Biosynthetic Gene Content of the ‘Perfume Lichens’ Evernia prunastri and Pseudevernia furfuracea


Abstract
:1. Introduction
- (I)
- What is the diversity of biosynthetic gene clusters in Evernia prunastri and Pseudevernia furfuracea and how does it compare to other lichenized fungi and non-lichenized fungi?
- (II)
- What is the architecture and gene content of those clusters with high homology between E. prunastri and P. furfuracea?
- (III)
- Where do PKSs from E. prunastri and P. furfuracea group phylogenetically in a phylogeny of PKSs with known functions?
2. Results & Discussion
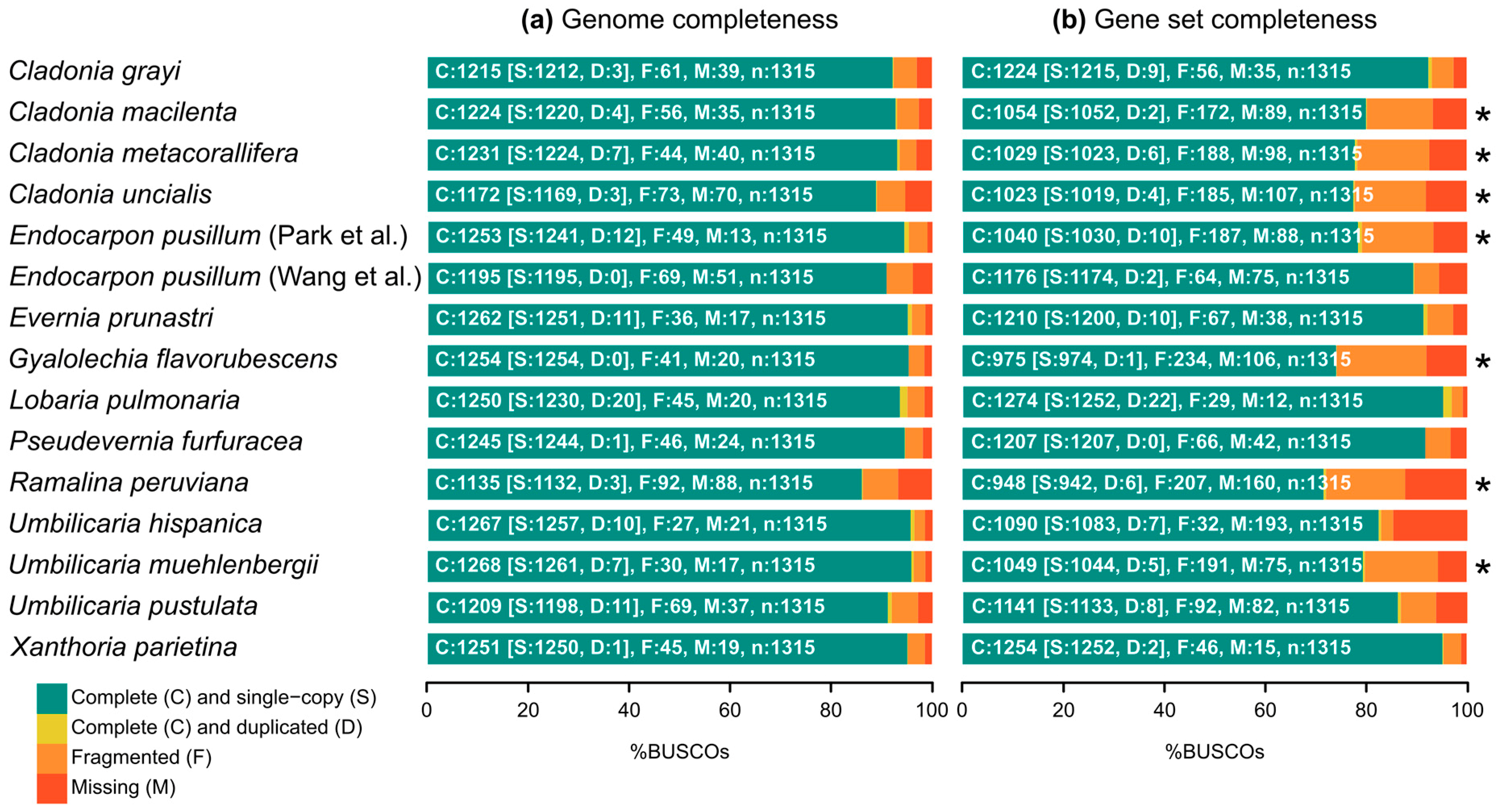
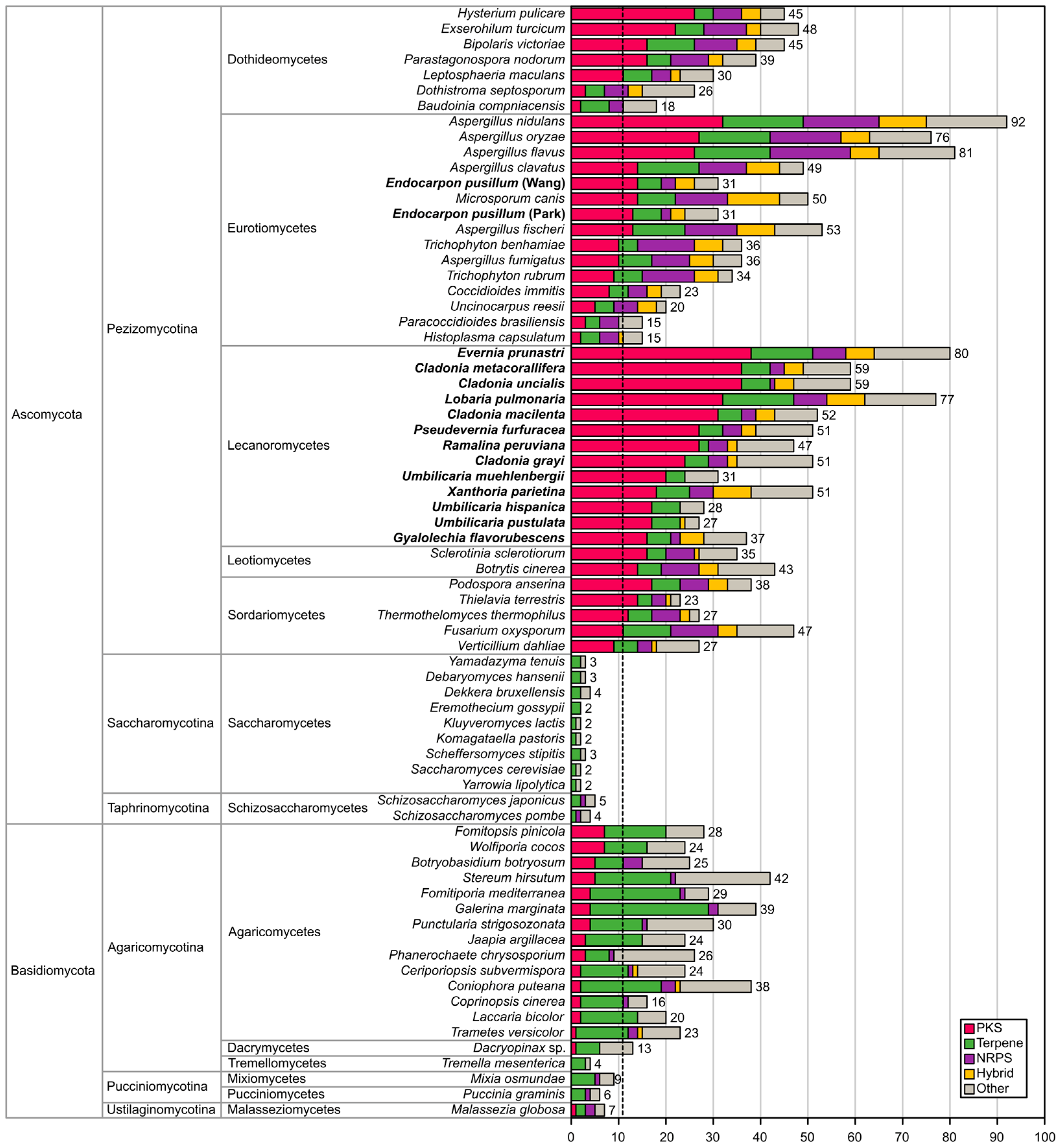
2.1. Biosynthetic Gene Richness in Fifteen Annotated Genomes of Lichen-Forming Fungi
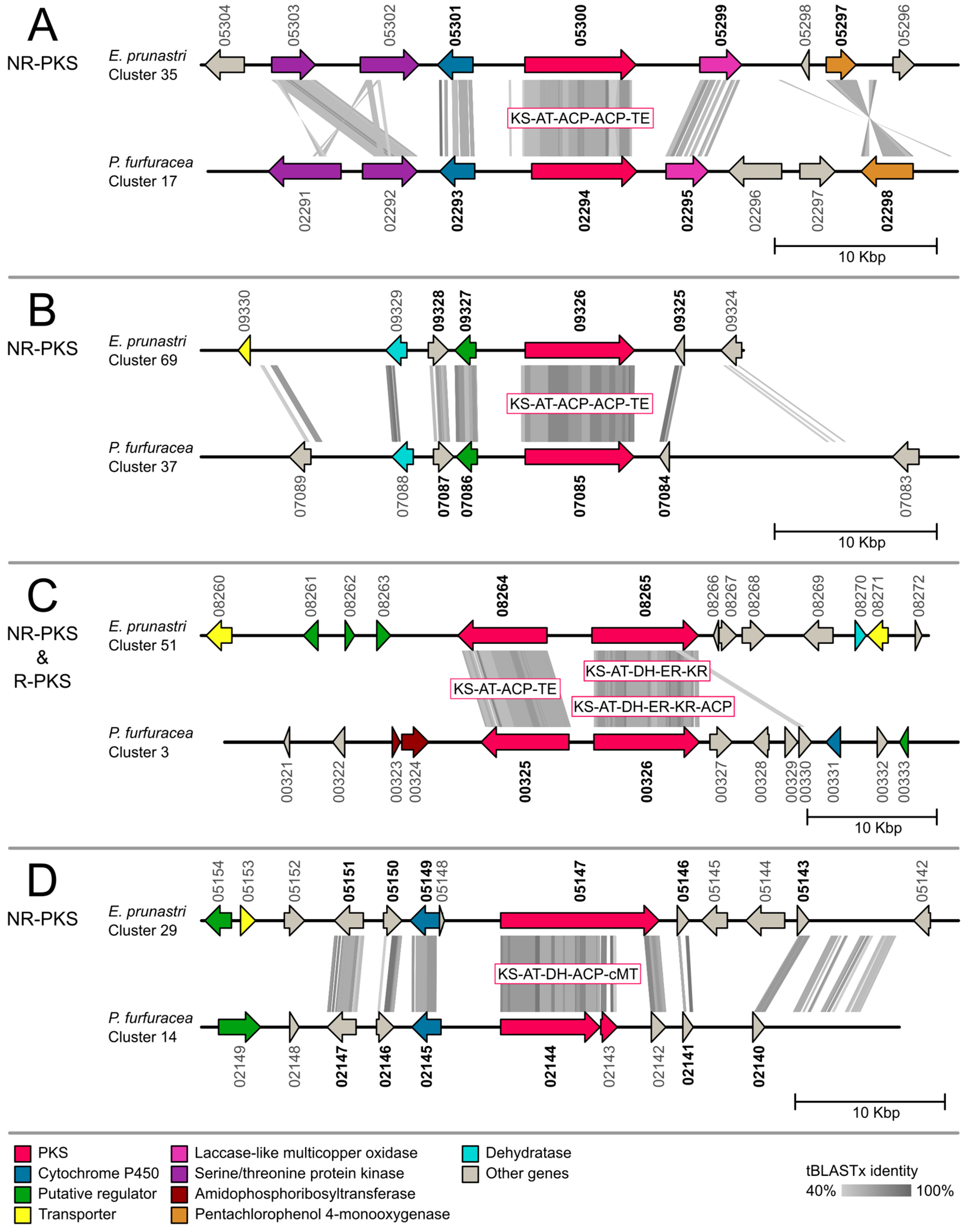
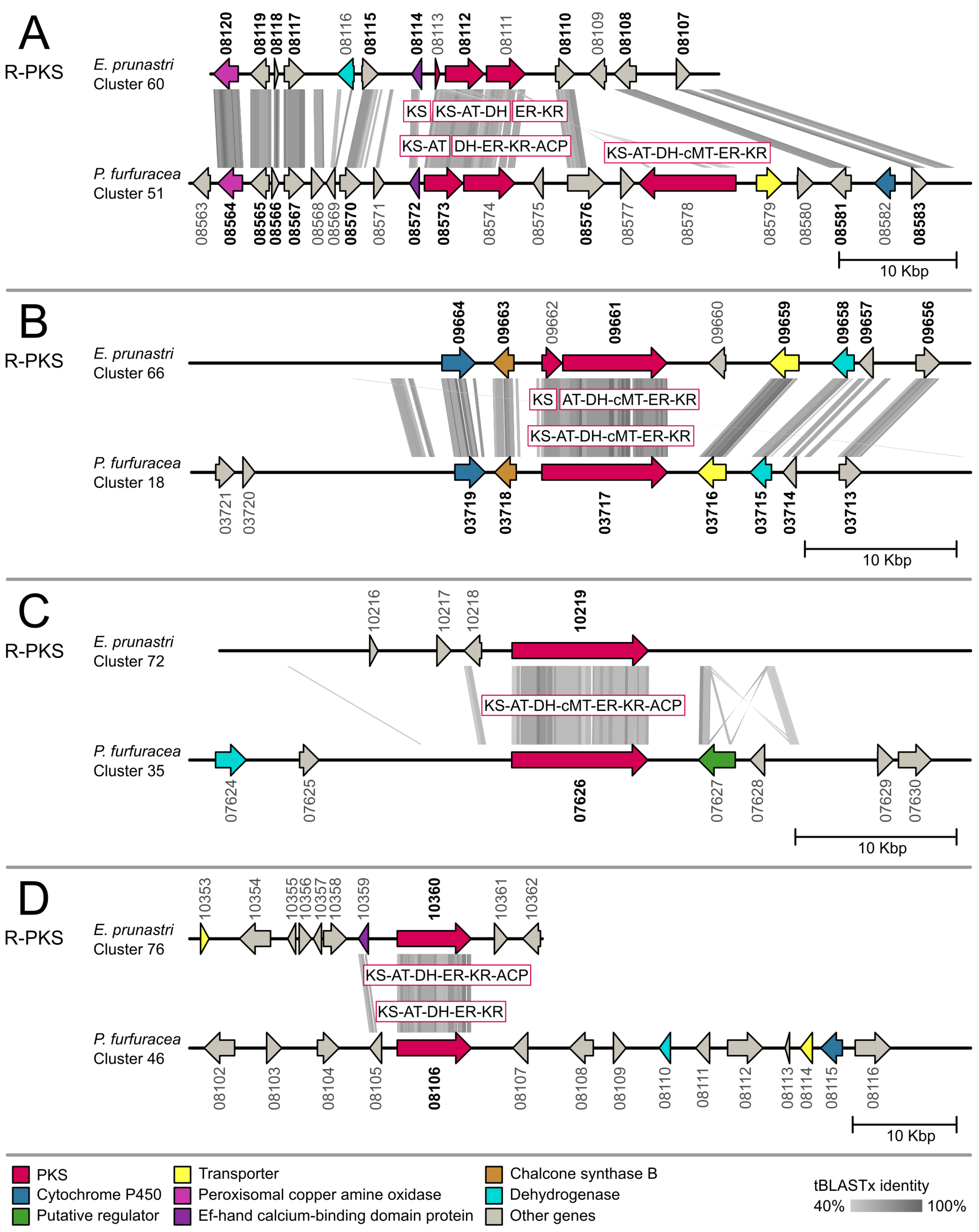
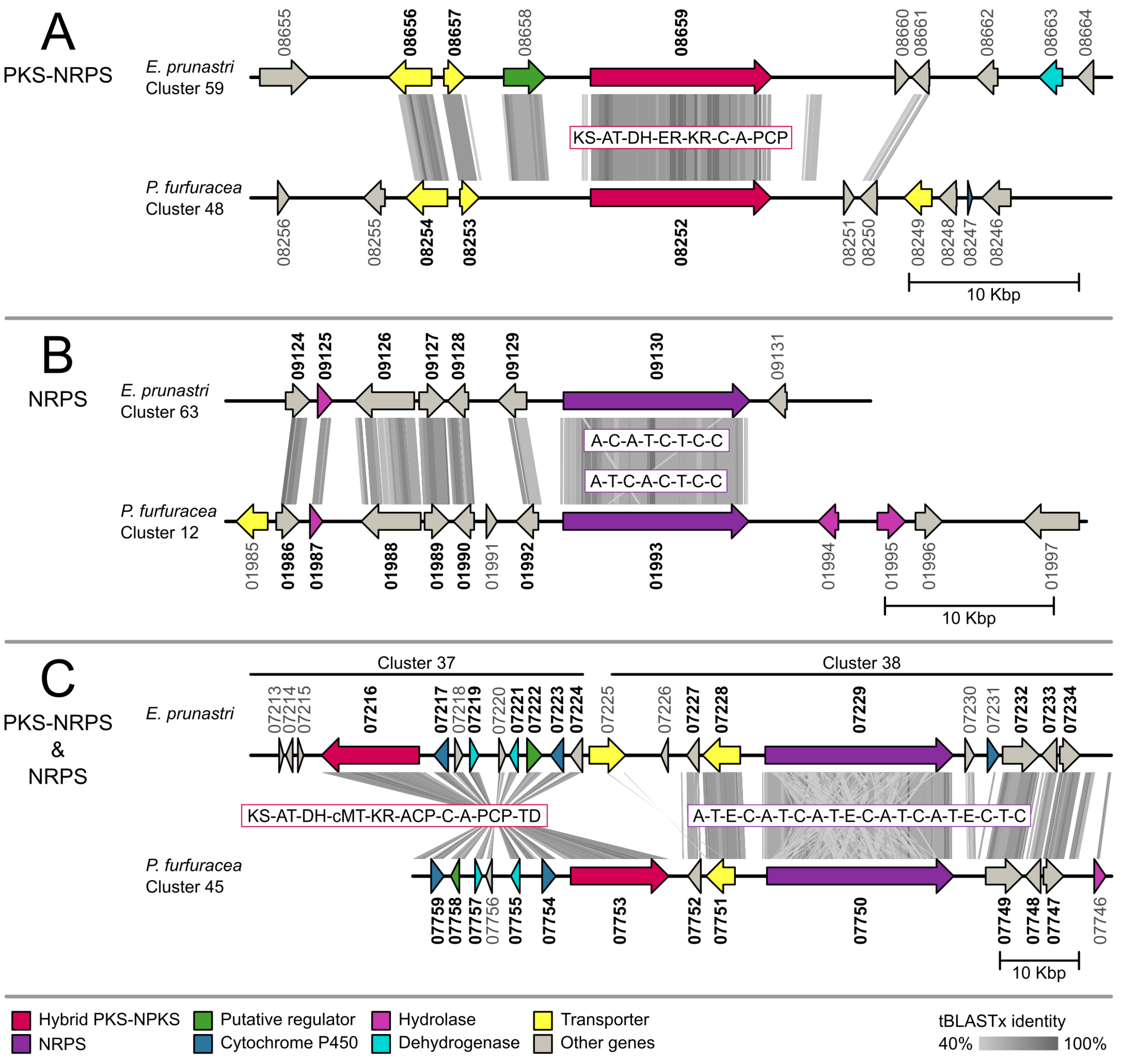
2.2. Gene Cluster Comparison
2.3. Putative Regulators of Biosynthetic Gene Clusters
2.4. Phylogenetic Analysis with Characterized Fungal Polyketides
3. Materials and Methods
3.1. Identification and Annotations of Biosynthetic Gene Clusters
3.2. Gene Cluster Comparison in Evernia prunastri and Pseudevernia furfuracea
3.3. Putative Regulators of Biosynthetic Gene Clusters
3.4. Biosynthetic Gene Richness in Other Genomes of Lichenized and Non-Lichenized Fungi
3.5. Phylogenetic Analysis with Characterized Fungal Polyketides
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nash, T.H. Lichen Biology, 2nd ed.; Nash, T.H., Ed.; Cambridge University Press: Cambridge, UK, 2008; ISBN 9780521871624. [Google Scholar]
- Lücking, R.; Hodkinson, B.P.; Leavitt, S.D. The 2016 classification of lichenized fungi in the Ascomycota and Basidiomycota—Approaching one thousand genera. Bryologist 2016, 119, 361–416. [Google Scholar] [CrossRef]
- Asplund, J.; Wardle, D.A. How lichens impact on terrestrial community and ecosystem properties. Biol. Rev. 2017, 92, 1720–1738. [Google Scholar] [CrossRef] [PubMed]
- Seaward, M.R.D. Environmental role of lichens. In Lichen Biology; Nash, T.H., Ed.; Cambridge University Press: Cambridge, UK, 2008; pp. 274–298. ISBN 9780521871624. [Google Scholar]
- Grube, M.; Wedin, M. Lichenized fungi and the evolution of symbiotic organization. Microbiol. Spectr. 2016, 4, 1–17. [Google Scholar] [CrossRef]
- Cernava, T.; Berg, G.; Grube, M. High life expectancy of bacteria on lichens. Microb. Ecol. 2016, 72, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, I.A.; Cernava, T.; Berg, G.; Grube, M. Understanding microbial multi-species symbioses. Front. Microbiol. 2016, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Spribille, T.; Tuovinen, V.; Resl, P.; Vanderpool, D.; Wolinski, H.; Aime, M.C.; Schneider, K.; Stabentheiner, E.; Toome-Heller, M.; Thor, G.; et al. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science 2016, 353, 488–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Mendoza, F.; Fleischhacker, A.; Kopun, T.; Grube, M.; Muggia, L. ITS1 metabarcoding highlights low specificity of lichen mycobiomes at a local scale. Mol. Ecol. 2017, 26, 4811–4830. [Google Scholar] [CrossRef] [PubMed]
- Dal Grande, F.; Rolshausen, G.; Divakar, P.K.; Crespo, A.; Otte, J.; Schleuning, M.; Schmitt, I. Environment and host identity structure communities of green algal symbionts in lichens. New Phytol. 2018, 217, 277–289. [Google Scholar] [CrossRef]
- Machado, H.; Tuttle, R.N.; Jensen, P.R. Omics-based natural product discovery and the lexicon of genome mining. Curr. Opin. Microbiol. 2017, 39, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Cernava, T.; Erlacher, A.; Aschenbrenner, I.A.; Krug, L.; Lassek, C.; Riedel, K.; Grube, M.; Berg, G. Deciphering functional diversification within the lichen microbiota by meta-omics. Microbiome 2017, 5, 82. [Google Scholar] [CrossRef]
- Moya, P.; Molins, A.; Martínez-Alberola, F.; Muggia, L.; Barreno, E. Unexpected associated microalgal diversity in the lichen Ramalina farinacea is uncovered by pyrosequencing analyses. PLoS ONE 2017, 12, e0175091. [Google Scholar] [CrossRef] [PubMed]
- Stocker-Wörgötter, E. Metabolic diversity of lichen-forming ascomycetous fungi: Culturing, polyketide and shikimatemetabolite production, and PKS genes. Nat. Prod. Rep. Prod. Rep. 2008, 25, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Molnár, K.; Farkas, E. Current results on biological activities of lichen secondary metabolites: A review. Z. Naturforsch. C 2010, 65, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Elix, J.A.; Stocker-Wörgötter, E. Biochemistry and secondary metabolites. In Lichen Biology; Nash, T.H., Ed.; Cambridge University Press: Cambridge, UK, 2008; pp. 104–133. ISBN 9780521871624. [Google Scholar]
- Huneck, S.; Yoshimura, I. Identification of Lichen Substances; Springer: Berlin/Heidelberg, Germany, 1996; ISBN 978-3-642-85245-9. [Google Scholar]
- Rundel, P.W. The ecological role of secondary lichen substances. Biochem. Syst. Ecol. 1978, 6, 157–170. [Google Scholar] [CrossRef]
- Huneck, S. The significance of lichens and their metabolites. Naturwissenschaften 1999, 86, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Lawrey, J.D. Biological role of lichen substances. Bryologist 1986, 89, 111. [Google Scholar] [CrossRef]
- Ranković, B.; Kosanić, M. Lichen Secondary Metabolites; Ranković, B., Ed.; Springer: Cham, Switzerland, 2015; ISBN 978-3-319-13373-7. [Google Scholar]
- Boustie, J.; Grube, M. Lichens—A promising source of bioactive secondary metabolites. Plant Genet. Resour. Charact. Util. 2005, 3, 273–287. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, Y.; Park, S.-Y.; Nguyen, T.T.; Seo, Y.-W.; Lee, K.H.; Lee, J.H.; Kim, K.K.; Hur, J.-S.; Kim, H. The lichen secondary metabolite atranorin suppresses lung cancer cell motility and tumorigenesis. Sci. Rep. 2017, 7, 8136. [Google Scholar] [CrossRef]
- Crawford, S.D. Lichens used in traditional medicine. In Lichen Secondary Metabolites; Springer: Cham, Switzerland, 2015; pp. 27–80. ISBN 9783319133744. [Google Scholar]
- Joulain, D.; Tabacchi, R. Lichen extracts as raw materials in perfumery. Part 1: Oakmoss. Flavour Fragr. J. 2009, 24, 49–61. [Google Scholar] [CrossRef]
- Joulain, D.; Tabacchi, R. Lichen extracts as raw materials in perfumery. Part 2: Treemoss. Flavour Fragr. J. 2009, 24, 105–116. [Google Scholar] [CrossRef]
- Calcott, M.J.; Ackerley, D.F.; Knight, A.; Keyzers, R.A.; Owen, J.G. Secondary metabolism in the lichen symbiosis. Chem. Soc. Rev. 2018, 47, 1730–1760. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.M.; Townsend, C.A. New insights into the formation of fungal aromatic polyketides. Nat. Rev. Microbiol. 2010, 8, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, I.; Martín, M.P.; Kautz, S.; Lumbsch, H.T. Diversity of non-reducing polyketide synthase genes in the Pertusariales (lichenized Ascomycota): A phylogenetic perspective. Phytochemistry 2005, 66, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P.; Turner, G.; Bennett, J.W. Fungal secondary metabolism—From biochemistry to genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.J.; Skellam, E.; Williams, K. Biosynthesis of fungal polyketides. In Physiology and Genetics; Springer: Cham, Switzerland, 2018; pp. 385–412. [Google Scholar]
- Rokas, A.; Wisecaver, J.H.; Lind, A.L. The birth, evolution and death of metabolic gene clusters in fungi. Nat. Rev. Microbiol. 2018, 16, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Cimermancic, P.; Medema, M.H.; Claesen, J.; Kurita, K.; Wieland Brown, L.C.; Mavrommatis, K.; Pati, A.; Godfrey, P.A.; Koehrsen, M.; Clardy, J.; et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 2014, 158, 412–421. [Google Scholar] [CrossRef]
- Doroghazi, J.R.; Albright, J.C.; Goering, A.W.; Ju, K.-S.; Haines, R.R.; Tchalukov, K.A.; Labeda, D.P.; Kelleher, N.L.; Metcalf, W.W. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol. 2014, 10, 963–968. [Google Scholar] [CrossRef]
- Zhao, H.; Medema, M.H. Standardization for natural product synthetic biology. Nat. Prod. Rep. 2016, 33, 920–924. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, J.C.; Grijseels, S.; Prigent, S.; Ji, B.; Dainat, J.; Nielsen, K.F.; Frisvad, J.C.; Workman, M.; Nielsen, J. Global analysis of biosynthetic gene clusters reveals vast potential of secondary metabolite production in Penicillium species. Nat. Microbiol. 2017, 2, 17044. [Google Scholar] [CrossRef]
- Brakhage, A.A. Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 2013, 11, 21–32. [Google Scholar] [CrossRef]
- Schümann, J.; Hertweck, C. Advances in cloning, functional analysis and heterologous expression of fungal polyketide synthase genes. J. Biotechnol. 2006, 124, 690–703. [Google Scholar] [CrossRef]
- Hertweck, C. Hidden biosynthetic treasures brought to light. Nat. Chem. Biol. 2009, 5, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, S.; Schümann, J.; Scherlach, K.; Lange, C.; Brakhage, A.A.; Hertweck, C. Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans. Nat. Chem. Biol. 2007, 3, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hameed, M.; Bertrand, R.L.; Piercey-Normore, M.D.; Sorensen, J.L. Putative identification of the usnic acid biosynthetic gene cluster by de novo whole-genome sequencing of a lichen-forming fungus. Fungal Biol. 2016, 120, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Muggia, L.; Schmitt, I.; Grube, M. Lichens as treasure chests of natural products. Sim News 2009, 59, 85–97. [Google Scholar]
- Armaleo, D.; Sun, X.; Culberson, C. Insights from the first putative biosynthetic gene cluster for a lichen depside and depsidone. Mycologia 2011, 103, 741–754. [Google Scholar] [CrossRef]
- Abdel-Hameed, M.; Bertrand, R.L.; Piercey-Normore, M.D.; Sorensen, J.L. Identification of 6-hydroxymellein synthase and accessory genes in the lichen Cladonia uncialis. J. Nat. Prod. 2016, 79, 1645–1650. [Google Scholar] [CrossRef]
- Bertrand, R.L.; Sorensen, J.L. A comprehensive catalogue of polyketide synthase gene clusters in lichenizing fungi. J. Ind. Microbiol. Biotechnol. 2018, 45, 1067–1081. [Google Scholar] [CrossRef]
- Meiser, A.; Otte, J.; Schmitt, I.; Dal Grande, F. Sequencing genomes from mixed DNA samples—Evaluating the metagenome skimming approach in lichenized fungi. Sci. Rep. 2017, 7, 14881. [Google Scholar] [CrossRef]
- Wang, Y.; Geng, C.; Yuan, X.; Hua, M.; Tian, F.; Li, C. Identification of a putative polyketide synthase gene involved in usnic acid biosynthesis in the lichen Nephromopsis pallescens. PLoS ONE 2018, 13, e0199110. [Google Scholar] [CrossRef]
- Taguchi, H.; Sankawa, U.; Shibata, S. Biosynthesis of natural products. VI. Biosynthesis of usnic acid in lichens. (1). A general scheme of biosynthesis of usnic acid. Chem. Pharm. Bull. (Tokyo) 1969, 17, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Barton, D.H.R.; Deflorin, A.M.; Edwards, O.E. 108. The synthesis of usnic acid. J. Chem. Soc. 1956, 108, 530–534. [Google Scholar] [CrossRef]
- Gagunashvili, A.N.; Davíðsson, S.P.; Jónsson, Z.O.; Andrésson, Ó.S. Cloning and heterologous transcription of a polyketide synthase gene from the lichen Solorina crocea. Mycol. Res. 2009, 113, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Chooi, Y.-H.; Stalker, D.M.; Davis, M.A.; Fujii, I.; Elix, J.A.; Louwhoff, S.H.J.J.; Lawrie, A.C. Cloning and sequence characterization of a non-reducing polyketide synthase gene from the lichen Xanthoparmelia semiviridis. Mycol. Res. 2008, 112, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Jensen, P.R. Phylogenetic approaches to natural product structure prediction. In Methods in Enzymology; Elsevier Inc.: San Diego, CA, USA, 2012; Volume 517, pp. 161–182. ISBN 9780124046344. [Google Scholar]
- Wang, Y.; Wang, J.; Cheong, Y.H.; Hur, J.-S. Three new non-reducing polyketide synthase genes from the lichen-forming fungus Usnea longissima. Mycobiology 2014, 42, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, R.L.; Abdel-Hameed, M.; Sorensen, J.L. Lichen biosynthetic gene clusters part II: Homology mapping suggests a functional diversity. J. Nat. Prod. 2018, 81, 732–748. [Google Scholar] [CrossRef] [PubMed]
- Purvis, O.W.; Coppins, B.J.; Hawksworth, D.L.; James, P.W.; Moore, D.M. (Eds.) The Lichen Flora of Great Britain and Ireland; Natural History Museum Publications in association with The British Lichen Society: London, UK, 1992; ISBN 0952304902. [Google Scholar]
- Brodo, I.M.; Sharnoff, S.D.; Sharnoff, S. (Eds.) Lichens of North America; Yale University Press: New Haven, CO, USA; London, UK, 2001; ISBN 0-300-08249-5. [Google Scholar]
- Stenroos, S.; Velmala, S.; Pykälä, J.; Ahti, T. (Eds.) Lichens of Finland; Finnish Museum of Natural History LUMOS, University of Helsinki: Helsinki, Finland, 2016; ISBN 978-951-51-2266-7. [Google Scholar]
- Posner, B.; Feige, G.B.; Leuckert, C. Beiträge zur Chemie der Flechtengattung Lasallia Mérat/On the Chemistry of the lichen genus Lasallia Merat. Z. Naturforsch. C 1991, 46, 19–27. [Google Scholar] [CrossRef]
- Dal Grande, F.; Meiser, A.; Greshake Tzovaras, B.; Otte, J.; Ebersberger, I.; Schmitt, I. The draft genome of the lichen-forming fungus Lasallia hispanica (Frey) Sancho & A. Crespo. Lichenologist 2018, 50, 329–340. [Google Scholar] [CrossRef]
- Park, S.-Y.; Choi, J.; Lee, G.-W.; Park, C.-H.; Kim, J.A.; Oh, S.-O.; Lee, Y.-H.; Hur, J.-S. Draft Genome Sequence of Endocarpon pusillum Strain KoLRILF000583. Genome Announc. 2014, 2, e00452-14. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Liu, B.; Zhang, X.-Y.; Zhou, Q.-M.; Zhang, T.; Li, H.; Yu, Y.-F.; Zhang, X.-L.; Hao, X.-Y.; Wang, M.; et al. Genome characteristics reveal the impact of lichenization on lichen-forming fungus Endocarpon pusillum Hedwig (Verrucariales, Ascomycota). BMC Genom. 2014, 15, 34. [Google Scholar] [CrossRef]
- Blin, K.; Kim, H.U.; Medema, M.H.; Weber, T. Recent development of antiSMASH and other computational approaches to mine secondary metabolite biosynthetic gene clusters. Brief. Bioinform. 2017, bbx146, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stocker-Wörgötter, E. Biochemical diversity and ecology of lichen-forming fungi: Lichen substances, chemosyndromic variation and origin of polyketide-type metabolites (biosynthetic pathways). In Recent Advances in Lichenology; Springer: New Delhi, India, 2015; pp. 161–179. ISBN 9788132222354. [Google Scholar]
- Wang, X.; Zhang, X.; Liu, L.; Xiang, M.; Wang, W.; Sun, X.; Che, Y.; Guo, L.; Liu, G.; Guo, L.; et al. Genomic and transcriptomic analysis of the endophytic fungus Pestalotiopsis fici reveals its lifestyle and high potential for synthesis of natural products. BMC Genom. 2015, 16, 28. [Google Scholar] [CrossRef]
- Crawford, J.M.; Dancy, B.C.R.; Hill, E.A.; Udwary, D.W.; Townsend, C.A. Identification of a starter unit acyl-carrier protein transacylase domain in an iterative type I polyketide synthase. Proc. Natl. Acad. Sci. USA 2006, 103, 16728–16733. [Google Scholar] [CrossRef]
- Crawford, J.M.; Thomas, P.M.; Scheerer, J.R.; Vagstad, A.L.; Kelleher, N.L.; Townsend, C.A. Deconstruction of iterative multidomain polyketide synthase function. Science 2008, 320, 243–246. [Google Scholar] [CrossRef]
- Crawford, J.M.; Korman, T.P.; Labonte, J.W.; Vagstad, A.L.; Hill, E.A.; Kamari-Bidkorpeh, O.; Tsai, S.-C.; Townsend, C.A. Structural basis for biosynthetic programming of fungal aromatic polyketide cyclization. Nature 2009, 461, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Cacho, R.A.; Tang, Y.; Chooi, Y.-H. Next-generation sequencing approach for connecting secondary metabolites to biosynthetic gene clusters in fungi. Front. Microbiol. 2015, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Knox, B.P.; Keller, N.P. Key players in the regulation of fungal secondary metabolism. In Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites, Volume 2; Zeilinger, S., Martín, J.-F., García-Estrada, C., Eds.; Springer: New York, NY, USA, 2015; Volume 2, pp. 13–28. ISBN 978-1-4939-2530-8. [Google Scholar]
- Kroken, S.; Glass, N.L.; Taylor, J.W.; Yoder, O.C.; Turgeon, B.G. Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc. Natl. Acad. Sci. USA 2003, 100, 15670–15675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenke-Kodama, H.; Sandmann, A.; Müller, R.; Dittmann, E. Evolutionary implications of bacterial polyketide synthases. Mol. Biol. Evol. 2005, 22, 2027–2039. [Google Scholar] [CrossRef]
- Li, Y.F.; Tsai, K.J.S.; Harvey, C.J.B.; Li, J.J.; Ary, B.E.; Berlew, E.E.; Boehman, B.L.; Findley, D.M.; Friant, A.G.; Gardner, C.A.; et al. Comprehensive curation and analysis of fungal biosynthetic gene clusters of published natural products. Fungal Genet. Biol. 2016, 89, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.-H.; Chollet-Krugler, M.; Gouault, N.; Tomasi, S. UV-protectant metabolites from lichens and their symbiotic partners. Nat. Prod. Rep. 2013, 30, 1490. [Google Scholar] [CrossRef]
- Fulton, T.R.; Ibrahim, N.; Losada, M.C.; Grzegorski, D.; Tkacz, J.S. A melanin polyketide synthase (PKS) gene from Nodulisporium sp. that shows homology to the pks1 gene of Colletotrichum lagenarium. Mol. Gen. Genet. 1999, 262, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Lu, P.; Dahl-Roshak, A.M.; Paress, P.S.; Kennedy, S.; Tkacz, J.S.; An, Z. Efficient disruption of a polyketide synthase gene (pks1) required for melanin synthesis through Agrobacterium-mediated transformation of Glarea lozoyensis. Mol. Genet. Genom. 2003, 268, 645–655. [Google Scholar] [CrossRef]
- Sanchez, J.F.; Chiang, Y.-M.; Szewczyk, E.; Davidson, A.D.; Ahuja, M.; Elizabeth Oakley, C.; Woo Bok, J.; Keller, N.; Oakley, B.R.; Wang, C.C.C. Molecular genetic analysis of the orsellinic acid/F9775 genecluster of Aspergillus nidulans. Mol. Biosyst. 2010, 6, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Wolf, T.; Chevrette, M.G.; Lu, X.; Schwalen, C.J.; Kautsar, S.A.; Suarez Duran, H.G.; de los Santos, E.L.C.; Kim, H.U.; Nave, M.; et al. antiSMASH 4.0—Improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017, 45, W36–W41. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2017, 45, D37–D42. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Ward, N.; Moreno-Hagelsieb, G. Quickly finding orthologs as reciprocal best hits with BLAT, LAST, and UBLAST: How much do we miss? PLoS ONE 2014, 9, e101850. [Google Scholar] [CrossRef]
- Moreno-Hagelsieb, G.; Latimer, K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 2008, 24, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Kjærbølling, I.; Vesth, T.C.; Frisvad, J.C.; Nybo, J.L.; Theobald, S.; Kuo, A.; Bowyer, P.; Matsuda, Y.; Mondo, S.; Lyhne, E.K.; et al. Linking secondary metabolites to gene clusters through genome sequencing of six diverse Aspergillus species. Proc. Natl. Acad. Sci. USA 2018, 115, E753–E761. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Dal Grande, F.; Sharma, R.; Meiser, A.; Rolshausen, G.; Büdel, B.; Mishra, B.; Thines, M.; Otte, J.; Pfenninger, M.; Schmitt, I. Adaptive differentiation coincides with local bioclimatic conditions along an elevational cline in populations of a lichen-forming fungus. BMC Evol. Biol. 2017, 17, 93. [Google Scholar] [CrossRef]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.S.; Holt, C.; Moore, B.; Yandell, M. Genome annotation and curation using MAKER and MAKER-P. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; Volume 48, pp. 4.11.1–4.11.39. ISBN 0471250953. [Google Scholar]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.A.; Vaughn, M.; McKay, S.; Lyons, E.; Stapleton, A.E.; Gessler, D.; Matasci, N.; Wang, L.; Hanlon, M.; Lenards, A.; et al. The iPlant collaborative: Cyberinfrastructure for plant biology. Front. Plant Sci. 2011, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Zdobnov, E.M.; Tegenfeldt, F.; Kuznetsov, D.; Waterhouse, R.M.; Simão, F.A.; Ioannidis, P.; Seppey, M.; Loetscher, A.; Kriventseva, E.V. OrthoDB v9.1: Cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2017, 45, D744–D749. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015; Available online: https://www.R-project.org/ (accessed on 29 March 2018).
- Bahram, M.; Hildebrand, F.; Forslund, S.K.; Anderson, J.L.; Soudzilovskaia, N.A.; Bodegom, P.M.; Bengtsson-Palme, J.; Anslan, S.; Coelho, L.P.; Harend, H.; et al. Structure and function of the global topsoil microbiome. Nature 2018, 560, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; de Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C.; et al. Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In 2010 Gateway Computing Environments Workshop (GCE); IEEE: New Orleans, LA, USA, 2010; pp. 1–8. [Google Scholar]
- Miller, M.A.; Schwartz, T.; Pickett, B.E.; He, S.; Klem, E.B.; Scheuermann, R.H.; Passarotti, M.; Kaufman, S.; O’Leary, M.A. A RESTful API for access to phylogenetic tools via the CIPRES Science Gateway. Evol. Bioinform. 2015, 11, EBO.S21501. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Taxonomic Group | Data Repository 1 | Gene Set Previously Available | Genome Size | Number of Scaffolds | Scaffold N50 | Number of Genes | Abbreviation | Metabolites Reported [55,56,57,58,59] |
|---|---|---|---|---|---|---|---|---|---|
| Cladonia grayi | Lecanoromycetes, Lecanorales | JGI Clagr3 v2.0 | yes | 34.6 Mb | 414 | 243,412 | 11,389 | CGRA | grayanic acid, fumarprotocetraric acid complex |
| Cladonia macilenta | Lecanoromycetes, Lecanorales | NCBI AUPP00000000.1 | no | 37.1 Mb | 240 | 1,469,036 | 10,559 | CMAC | thamnolic acid, barbatic acid, didymic acid, squamatic acid, usnic acid, rhodocladonic acid |
| Cladonia metacorallifera | Lecanoromycetes, Lecanorales | NCBI AXCT00000000.2 | no | 36.7 Mb | 30 | 1,591,850 | 10,497 | CMET | usnic acid, didymic acid, squamatic acid, rhodocladonic acid |
| Cladonia uncialis | Lecanoromycetes, Lecanorales | NCBI NAPT00000000.1 | no | 32.9 Mb | 2124 | 34,871 | 10,902 | CUNC | usnic acid, squamatic acid |
| Endocarpon pusillum (Park et al.) [60] | Eurotiomycetes, Verrucariales | NCBI JFDM00000000.1 | no | 37.2 Mb | 40 | 1,340,794 | 11,756 | EPUP | (none reported) |
| Endocarpon pusillum (Wang et al.) [61] | Eurotiomycetes, Verrucariales | NCBI APWS00000000.1 | yes | 37.1 Mb | 908 | 178,225 | 9238 | EPUW | (none reported) |
| Evernia prunastri | Lecanoromycetes, Lecanorales | NCBI NKYR00000000.1 | yes | 40.3 Mb | 277 | 264,454 | 10,992 | EPRU | evernic acid, atranorin, usnic acid |
| Gyalolechia flavorubescens | Lecanoromycetes, Teloschistales | NCBI AUPK00000000.1 | no | 34.5 Mb | 36 | 1,693,300 | 10,460 | GFLA | parietin, emodin, fallacinal, fragilin |
| Lobaria pulmonaria | Lecanoromycetes, Peltigerales | JGI Lobpul1 v1.0 | yes | 56.1 Mb | 1911 | 50,541 | 15,607 | LPUL | stictic acid, norstictic acid, constictic acid |
| Pseudevernia furfuracea | Lecanoromycetes, Lecanorales | NCBI NKYQ00000000.1 | yes | 37.8 Mb | 46 | 1,178,799 | 8842 | PFUR | atranorin, olivetoric acid, physodic acid |
| Ramalina peruviana | Lecanoromycetes, Lecanorales | NCBI MSTJ00000000.1 | no | 27.0 Mb | 1657 | 40,431 | 9338 | RPER | sekikaic acid complex |
| Umbilicaria hispanica | Lecanoromycetes, Umbilicariales | NCBI PKMA00000000.1 | yes | 41.2 Mb | 1619 | 145,035 | 8488 | LHIS | gyrophoric acid, lecanoric acid, umbilicaric acid, skyrin |
| Umbilicaria muehlenbergii | Lecanoromycetes, Umbilicariales | NCBI JFDN00000000.1 | no | 34.8 Mb | 7 | 7,009,248 | 8968 | UMUE | gyrophoric acid |
| Umbilicaria pustulata | Lecanoromycetes, Umbilicariales | NCBI FWEW00000000.1 | yes | 39.2 Mb | 3891 | 104,297 | 8268 | LPUS | gyrophoric acid, lecanoric acid, hiascinic acid, skyrin |
| Xanthoria parietina | Lecanoromycetes, Teloschistales | JGI Xanpa2 v1.1 | yes | 31.9 Mb | 39 | 1,731,186 | 11,065 | XPAR | physcion, parietinic acid, teloschistin, emodin |
| Species | Abbreviation | Number of Clusters | Type I NR-PKS | Type I R-PKS | Type I PKS | Type III PKS | Hybrid PKS-NRPS | NRPS | Terpene Synthases | Total KS Sequences for Phylogeny | Complete PKS (KS + AT + ACP) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cladonia grayi | CGRA | 51 | 8 | 17 | 1 | 3 | 1 | 2 | 5 | 27 | 15 |
| Cladonia macilenta | CMAC | 52 | 11 | 23 | - | 2 | 4 | 2 | 5 | 38 | 25 |
| Cladonia metacorallifera | CMET | 59 | 13 | 26 | 1 | 2 | 2 | 2 | 8 | 42 | 29 |
| Cladonia uncialis | CUNC | 59 | 10 | 25 | 1 | 2 | 3 | 1 | 8 | 39 | 25 |
| Endocarpon pusillum [8] (Park et al.) | EPUP | 31 | 4 | 9 | - | 1 | 2 | 2 | 6 | 15 | 9 |
| Endocarpon pusillum [9] (Wang et al.) | EPUW | 31 | 5 | 12 | - | 1 | 2 | 1 | 6 | 19 | 12 |
| Evernia prunastri | EPRU | 80 | 9 | 29 | 1 | 2 | 4 | 4 | 13 | 43 | 30 |
| Gyalolechia flavorubescens | GFLA | 37 | 7 | 12 | - | 1 | 1 | 3 | 6 | 20 | 10 |
| Lobaria pulmonaria | LPUL | 77 | 8 | 28 | - | - | 4 | 9 | 16 | 40 | 22 |
| Pseudevernia furfuracea | PFUR | 51 | 5 | 23 | - | 2 | 3 | 4 | 5 | 31 | 17 |
| Ramalina peruviana | RPER | 47 | 9 | 18 | 3 | 1 | 1 | 3 | 2 | 31 | 15 |
| Umbilicaria hispanica | LHIS | 28 | 7 | 10 | 1 | 1 | - | - | 6 | 18 | 14 |
| Umbilicaria muehlenbergii | UMUE | 31 | 5 | 15 | - | 1 | - | - | 4 | 20 | 17 |
| Umbilicaria pustulata | LPUS | 27 | 6 | 9 | - | 1 | 1 | - | 6 | 16 | 13 |
| Xanthoria parietina | XPAR | 51 | 5 | 18 | 1 | 1 | 2 | 5 | 7 | 26 | 13 |
| Sum | 712 | 112 | 274 | 9 | 25 | 30 | 38 | 125 | 425 | 266 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calchera, A.; Dal Grande, F.; Bode, H.B.; Schmitt, I. Biosynthetic Gene Content of the ‘Perfume Lichens’ Evernia prunastri and Pseudevernia furfuracea. Molecules 2019, 24, 203. https://doi.org/10.3390/molecules24010203
Calchera A, Dal Grande F, Bode HB, Schmitt I. Biosynthetic Gene Content of the ‘Perfume Lichens’ Evernia prunastri and Pseudevernia furfuracea. Molecules. 2019; 24(1):203. https://doi.org/10.3390/molecules24010203
Chicago/Turabian StyleCalchera, Anjuli, Francesco Dal Grande, Helge B. Bode, and Imke Schmitt. 2019. "Biosynthetic Gene Content of the ‘Perfume Lichens’ Evernia prunastri and Pseudevernia furfuracea" Molecules 24, no. 1: 203. https://doi.org/10.3390/molecules24010203
APA StyleCalchera, A., Dal Grande, F., Bode, H. B., & Schmitt, I. (2019). Biosynthetic Gene Content of the ‘Perfume Lichens’ Evernia prunastri and Pseudevernia furfuracea. Molecules, 24(1), 203. https://doi.org/10.3390/molecules24010203