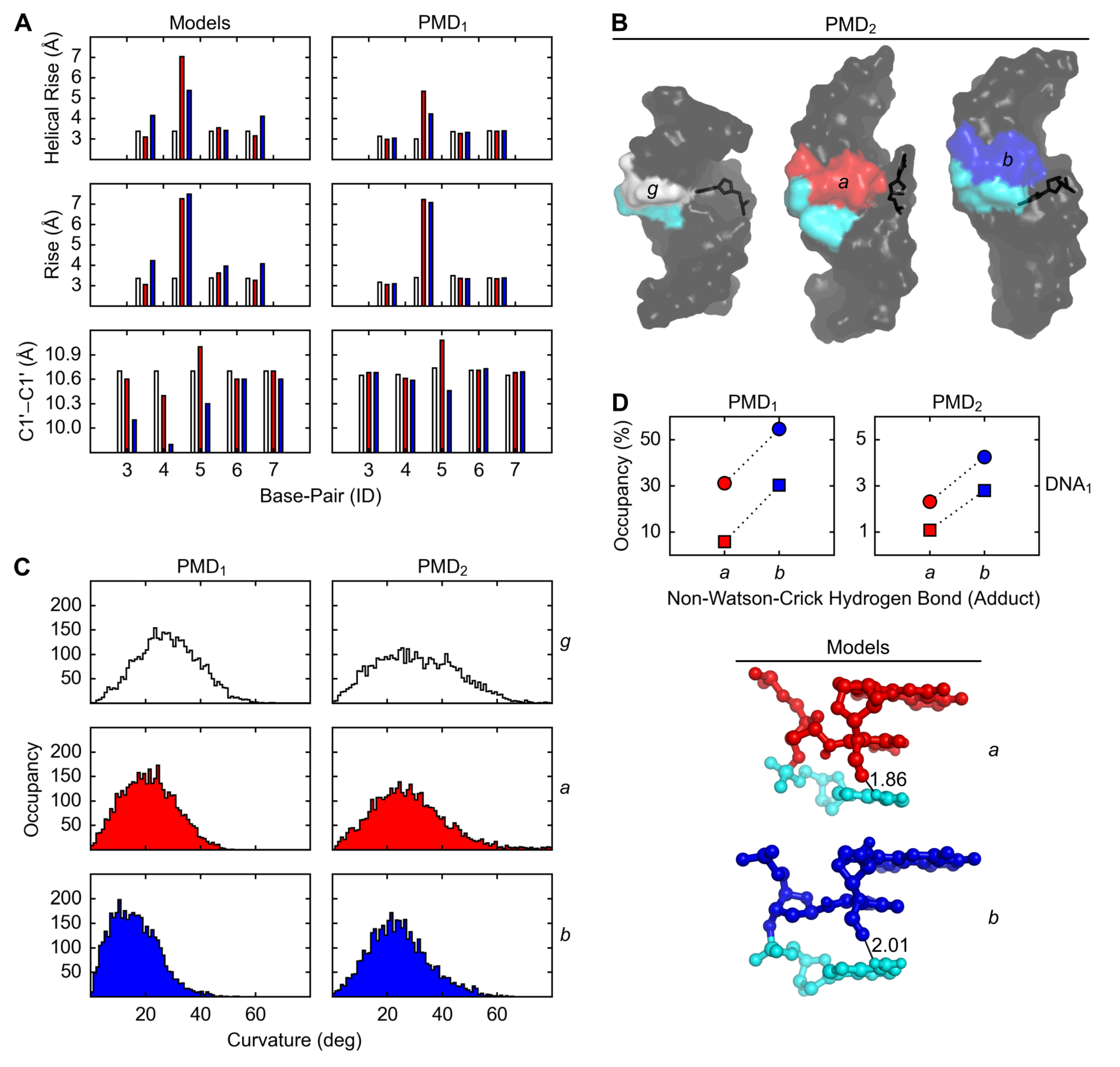
2.2. Structures
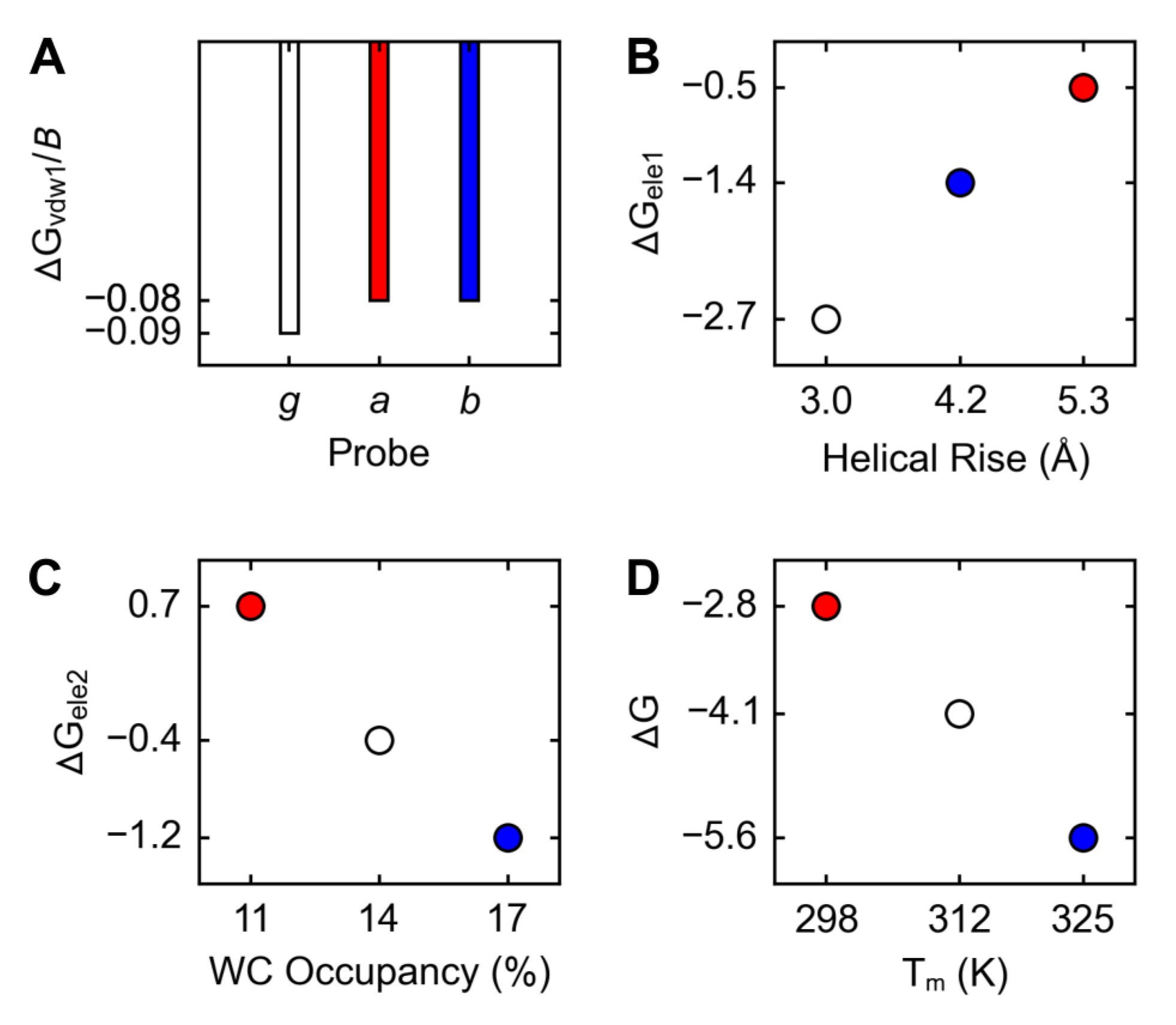
2.3. Free Energies
2.4. Correlations
3. Discussion
3.1. Relationships between Structures, Free Energies, and Melting Temperatures
3.2. Errors
3.3. Strengths and Weaknesses
4. Materials and Methods
4.1. Structural Models
4.2. Energetical Models
4.3. Solvation
4.4. Simulation
4.5. Visualization
4.6. Measurement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AFB1 | aflatoxin (Aspergillus flavus toxin) B1 |
| a | α-FAPy-N7-9-hydroxy-AFB1 |
| b | β-FAPy-N7-9-hydroxy-AFB1 |
| g | Gua |
| AFB1-E | exo-8,9-epoxide of AFB1 |
| dsDNA | double-stranded DNA |
| ΔΔG | relative free energy of dsDNA formation |
| ΔG | contribution of a probe (g, a, or b) to the absolute free energy of dsDNA formation |
| EMD | equilibrating MD simulation |
| FAPy | formamidopyrimidine |
| LIE | linear interaction energy |
| LRA | linear response approximation |
| MD | molecular dynamics |
| PMD | producing MD simulation |
| ssDNA | single-stranded DNA |
| Tm | melting temperature |
| WC | Watson-Crick |
References
- Asao, T.; Buchi, G.; Abdel-Kader, M.M.; Chang, S.B.; Wick, E.L.; Wogan, G.N. Aflatoxins B and G. J. Am. Chem. Soc. 1963, 85, 1706–1707. [Google Scholar] [CrossRef]
- Asao, T.; Buechi, G.; Abdel-Kader, M.M.; Chang, S.B.; Wick, E.L.; Wogan, G.N. The structures of aflatoxins B and G. J. Am. Chem. Soc. 1965, 87, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, B.F.; O’Kelly, J.; Sargeant, K.; Sheridan, A. Aspergillus flavus and turkey X disease. Toxic metabolites of Aspergillus flavus. Nature 1962, 195, 1062–1063. [Google Scholar] [CrossRef]
- Sargeant, K.; Sheridan, A.; O’Kelly, J.; Carnaghan, R.B.A. Toxicity associated with certain samples of groundnuts. Nature 1961, 192, 1096–1097. [Google Scholar] [CrossRef]
- Codner, R.C.; Sargeant, K.; Yeo, R. Production of aflatoxin by the culture of strains of Aspergillus flavus–oryzae on sterilized peanuts. Biotechnol. Bioeng. 1963, 5, 185–192. [Google Scholar] [CrossRef]
- Kurtzman, C.P.; Horn, B.W.; Hesseltine, C.W. Aspergillus nomius, a new aflatoxin-producing species related to Aspergillus flavus and Aspergillus tamarii. Antonie van Leeuwenhoek 1987, 53, 147–158. [Google Scholar] [CrossRef]
- Klich, M.A.; Mullaney, E.J.; Daly, C.B.; Cary, J.W. Molecular and physiological aspects of aflatoxin and sterigmatocystin biosynthesis by Aspergillus tamarii and A. ochraceoroseus. Appl. Microbiol. Biotechnol. 2000, 53, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Frisvad, J.C.; Skouboe, P.; Samson, R.A. Taxonomic comparison of three different groups of aflatoxin producers and a new efficient producer of aflatoxin B1, sterigmatocystin and 3-O-methylsterigmatocystin, Aspergillus rambellii sp. nov. Syst. Appl. Microbiol. 2005, 28, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Frisvad, J.C.; Samson, R.A. Two new aflatoxin producing species, and an overview of Aspergillus section Flavi. Stud. Mycol. 2011, 69, 57–80. [Google Scholar] [CrossRef]
- Carvajal-Campos, A.; Manizan, A.L.; Tadrist, S.; Akaki, D.K.; Koffi-Nevry, R.; Moore, G.G.; Fapohunda, S.O.; Bailly, S.; Montet, D.; Oswald, I.P.; et al. Aspergillus korhogoensis, a novel aflatoxin producing species from the Côte d’Ivoire. Toxins 2017, 9, 353. [Google Scholar] [CrossRef]
- Hesseltine, C.W.; Shotwell, O.L.; Ellis, J.J.; Stubblefield, R.D. Aflatoxin formation by Aspergillus flavus. Bacteriol. Rev. 1966, 30, 795–805. [Google Scholar] [PubMed]
- Battilani, P.; Toscano, P.; Van der Fels-Klerx, H.J.; Moretti, A.; Camardo Leggieri, M.; Brera, C.; Rortais, A.; Goumperis, T.; Robinson, T. Aflatoxin B1 contamination in maize in Europe increases due to climate change. Sci. Rep. 2016, 6, 24328. [Google Scholar] [CrossRef] [PubMed]
- Legator, M.S.; Zuffante, S.M.; Harp, A.R. Aflatoxin: Effect on cultured heteroploid human embryonic lung cells. Nature 1965, 208, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Schoental, R. Hepatotoxic activity of retrorsine, senkirkine and hydroxysenkirkine in newborn rats, and the role of epoxides in carcinogenesis by pyrrolizidine alkaloids and aflatoxins. Nature 1970, 227, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Garner, R.C. Chemical evidence for the formation of a reactive aflatoxin B1 metabolite, by hamster liver microsomes. FEBS Lett. 1973, 36, 261–264. [Google Scholar] [CrossRef]
- Swenson, D.H.; Miller, J.A.; Miller, E.C. 2,3-Dihydro-2,3-dihydroxy-aflatoxin B1: An acid hydrolysis product of an RNA–aflatoxin B1 adduct formed by hamster and rat liver microsomes in vitro. Biochem. Biophys. Res. Commun. 1973, 53, 1260–1267. [Google Scholar] [CrossRef]
- Swenson, D.H.; Miller, E.C.; Miller, J.A. Aflatoxin B1-2,3-oxide: Evidence for its formation in rat liver in vivo and by human liver microsomes in vitro. Biochem. Biophys. Res. Commun. 1974, 60, 1036–1043. [Google Scholar] [CrossRef]
- Baertschi, S.W.; Raney, K.D.; Shimada, T.; Harris, T.M.; Guengerich, F.P. Comparison of rates of enzymatic oxidation of aflatoxin B1, aflatoxin G1, and sterigmatocystin and activities of the epoxides in forming guanyl-N7 adducts and inducing different genetic responses. Chem. Res. Toxicol. 1989, 2, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Guengerich, F.P. Evidence for cytochrome P-450NF, the nifedipine oxidase, being the principal enzyme involved in the bioactivation of aflatoxins in human liver. Proc. Natl. Acad. Sci. USA 1989, 86, 462–465. [Google Scholar] [CrossRef]
- Forrester, L.M.; Neal, G.E.; Judah, D.J.; Glancey, M.J.; Wolf, C.R. Evidence for involvement of multiple forms of cytochrome P-450 in aflatoxin B1 metabolism in human liver. Proc. Natl. Acad. Sci. USA 1990, 87, 8306–8310. [Google Scholar] [CrossRef]
- Kamdem, L.K.; Meineke, I.; Gödtel-Armbrust, U.; Brockmöller, J.; Wojnowski, L. Dominant contribution of P450 3A4 to the hepatic carcinogenic activation of aflatoxin B1. Chem. Res. Toxicol. 2006, 19, 577–586. [Google Scholar] [CrossRef] [PubMed]
- He, X.Y.; Tang, L.; Wang, S.L.; Cai, Q.S.; Wang, J.S.; Hong, J.Y. Efficient activation of aflatoxin B1 by cytochrome P450 2A13, an enzyme predominantly expressed in human respiratory tract. Int. J. Cancer 2006, 118, 2665–2671. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.K. Effects of aflatoxin B1 on polysomal profiles and RNA synthesis in rat liver. Biochim. Biophys. Acta 1968, 169, 206–211. [Google Scholar] [CrossRef]
- Goodall, C.M.; Butler, W.H. Aflatoxin carcinogenesis: Inhibition of liver cancer induction in hypophysectomized rats. Int. J. Cancer 1969, 4, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.S.; Wogan, G.N. Aflatoxin inhibition of template activity of rat liver chromatin. Biochim. Biophys. Acta 1970, 224, 597–607. [Google Scholar] [CrossRef]
- Saunders, F.C.; Barker, E.A.; Smuckler, E.A. Selective inhibition of nucleoplasmic rat liver DNA-dependent RNA polymerase by aflatoxin B1. Cancer Res. 1972, 32, 2487–2494. [Google Scholar] [PubMed]
- Ames, B.N.; Durston, W.E.; Yamasaki, E.; Lee, F.D. Carcinogens are mutagens: A simple test system combining liver homogenates for activation and bacteria for detection. Proc. Natl. Acad. Sci. USA 1973, 70, 2281–2285. [Google Scholar] [CrossRef]
- Raney, V.M.; Harris, T.M.; Stone, M.P. DNA conformation mediates aflatoxin B1-DNA binding and the formation of guanine N7 adducts by aflatoxin B1 8,9-exo-epoxide. Chem. Res. Toxicol. 1993, 6, 64–68. [Google Scholar] [CrossRef]
- Johnson, W.W.; Guengerich, F.P. Reaction of aflatoxin B1 exo-8,9-epoxide with DNA: Kinetic analysis of covalent binding and DNA-induced hydrolysis. Proc. Natl. Acad. Sci. USA 1997, 94, 6121–6125. [Google Scholar] [CrossRef]
- Bren, U.; Guengerich, F.P.; Mavri, J. Guanine alkylation by the potent carcinogen aflatoxin B1: Quantum chemical calculations. Chem. Res. Toxicol. 2007, 20, 1134–1140. [Google Scholar] [CrossRef]
- Brown, K.L.; Bren, U.; Stone, M.P.; Guengerich, F.P. Inherent stereospecificity in the reaction of aflatoxin B1 8,9-epoxide with deoxyguanosine and efficiency of DNA catalysis. Chem. Res. Toxicol. 2009, 22, 913–917. [Google Scholar] [CrossRef]
- Bhat, N.K.; Emeh, J.K.; Niranjan, B.G.; Avadhani, N.G. Inhibition of mitochondrial protein synthesis during early stages of aflatoxin B1-induced hepatocarcinogenesis. Cancer Res. 1982, 42, 1876–1880. [Google Scholar] [PubMed]
- Gopalakrishnan, S.; Byrd, S.; Stone, M.P.; Harris, T.M. Carcinogen-nucleic acid interactions: Equilibrium binding studies of aflatoxin B1 with the oligodeoxynucleotide d(ATGCAT)2 and with plasmid pBR322 support intercalative association with the B-DNA helix. Biochemistry 1989, 28, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, S.; Harris, T.M.; Stone, M.P. Intercalation of aflatoxin B1 in two oligodeoxynucleotide adducts: Comparative 1H NMR analysis of d(ATCAFBGAT)·d(ATCGAT) and d(ATAFBGCAT)2. Biochemistry 1990, 29, 10438–10448. [Google Scholar] [CrossRef]
- Johnston, D.S.; Stone, M.P. Refined solution structure of 8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1 opposite CpA in the complementary strand of an oligodeoxynucleotide duplex as determined by 1H NMR. Biochemistry 1995, 34, 14037–14050. [Google Scholar] [CrossRef]
- Mao, H.; Deng, Z.; Wang, F.; Harris, T.M.; Stone, M.P. An intercalated and thermally stable FAPY adduct of aflatoxin B1 in a DNA duplex: Structural refinement from 1H NMR. Biochemistry 1998, 37, 4374–4387. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, K.; Matsuno, T.; Adachi, K.; Hagihara, S.; Saito, I. Selective intercalation of charge neutral intercalators into GG and CG steps: Implication of HOMO-LUMO interaction for sequence-selective drug intercalation into DNA. J. Am. Chem. Soc. 2001, 123, 5695–5702. [Google Scholar] [CrossRef]
- Giri, I.; Stone, M.P. Thermal stabilization of the DNA duplex by adducts of aflatoxin B1. Biopolymers 2002, 65, 190–201. [Google Scholar] [CrossRef]
- Brown, K.L.; Voehler, M.W.; Magee, S.M.; Harris, C.M.; Harris, T.M.; Stone, M.P. Structural perturbations induced by the α-anomer of the aflatoxin B1 formamidopyrimidine adduct in duplex and single-strand DNA. J. Am. Chem. Soc. 2009, 131, 16096–16107. [Google Scholar] [CrossRef]
- Croy, R.G.; Essigmann, J.M.; Reinhold, V.N.; Wogan, G.N. Identification of the principal aflatoxin B1–DNA adduct formed in vivo in rat liver. Proc. Natl. Acad. Sci. USA 1978, 75, 1745–1749. [Google Scholar] [CrossRef]
- Lamm, G.; Pack, G.R. Acidic domains around nucleic acids. Proc. Natl. Acad. Sci. USA 1990, 87, 9033–9036. [Google Scholar] [CrossRef] [PubMed]
- Essigmann, J.M.; Croy, R.G.; Nadzan, A.M.; Busby, W.F., Jr.; Reinhold, V.N.; Büchi, G.; Wogan, G.N. Structural identification of the major DNA adduct formed by aflatoxin B1 in vitro. Proc. Natl. Acad. Sci. USA 1977, 74, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.K.; Miller, J.A.; Miller, E.C. 2,3-Dihydro-2-(guan-7-yl)-3-hydroxy-aflatoxin B1, a major acid hydrolysis product of aflatoxin B1–DNA or –ribosomal RNA adducts formed in hepatic microsome-mediated reactions and in rat liver in vivo. Cancer Res. 1977, 37, 4430–4438. [Google Scholar] [PubMed]
- Martin, C.N.; Garner, R.C. Aflatoxin B-oxide generated by chemical or enzymic oxidation of aflatoxin B1 causes guanine substitution in nucleic acids. Nature 1977, 267, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.A.; Essigmann, J.M.; Demain, A.L.; Skopek, T.R.; Wogan, G.N. Aflatoxin B1 mutagenesis, DNA binding, and adduct formation in Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 1979, 76, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Bailey, E.A.; Iyer, R.S.; Stone, M.P.; Harris, T.M.; Essigmann, J.M. Mutational properties of the primary aflatoxin B1–DNA adduct. Proc. Natl. Acad. Sci. USA 1996, 93, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Brown, K.L.; Ma, R.; Stone, M.P. DNA sequence modulates geometrical isomerism of the trans-8,9-dihydro-8-(2,6-diamino-4-oxo-3,4-dihydropyrimid-5-yl-formamido)-9-hydroxy aflatoxin B1 adduct. Chem. Res. Toxicol. 2015, 28, 225–237. [Google Scholar] [CrossRef]
- Chu, Y.H.; Saffhill, R. Errors in DNA synthesis induced by aflatoxin B1 modification of poly(dC-dG). Carcinogenesis 1983, 4, 643–646. [Google Scholar] [CrossRef]
- Foster, P.L.; Eisenstadt, E.; Miller, J.H. Base substitution mutations induced by metabolically activated aflatoxin B1. Proc. Natl. Acad. Sci. USA 1983, 80, 2695–2698. [Google Scholar] [CrossRef]
- Refolo, L.M.; Conley, M.P.; Sambamurti, K.; Jacobsen, J.S.; Humayun, M.Z. Sequence context effects in DNA replication blocks induced by aflatoxin B1. Proc. Natl. Acad. Sci. USA 1985, 82, 3096–3100. [Google Scholar] [CrossRef]
- Leadon, S.A.; Tyrrell, R.M.; Cerutti, P.A. Excision repair of aflatoxin B1–DNA adducts in human fibroblasts. Cancer Res. 1981, 41, 5125–5129. [Google Scholar] [PubMed]
- Chetsanga, C.J.; Frenette, G.P. Excision of aflatoxin B1–imidazole ring opened guanine adducts from DNA by formamidopyrimidine-DNA glycosylase. Carcinogenesis 1983, 4, 997–1000. [Google Scholar] [CrossRef]
- Smela, M.E.; Hamm, M.L.; Henderson, P.T.; Harris, C.M.; Thomas, H.M.; Essigmann, J.M. The aflatoxin B1 formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2002, 99, 6655–6660. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Deng, J.Z.; Iyer, R.S.; Iyer, L.G.; Voehler, M.W.; Stone, M.P.; Harris, C.M.; Harris, T.M. Unraveling the aflatoxin–FAPY conundrum: Structural basis for differential replicative processing of isomeric forms of the formamidopyrimidine-type DNA adduct of aflatoxin B1. J. Am. Chem. Soc. 2006, 128, 15188–15199. [Google Scholar] [CrossRef] [PubMed]
- Gabliks, J.; Schaeffer, W.; Friedman, L.; Wogan, G. Effect of aflatoxin B1 on cell cultures. J. Bacteriol. 1965, 90, 720–723. [Google Scholar] [PubMed]
- Legator, M. Biological effects of aflatoxin in cell culture. Bacteriol. Rev. 1966, 30, 471–477. [Google Scholar] [PubMed]
- Lillehoj, E.B.; Ciegler, A. Inhibition of deoxyribonucleic acid synthesis in Flavobacterium aurantiacum by aflatoxin B1. J. Bacteriol. 1967, 94, 787–788. [Google Scholar] [PubMed]
- Wragg, J.B.; Ross, V.C.; Legator, M.S. Effect of aflatoxin B1 on the deoxyribonucleic acid polymerase of Escherichia coli. Proc. Soc. Exp. Biol. Med. 1967, 125, 1052–1055. [Google Scholar] [CrossRef] [PubMed]
- Harley, E.H.; Rees, K.R.; Cohen, A. A comparative study of the effect of aflatoxin B1 and actinomycin D on HeLa cells. Biochem. J. 1969, 114, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Lafarge, C.; Frayssinet, C. The reversibility of inhibition of RNA and DNA synthesis induced by aflatoxin in rat liver. A tentative explanation for carcinogenic mechanism. Int. J. Cancer 1970, 6, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Maher, V.M.; Summers, W.C. Mutagenic action of aflatoxin B1 on transforming DNA and inhibition of DNA template activity in vitro. Nature 1970, 225, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Shieh, J.C.; Song, P.S. Photochemically induced binding of aflatoxins to DNA and its effects on template activity. Cancer Res. 1980, 40, 689–695. [Google Scholar] [PubMed]
- Johnston, D.S.; Stone, M.P. Replication of a site-specific trans-8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1 adduct by the exonuclease deficient Klenow fragment of DNA polymerase I. Chem. Res. Toxicol. 2000, 13, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Li, L.; Makarova, A.V.; Burgers, P.M.; Stone, M.P.; Lloyd, R.S. Molecular basis of aflatoxin-induced mutagenesis-role of the aflatoxin B1–formamidopyrimidine adduct. Carcinogenesis 2014, 35, 1461–1468. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Owen, N.; Minko, I.G.; Lange, S.S.; Li, L.; Stone, M.P.; Wood, R.D.; McCullough, A.K.; Lloyd, R.S. DNA polymerase ζ limits chromosomal damage and promotes cell survival following aflatoxin exposure. Proc. Natl. Acad. Sci. USA 2016, 113, 13774–13779. [Google Scholar] [CrossRef] [PubMed]
- Gelboin, H.V.; Wortham, J.S.; Wilson, R.G.; Friedman, M.; Wogan, G.N. Rapid and marked inhibition of rat-liver RNA polymerase by aflatoxin B1. Science 1966, 154, 1205–1206. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Dingman, C.W.; Phelps, H.L.; Wogan, G.N. Aflatoxin B1: Binding to DNA in vitro and alteration of RNA metabolism in vivo. Science 1966, 151, 1539–1541. [Google Scholar] [CrossRef]
- Clifford, J.I.; Rees, K.R. The action of aflatoxin B1 on the rat liver. Biochem. J. 1967, 102, 65–75. [Google Scholar] [CrossRef]
- Neal, G.E. The effect of aflatoxin B1 on normal and cortisol-stimulated rat liver ribonucleic acid synthesis. Biochem. J. 1972, 130, 619–629. [Google Scholar] [CrossRef]
- Bressac, B.; Kew, M.; Wands, J.; Ozturk, M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 1991, 350, 429–431. [Google Scholar] [CrossRef]
- Hsu, I.C.; Metcalf, R.A.; Sun, T.; Welsh, J.A.; Wang, N.J.; Harris, C.C. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 1991, 350, 427–428. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Hampton, L.L.; Luo, L.D.; Wirth, P.J.; Thorgeirsson, S.S. Low frequency of p53 gene mutation in tumors induced by aflatoxin B1 in nonhuman primates. Cancer Res. 1992, 52, 1044–1046. [Google Scholar] [PubMed]
- Aguilar, F.; Hussain, S.P.; Cerutti, P. Aflatoxin B1 induces the transversion of G→T in codon 249 of the p53 tumor suppressor gene in human hepatocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 8586–8590. [Google Scholar] [CrossRef] [PubMed]
- Cariello, N.F.; Cui, L.; Skopek, T.R. In vitro mutational spectrum of aflatoxin B1 in the human hypoxanthine guanine phosphoribosyltransferase gene. Cancer Res. 1994, 54, 4436–4441. [Google Scholar] [PubMed]
- Yang, M.; Zhou, H.; Kong, R.Y.; Fong, W.F.; Ren, L.Q.; Liao, X.H.; Wang, Y.; Zhuang, W.; Yang, S. Mutations at codon 249 of p53 gene in human hepatocellular carcinomas from Tongan, China. Mutat. Res. 1997, 381, 25–29. [Google Scholar] [CrossRef]
- Courtemanche, C.; Anderson, A. Multiple mutations in a shuttle vector modified by ultraviolet irradiation, (±)-7β,8α-dihydroxy-9α,10α-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene, and aflatoxin B1 have different properties than single mutations and may be generated during translesion synthesis. Mutat. Res. 1999, 430, 23–36. [Google Scholar] [PubMed]
- Denissenko, M.F.; Cahill, J.; Koudriakova, T.B.; Gerber, N.; Pfeifer, G.P. Quantitation and mapping of aflatoxin B1-induced DNA damage in genomic DNA using aflatoxin B1-8,9-epoxide and microsomal activation systems. Mutat. Res. 1999, 425, 205–211. [Google Scholar] [CrossRef]
- Pineau, P.; Marchio, A.; Battiston, C.; Cordina, E.; Russo, A.; Terris, B.; Qin, L.X.; Turlin, B.; Tang, Z.Y.; Mazzaferro, V.; et al. Chromosome instability in human hepatocellular carcinoma depends on p53 status and aflatoxin exposure. Mutat. Res. 2008, 653, 6–13. [Google Scholar] [CrossRef]
- Paget, V.; Lechevrel, M.; Andre, V.; Goff, J.L.; Pottier, D.; Billet, S.; Garçon, G.; Shirali, P.; Sichel, F. Benzo[a]pyrene, aflatoxin B1 and acetaldehyde mutational patterns in TP53 gene using a functional assay: Relevance to human cancer aetiology. PLoS ONE 2012, 7, e30921. [Google Scholar] [CrossRef]
- Chawanthayatham, S.; Valentine, C.C.; Fedeles, B.I.; Fox, E.J.; Loeb, L.A.; Levine, S.S.; Slocum, S.L.; Wogan, G.N.; Croy, R.G.; Essigmann, J.M. Mutational spectra of aflatoxin B1 in vivo establish biomarkers of exposure for human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, E3101–E3109. [Google Scholar] [CrossRef]
- Letouzé, E.; Shinde, J.; Renault, V.; Couchy, G.; Blanc, J.F.; Tubacher, E.; Bayard, Q.; Bacq, D.; Meyer, V.; Semhoun, J.; et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat. Commun. 2017, 8, 1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, M.W.; Lee, H.W.; Choi, B.; Wang, H.T.; Hu, Y.; Mehta, M.; Desai, D.; Amin, S.; Zheng, Y.; Tang, M.S. AFB1 hepatocarcinogenesis is via lipid peroxidation that inhibits DNA repair, sensitizes mutation susceptibility and induces aldehyde–DNA adducts at p53 mutational hotspot codon 249. Oncotarget 2017, 8, 18213–18226. [Google Scholar] [CrossRef] [PubMed]
- Sarasin, A.R.; Smith, C.A.; Hanawalt, P.C. Repair of DNA in human cells after treatment with activated aflatoxin B1. Cancer Res. 1977, 37, 1786–1793. [Google Scholar] [PubMed]
- Oleykowski, C.A.; Mayernik, J.A.; Lim, S.E.; Groopman, J.D.; Grossman, L.; Wogan, G.N.; Yeung, A.T. Repair of aflatoxin B1 DNA adducts by the UvrABC endonuclease of Escherichia coli. J. Biol. Chem. 1993, 268, 7990–8002. [Google Scholar] [PubMed]
- Alekseyev, Y.O.; Hamm, M.L.; Essigmann, J.M. Aflatoxin B1 formamidopyrimidine adducts are preferentially repaired by the nucleotide excision repair pathway in vivo. Carcinogenesis 2004, 25, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Bedard, L.L.; Alessi, M.; Davey, S.; Massey, T.E. Susceptibility to aflatoxin B1-induced carcinogenesis correlates with tissue-specific differences in DNA repair activity in mouse and in rat. Cancer Res. 2005, 65, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Breeden, L.L.; Zarbl, H.; Preston, B.D.; Eaton, D.L. Expression of a human cytochrome P450 in yeast permits analysis of pathways for response to and repair of aflatoxin-induced DNA damage. Mol. Cell. Biol. 2005, 25, 5823–5833. [Google Scholar] [CrossRef] [PubMed]
- Bedard, L.L.; Massey, T.E. Aflatoxin B1-induced DNA damage and its repair. Cancer Lett. 2006, 241, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Irvin, T.R.; Wogan, G.N. Quantitation of aflatoxin B1 adduction within the ribosomal RNA gene sequences of rat liver DNA. Proc. Natl. Acad. Sci. USA 1984, 81, 664–668. [Google Scholar] [CrossRef]
- Vartanian, V.; Minko, I.G.; Chawanthayatham, S.; Egner, P.A.; Lin, Y.C.; Earley, L.F.; Makar, R.; Eng, J.R.; Camp, M.T.; Li, L.; et al. NEIL1 protects against aflatoxin-induced hepatocellular carcinoma in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 4207–4212. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.M.; Shi, C.Y.; Lee, H.P.; Ong, C.N. Aflatoxin B1-induced lipid peroxidation in rat liver. Toxicol. Appl. Pharmacol. 1994, 127, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.M.; Ong, C.N.; Lee, B.L.; Shi, C.Y. Aflatoxin B1-induced 8-hydroxydeoxyguanosine formation in rat hepatic DNA. Carcinogenesis 1995, 16, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.C.; Jenkins, F.P.; Philp, J.M. Toxicity associated with certain samples of groundnuts. Nature 1961, 192, 1095–1096. [Google Scholar] [CrossRef]
- Butler, W.H.; Barnes, J.M. Toxic effects of groundnut meal containing aflatoxin to rats and guinea-pigs. Br. J. Cancer 1963, 17, 699–710. [Google Scholar] [CrossRef]
- Carnaghan, R.B.A.; Hartley, R.D.; O’Kelly, J. Toxicity and fluorescence properties of the aflatoxins. Nature 1963, 200, 1101. [Google Scholar] [CrossRef]
- Tulpule, P.G.; Madhavan, T.V.; Gopalan, C. Effect of feeding aflatoxin to young monkeys. Lancet 1964, 1, 962–963. [Google Scholar] [CrossRef]
- Judah, D.J.; Legg, R.F.; Neal, G.E. Development of resistance to cytotoxicity during aflatoxin carcinogenesis. Nature 1977, 265, 343–345. [Google Scholar] [CrossRef]
- Paini, A.; Scholz, G.; Marin-Kuan, M.; Schilter, B.; O’Brien, J.; van Bladeren, P.J.; Rietjens, I.M.C.M. Quantitative comparison between in vivo DNA adduct formation from exposure to selected DNA-reactive carcinogens, natural background levels of DNA adduct formation and tumour incidence in rodent bioassays. Mutagenesis 2011, 26, 605–618. [Google Scholar] [CrossRef] [Green Version]
- Barnes, J.; Butler, W.H. Carcinogenic activity of aflatoxin to rats. Nature 1964, 202, 1016. [Google Scholar] [CrossRef]
- Butler, W.H. Acute toxicity of aflatoxin B1 in rats. Br. J. Cancer 1964, 18, 756–762. [Google Scholar] [CrossRef]
- Carnaghan, R.B. Hepatic tumours and other chronic liver changes in rats following a single oral administration of aflatoxin. Br. J. Cancer 1967, 21, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Epstein, S.M.; Bartus, B.; Farber, E. Renal epithelial neoplasms induced in male Wistar rats by oral aflatoxin B1. Cancer Res. 1969, 29, 1045–1050. [Google Scholar] [PubMed]
- Alpert, M.E.; Hutt, M.S.; Wogan, G.N.; Davidson, C.S. Association between aflatoxin content of food and hepatoma frequency in Uganda. Cancer 1971, 28, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Lutwick, L.I. Relation between aflatoxin, hepatitis-B virus, and hepatocellular carcinoma. Lancet 1979, 313, 755–757. [Google Scholar] [CrossRef]
- Grosman, M.E.; Elías, M.M.; Comin, E.J.; Rodriguez Garay, E.A. Alterations in renal function induced by aflatoxin B1 in the rat. Toxicol. Appl. Pharmacol. 1983, 69, 319–325. [Google Scholar] [CrossRef]
- Wieder, R.; Wogan, G.N.; Shimkin, M.B. Pulmonary tumors in strain A mice given injections of aflatoxin B1. J. Natl. Cancer Inst. 1968, 40, 1195–1197. [Google Scholar] [PubMed]
- Paget, V.; Sichel, F.; Garon, D.; Lechevrel, M. Aflatoxin B1-induced TP53 mutational pattern in normal human cells using the FASAY (Functional Analysis of Separated Alleles in Yeast). Mutat. Res. 2008, 656, 55–61. [Google Scholar] [CrossRef]
- Pier, A.C.; Heddleston, K.L. The effect of aflatoxin on immunity in turkeys. I. Impairment of actively acquired resistance to bacterial challenge. Avian Dis. 1970, 14, 797–809. [Google Scholar] [CrossRef]
- Ikegwuonu, F.I. The neurotoxicity of aflatoxin B1 in the rat. Toxicology 1983, 28, 247–259. [Google Scholar] [CrossRef]
- DiPaolo, J.A.; Elis, J.; Erwin, H. Teratogenic response by hamsters, rats and mice to aflatoxin B1. Nature 1967, 215, 638–639. [Google Scholar] [CrossRef]
- Dickens, F.; Jones, H.E. The carcinogenic action of aflatoxin after its subcutaneous injection in the rat. Br. J. Cancer 1963, 17, 691–698. [Google Scholar] [CrossRef]
- Carnaghan, R.B.A. Hepatic tumours in ducks fed a low level of toxic groundnut meal. Nature 1965, 208, 308. [Google Scholar] [CrossRef] [PubMed]
- Callen, D.F.; Mohn, G.R.; Ong, T.M. Comparison of the genetic activity of aflatoxins B1 and G1 in Escherichia coli and Saccharomyces cerevisiae. Mutat. Res. 1977, 45, 7–11. [Google Scholar] [CrossRef]
- Aguilar, F.; Harris, C.C.; Sun, T.; Hollstein, M.; Cerutti, P. Geographic variation of p53 mutational profile in nonmalignant human liver. Science 1994, 264, 1317–1319. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, F. Global burden of aflatoxin-induced hepatocellular carcinoma: A risk assessment. Environ. Health Perspect. 2010, 118, 818–824. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; Rosvold, E.A.; Lustbader, E.D.; Hu, Y.; Clapper, M.L.; Zhou, T.; Wild, C.P.; Xia, X.L.; Baffoe-Bonnie, A.; Ofori-Adjei, D. Susceptibility to hepatocellular carcinoma is associated with genetic variation in the enzymatic detoxification of aflatoxin B1. Proc. Natl. Acad. Sci. USA 1995, 92, 2384–2387. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.C. Comparative aspects of aflatoxin-induced hepatic tumors. Cancer Res. 1968, 28, 2288–2292. [Google Scholar]
- Portman, R.S.; Plowman, K.M.; Campbell, T.C. Aflatoxin metabolism by liver microsomal preparations of two different species. Biochem. Biophys. Res. Commun. 1968, 33, 711–715. [Google Scholar] [CrossRef]
- Newberne, P.M.; Butler, W.H. Acute and chronic effects of aflatoxin on the liver of domestic and laboratory animals: A review. Cancer Res. 1969, 29, 236–250. [Google Scholar]
- Vologodskii, A.; Frank-Kamenetskii, M.D. DNA melting and energetics of the double helix. Phys. Life Rev. 2018, 25, 1–21. [Google Scholar] [CrossRef]
- Chandrasekaran, R.; Arnott, S. The structure of B-DNA in oriented fibers. J. Biomol. Struct. Dyn. 1996, 13, 1015–1027. [Google Scholar] [CrossRef]
- Lu, X.J.; Olson, W.K. 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucl. Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.J.; Olson, W.K. 3DNA: A versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008, 3, 1213–1227. [Google Scholar] [CrossRef] [PubMed]
- Marelius, J.; Kolmodin, K.; Feierberg, I.; Åqvist, J. Q: A molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 1998, 16, 213–225. [Google Scholar] [CrossRef]
- Ishikita, H.; Warshel, A. Predicting drug-resistant mutations of HIV protease. Angew. Chem. Int. Ed. Engl. 2008, 47, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.S.; Chu, Z.T.; Bolger, M.B.; Warshel, A. Calculations of antibody-antigen interactions: Microscopic and semi-microscopic evaluation of the free energies of binding of phosphorylcholine analogs to McPC603. Protein Eng. 1992, 5, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Åqvist, J.; Medina, C.; Samuelsson, J.E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Šponer, J.E.; Mládek, A.; Jurečka, P.; Banáš, P.; Otyepka, M. Nature and magnitude of aromatic base stacking in DNA and RNA: quantum chemistry, molecular mechanics, and experiment. Biopolymers 2013, 99, 978–988. [Google Scholar] [CrossRef]
- Singh, N.; Warshel, A. Absolute binding free energy calculations: On the accuracy of computational scoring of protein-ligand interactions. Proteins 2010, 78, 1705–1723. [Google Scholar] [CrossRef]
- Gutiérrez-de Terán, H.; Åqvist, J. Linear interaction energy: Method and applications in drug design. Methods Mol. Biol. 2012, 819, 305–323. [Google Scholar] [CrossRef]
- Sham, Y.Y.; Chu, Z.T.; Tao, H.; Warshel, A. Examining methods for calculations of binding free energies: LRA, LIE, PDLD-LRA, and PDLD/S-LRA calculations of ligands binding to an HIV protease. Proteins 2000, 39, 393–407. [Google Scholar] [CrossRef]
- Díaz, L.; Bujons, J.; Delgado, A.; Gutiérrez-de Terán, H.; Åqvist, J. Computational prediction of structure-activity relationships for the binding of aminocyclitols to β-glucocerebrosidase. J. Chem. Inf. Model. 2011, 51, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Hansson, T.; Marelius, J.; Åqvist, J. Ligand binding affinity prediction by linear interaction energy methods. J. Comput. Aided Mol. Des. 1998, 12, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florián, J.; Goodman, M.F.; Warshel, A. Free-energy perturbation calculations of DNA destabilization by base substitutions: The effect of neutral guanine·thymine, adenine·cytosine and adenine·difluorotoluene mismatches. J. Phys. Chem. B 2000, 104, 10092–10099. [Google Scholar] [CrossRef]
- Bren, U.; Lah, J.; Bren, M.; Martínek, V.; Florián, J. DNA duplex stability: The role of preorganized electrostatics. J. Phys. Chem. B 2010, 114, 2876–2885. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER; University of California: San Francisco, CA, USA, 2010. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Klvaňa, M.; Bren, U.; Florián, J. Uniform free-energy profiles of the P–O Bond formation and cleavage reactions catalyzed by DNA polymerases β and λ. J. Phys. Chem. B 2016, 120, 13017–13030. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific, LLC: Palo Alto, CA, USA, 2006. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef] [Green Version]
- Svoboda, D.L.; Taylor, J.S.; Hearst, J.E.; Sancar, A. DNA repair by eukaryotic nucleotide excision nuclease. Removal of thymine dimer and psoralen monoadduct by HeLa cell-free extract and of thymine dimer by Xenopus laevis oocytes. J. Biol. Chem. 1993, 268, 1931–1936. [Google Scholar] [PubMed]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| αEvdw1 | βEele1 | βEele2 | βEele | |
|---|---|---|---|---|
| dsDNA1g | −4.4 (0.0, 0.0, 0.1) | −26.7 (0.4, 0.1, 0.2) | 0.2 (0.1, 0.4, 0.8) | −26.5 (0.4, 0.4, 0.9) |
| dsDNA1a | −10.3 (0.1, 0.2, 0.4) | −50.6 (0.1, 2.1, 4.5) | −3.9 (0.6, 1.3, 3.1) | −54.4 (0.7, 2.5, 5.7) |
| dsDNA1b | −9.8 (0.0, 0.5, 1.0) | −50.6 (0.5, 2.5, 5.7) | −5.1 (0.6, 0.9, 1.9) | −55.8 (0.8, 3.0, 6.6) |
| ssDNA1g | −3.3 (0.2, 0.4, 0.8) | −24.3 (0.3, 0.5, 1.1) | 0.9 (0.3, 0.1, 0.2) | −23.4 (0.4, 0.5, 1.1) |
| ssDNA1a | −6.8 (0.6, 0.4, 0.8) | −51.5 (1.1, 0.9, 2.1) | −3.8 (0.3, 0.4, 0.8) | −55.3 (1.0, 1.2, 2.7) |
| ssDNA1b | −7.1 (0.3, 0.3, 0.6) | −48.8 (0.9, 0.4, 0.8) | −3.4 (0.3, 0.2, 0.6) | −52.2 (1.2, 0.4, 0.9) |
| dsDNA2g | −4.4 (0.0, 0.0, 0.0) | −26.8 (0.3, 0.2, 0.6) | 0.6 (1.3, 1.1, 2.7) | −26.1 (1.5, 1.1, 2.3) |
| dsDNA2a | −9.5 (0.1, 0.4, 0.7) | −51.8 (0.6, 1.6, 2.8) | −1.9 (1.3, 0.6, 1.3) | −53.7 (1.7, 1.7, 3.8) |
| dsDNA2b | −9.7 (0.2, 0.1, 0.3) | −50.2 (0.8, 0.8, 2.0) | −3.7 (0.7, 1.2, 2.5) | −53.9 (1.5, 1.3, 2.7) |
| ssDNA2g | −3.5 (0.3, 0.2, 0.4) | −23.8 (0.4, 0.2, 0.4) | 0.7 (0.1, 0.2, 0.5) | −23.1 (0.4, 0.1, 0.1) |
| ssDNA2a | −7.2 (0.4, 0.1, 0.4) | −49.9 (0.7, 2.0, 4.8) | −3.3 (0.9, 0.6, 1.4) | −53.2 (1.5, 2.5, 5.9) |
| ssDNA2b | −6.5 (0.4, 0.4, 0.9) | −49.2 (0.9, 0.9, 2.0) | −3.1 (0.1, 0.2, 0.4) | −52.3 (0.8, 1.0, 2.3) |
| dsDNAg | −4.4 (0.0, 0.0, 0.0) | −26.7 (0.2, 0.1, 0.3) | 0.4 (0.7, 0.5, 0.9) | −26.3 (0.6, 0.4, 0.9) |
| dsDNAa | −9.9 (0.1, 0.1, 0.2) | −51.2 (0.3, 1.8, 3.6) | −2.9 (0.6, 0.9, 1.9) | −54.1 (0.8, 2.0, 4.8) |
| dsDNAb | −9.8 (0.1, 0.2, 0.5) | −50.4 (0.7, 1.4, 3.2) | −4.4 (0.6, 0.9, 2.2) | −54.8 (1.1, 1.4, 3.4) |
| ssDNAg | −3.4 (0.2, 0.3, 0.6) | −24.0 (0.2, 0.3, 0.7) | 0.8 (0.2, 0.1, 0.3) | −23.2 (0.4, 0.3, 0.5) |
| ssDNAa | −7.0 (0.3, 0.2, 0.5) | −50.7 (0.8, 0.8, 1.7) | −3.6 (0.5, 0.4, 0.8) | −54.2 (1.1, 1.1, 2.4) |
| ssDNAb | −6.8 (0.3, 0.3, 0.6) | −49.0 (0.8, 0.5, 1.2) | −3.2 (0.2, 0.1, 0.2) | −52.2 (0.9, 0.4, 1.1) |
| ΔΔGvdw1 | ΔΔGele1 | ΔΔGele2 | ΔΔGele | ΔΔG | |
|---|---|---|---|---|---|
| DNA1a | −2.3 (0.6, 0.5, 1.3) | 3.3 (0.9, 3.0, 6.8) | 0.7 (0.3, 1.7, 4.2) | 4.0 (1.1, 4.0, 8.7) | 1.7 (1.3, 3.6, 7.7) |
| DNA1b | −1.6 (0.4, 0.6, 1.4) | 0.6 (1.1, 2.5, 5.3) | −1.0 (0.9, 1.0, 2.2) | −0.4 (1.5, 2.8, 6.2) | −2.0 (1.6, 2.3, 5.3) |
| DNA2a | −1.5 (0.5, 0.6, 1.3) | 1.0 (1.4, 1.2, 2.5) | 1.6 (1.5, 2.3, 5.3) | 2.6 (2.3, 3.0, 7.3) | 1.1 (2.8, 3.2, 7.8) |
| DNA2b | −2.3 (0.6, 0.3, 0.7) | 1.9 (1.8, 1.3, 3.1) | −0.5 (1.6, 2.2, 4.6) | 1.4 (3.0, 2.8, 6.7) | −0.9 (2.5, 2.5, 6.0) |
| DNAa | −1.9 (0.4, 0.6, 1.3) | 2.2 (1.1, 1.6, 3.7) | 1.1 (0.7, 1.6, 3.1) | 3.3 (1.6, 2.6, 6.2) | 1.4 (1.9, 2.4, 5.2) |
| DNAb | −1.9 (0.5, 0.4, 0.9) | 1.2 (1.4, 1.5, 3.6) | −0.7 (1.1, 1.3, 3.1) | 0.5 (1.9, 1.7, 4.0) | −1.4 (1.4, 1.4, 3.1) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klvana, M.; Bren, U. Aflatoxin B1–Formamidopyrimidine DNA Adducts: Relationships between Structures, Free Energies, and Melting Temperatures. Molecules 2019, 24, 150. https://doi.org/10.3390/molecules24010150
Klvana M, Bren U. Aflatoxin B1–Formamidopyrimidine DNA Adducts: Relationships between Structures, Free Energies, and Melting Temperatures. Molecules. 2019; 24(1):150. https://doi.org/10.3390/molecules24010150
Chicago/Turabian StyleKlvana, Martin, and Urban Bren. 2019. "Aflatoxin B1–Formamidopyrimidine DNA Adducts: Relationships between Structures, Free Energies, and Melting Temperatures" Molecules 24, no. 1: 150. https://doi.org/10.3390/molecules24010150
APA StyleKlvana, M., & Bren, U. (2019). Aflatoxin B1–Formamidopyrimidine DNA Adducts: Relationships between Structures, Free Energies, and Melting Temperatures. Molecules, 24(1), 150. https://doi.org/10.3390/molecules24010150