
Polyoxometalate-Based Catalysts for CO2 Conversion


Abstract
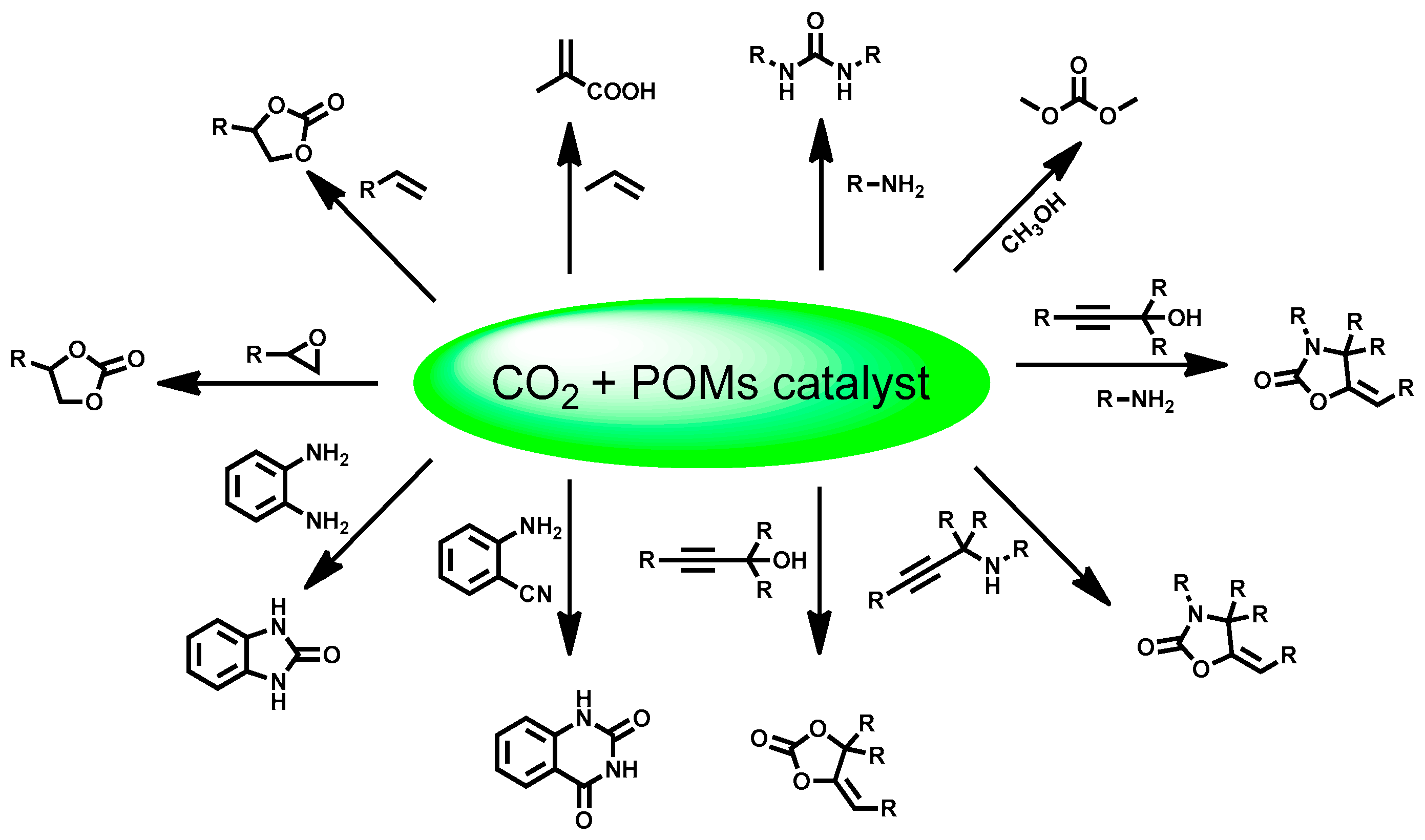
:1. Introduction
2. Photocatalytic CO2 Reduction
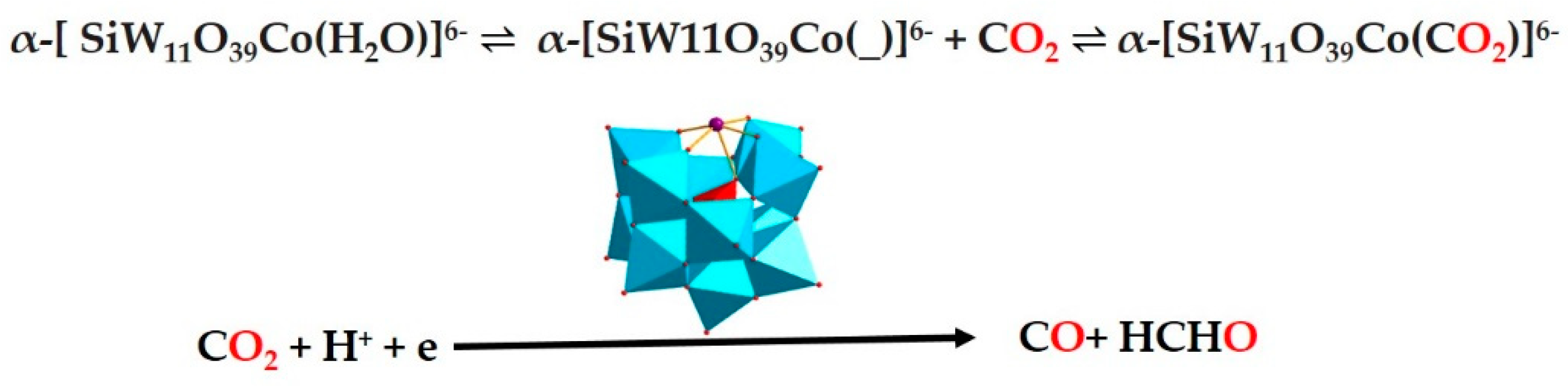
2.1. CO2 to CO
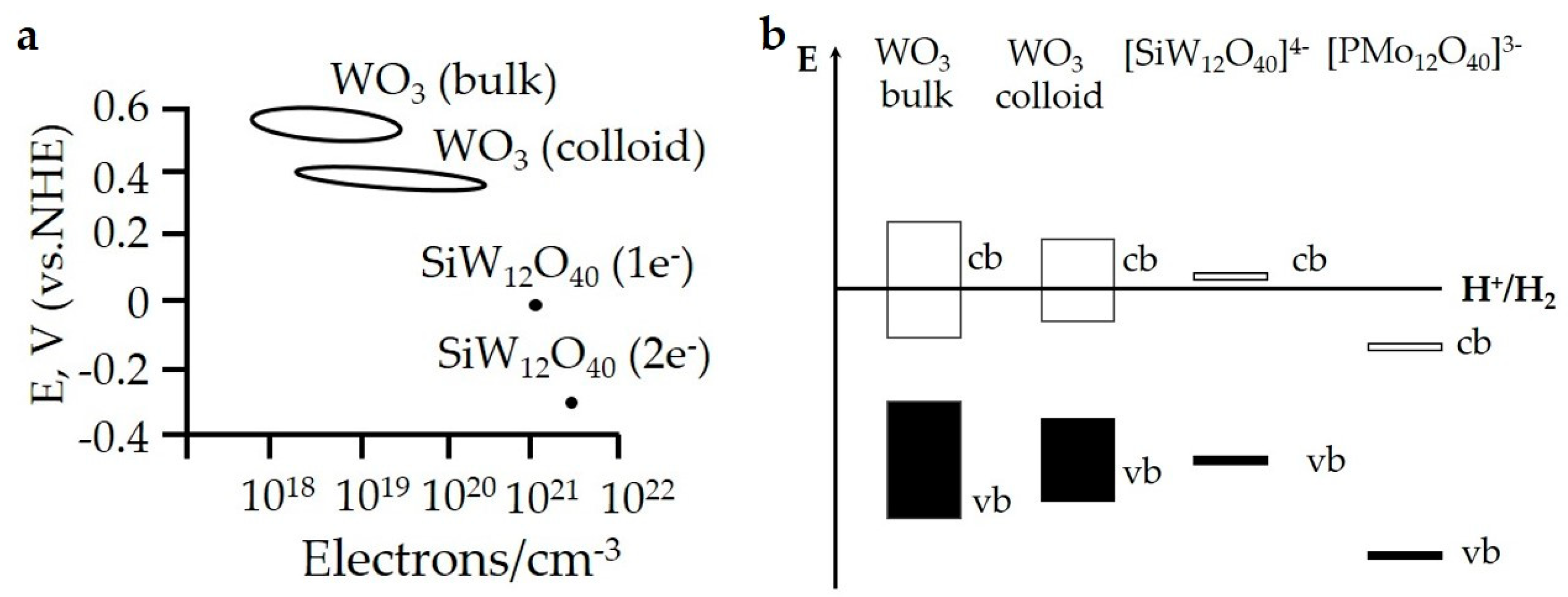
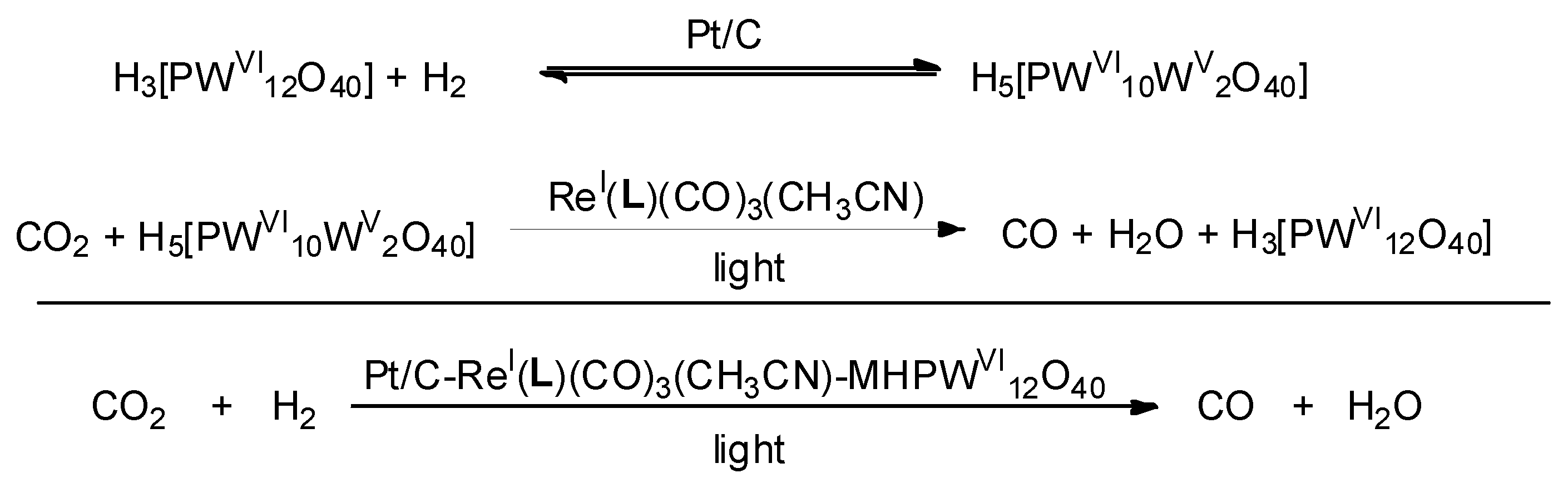
2.1.1. Homogeneous Catalysts
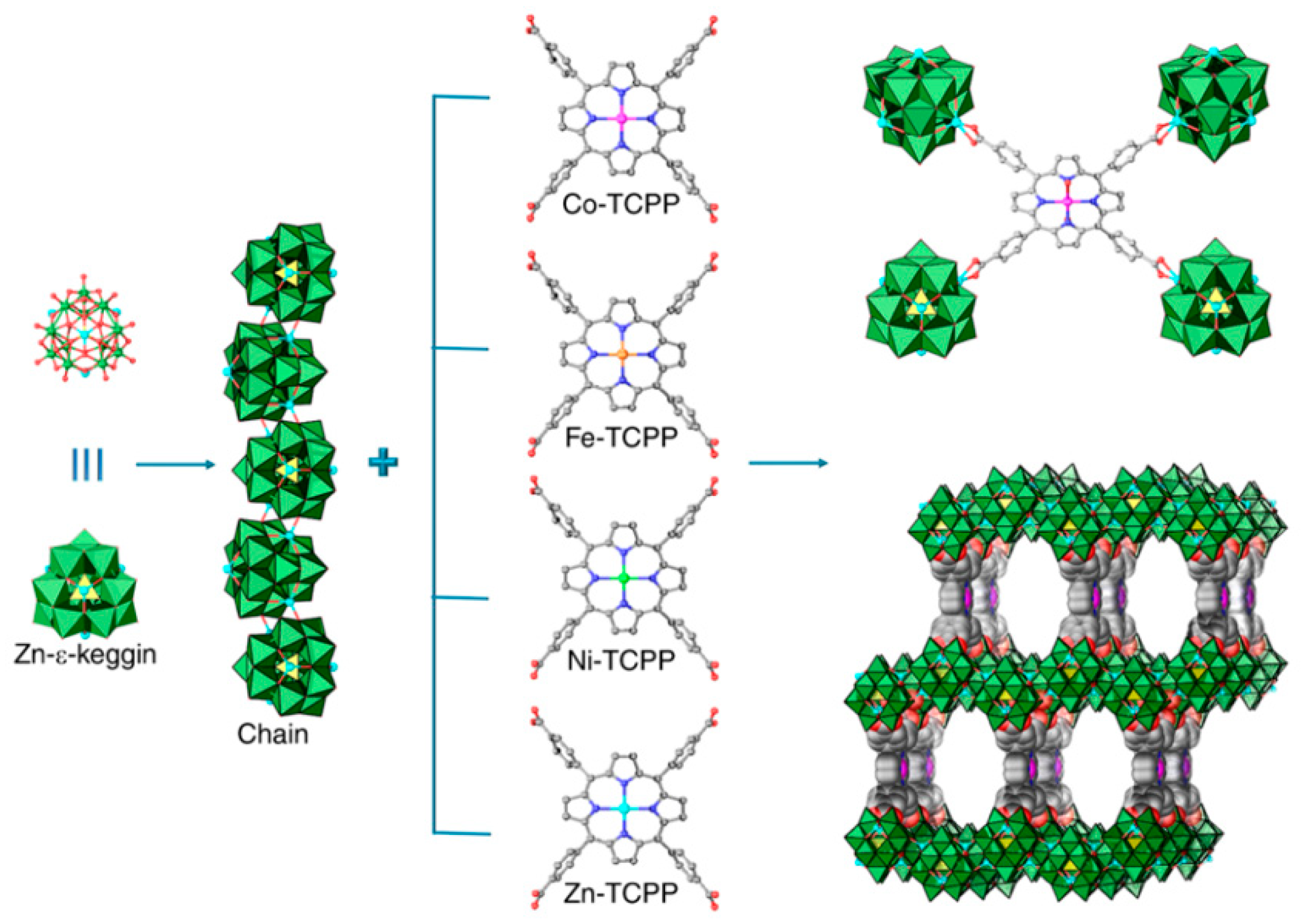
2.1.2. Heterogeneous Catalysts
2.2. CO2 to HCOOH
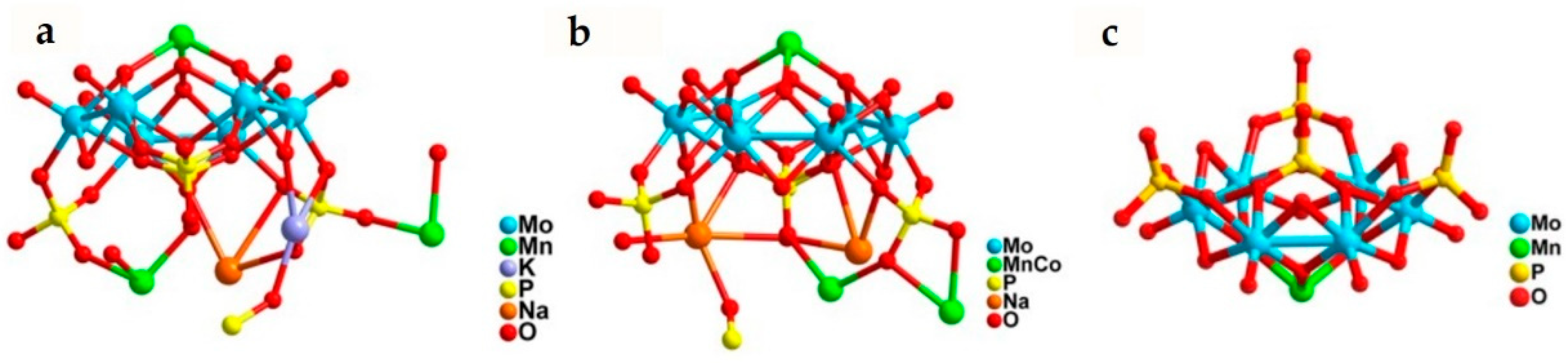
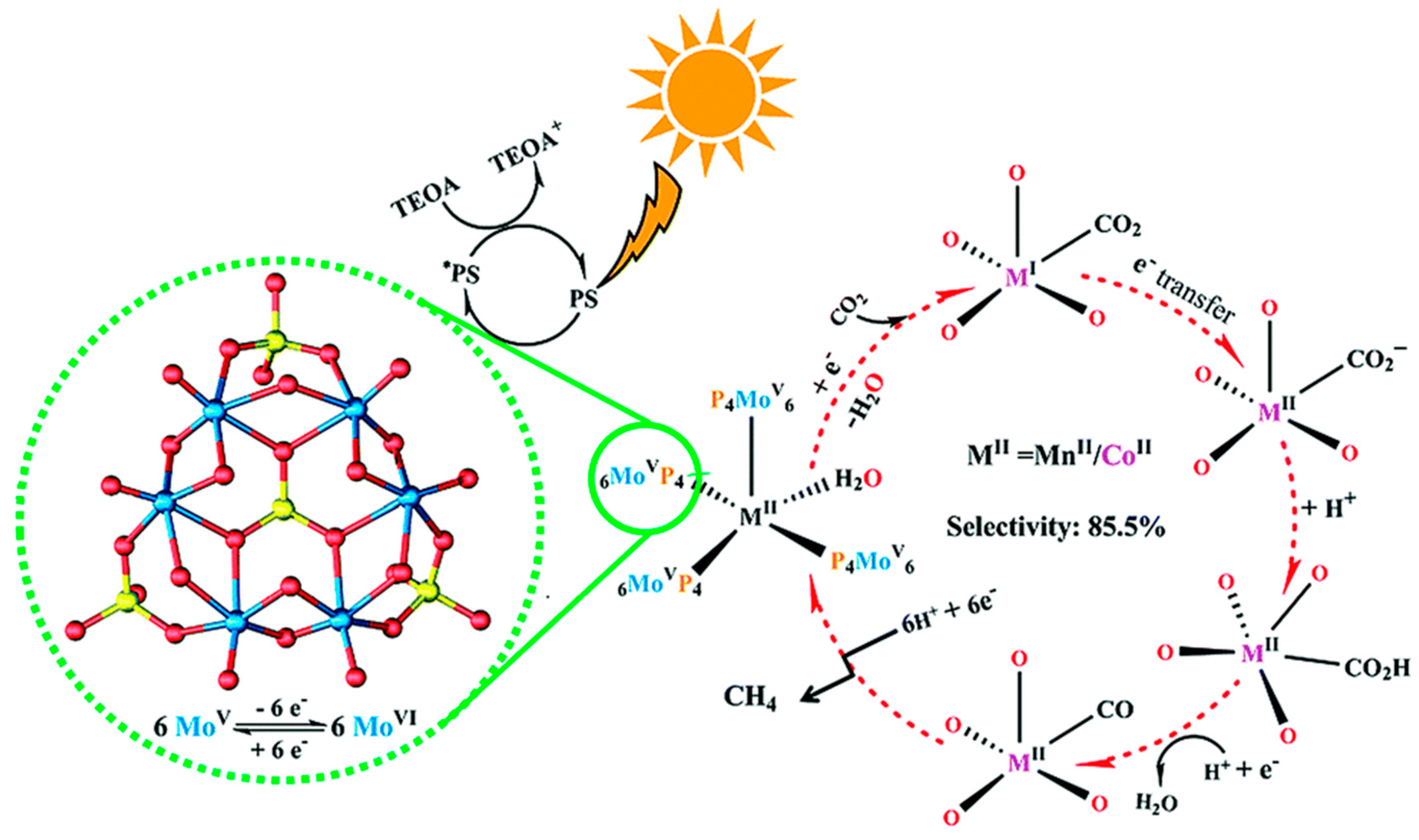
2.3. CO2 to CH4
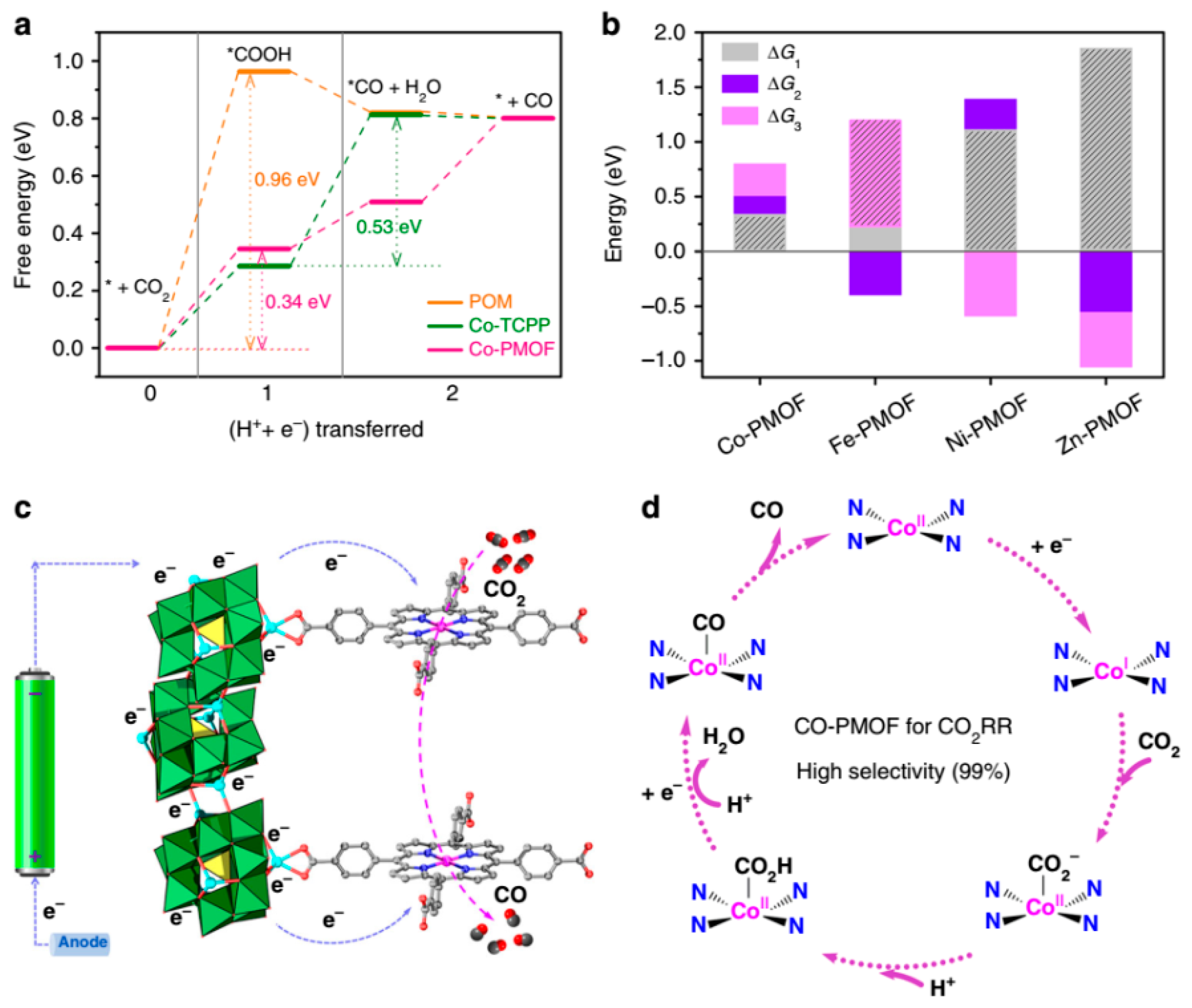
3. Electrocatalytic CO2 Reduction
4. Electromicrobial Conversion of CO2
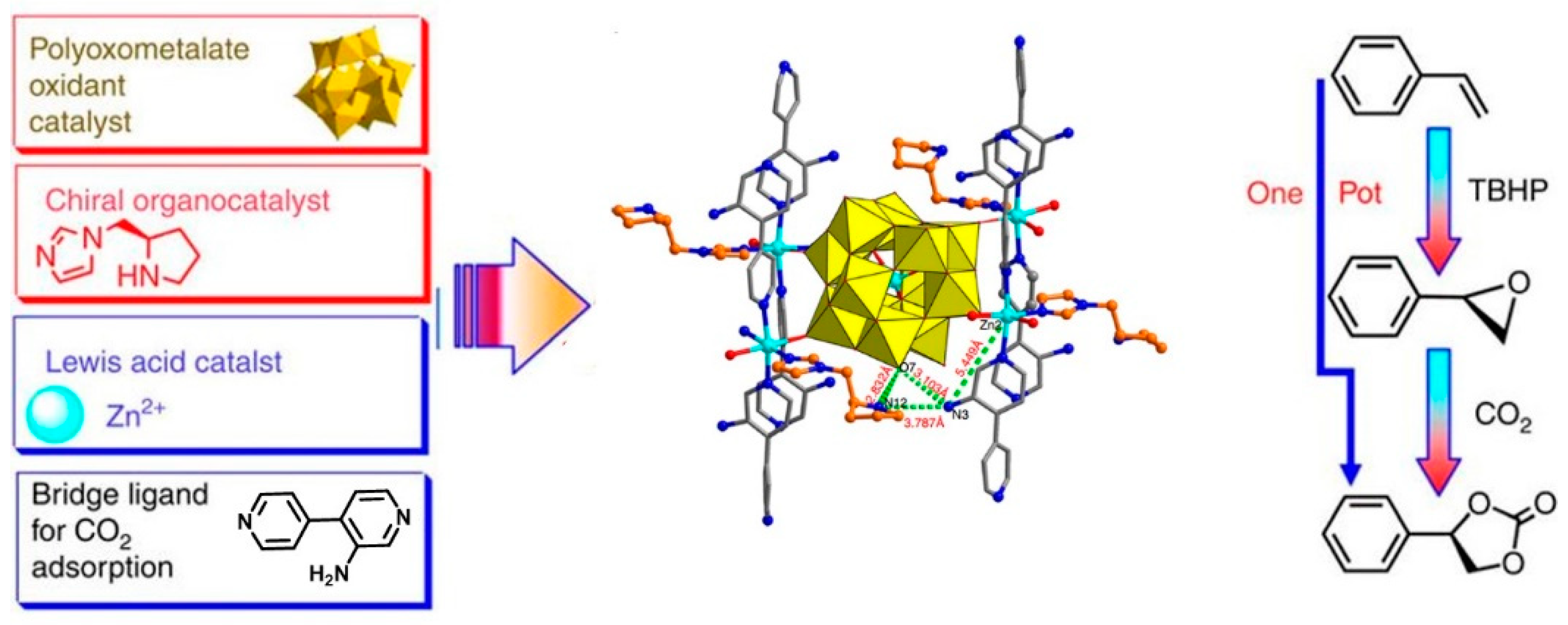
5. Non-Reductive CO2 Conversion to Carbonyl-Contained Organic Chemicals
6. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Le Quéré, C.; Andrew, R.M.; Friedlingstein, P.; Sitch, S.; Hauck, J.; Pongratz, J.; Pickers, P.A.; Korsbakken, J.I.; Peters, G.P.; Canadell, J.G.; et al. Global carbon budget 2018. Earth. Syst. Sci. Data 2018, 10, 2141–2194. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Berzelius, J. The preparation of the phosphomolybdate ion [PMo12O40]3−. Pogg. Ann. 1826, 6, 369–371. [Google Scholar]
- Song, Y.-F. Polyoxometalate-Based Assemblies and Functional Materials; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Van Eldik, R.; Cronin, L. Polyoxometalate Chemistry; Academic Press: Burlington, VT, USA, 2017. [Google Scholar]
- Liu, S.; Tang, Z. Polyoxometalate-based functional nanostructured films: Current progress and future prospects. Nano Today 2010, 5, 267–281. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, Y.; Li, G.; Wei, Y. Recent advances in alkoxylation chemistry of polyoxometalates: From synthetic strategies, structural overviews to functional applications. Coord. Chem. Rev. 2019, 378, 395–414. [Google Scholar] [CrossRef]
- Chen, W.; Wang, E. Polyoxometalate Chemistry; Science press: Beijing, China, 2013; (In Chinese edition). [Google Scholar]
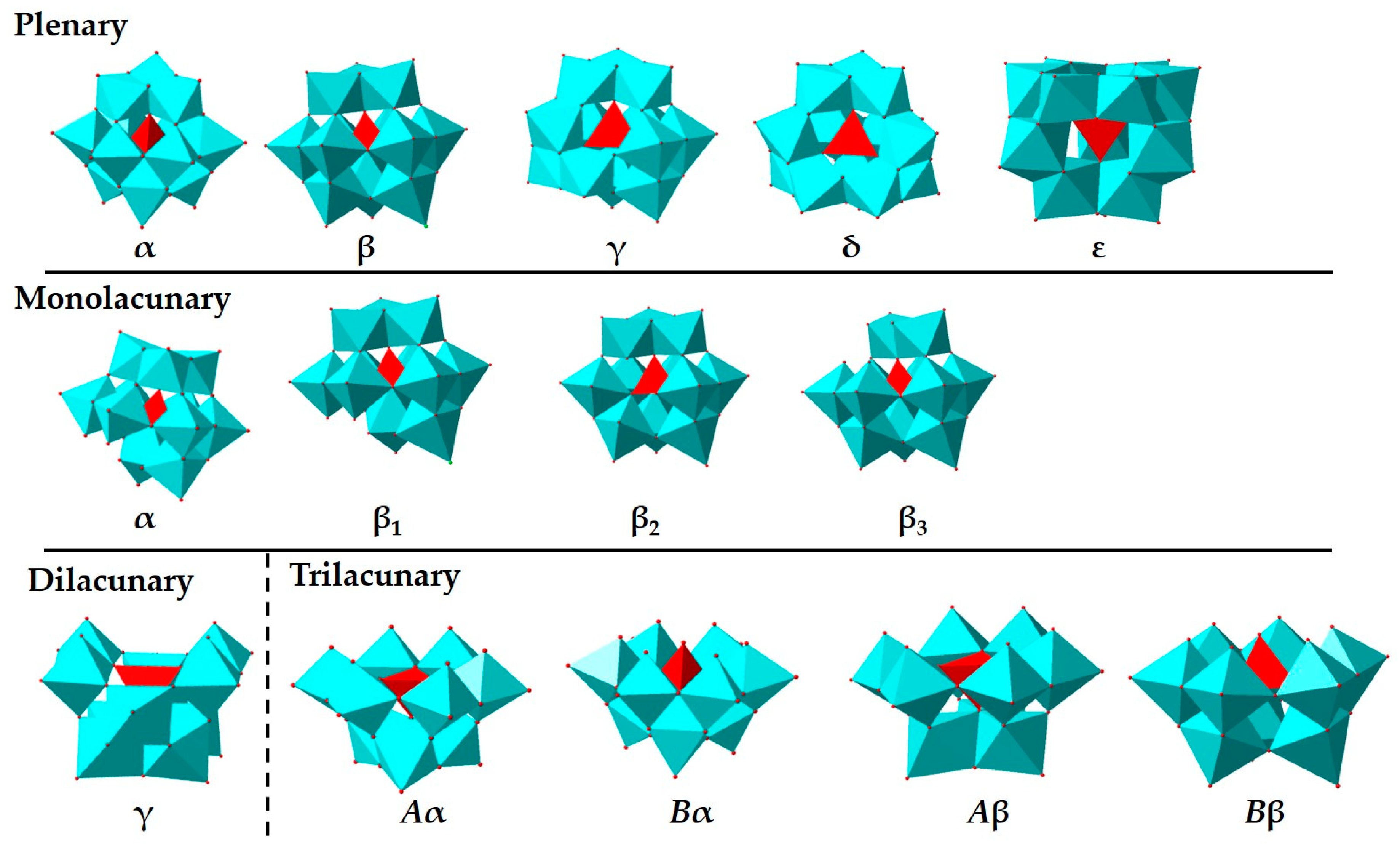
- Sartzi, H.; Miras, H.N.; Vila-Nadal, L.; Long, D.L.; Cronin, L. Trapping the delta isomer of the polyoxometalate-based Keggin cluster with a tripodal ligand. Angew. Chem. Int. Ed. 2015, 54, 15488–15492. [Google Scholar] [CrossRef]
- Bassil, B.S.; Kortz, U. Recent advances in lanthanide-containing polyoxotungstates. Z. Anorg. Allg. Chem. 2010, 636, 2222–2231. [Google Scholar] [CrossRef]
- Guo, Y.-H.; Hu, C.-W. Porous hybrid photocatalysts based on polyoxometalates. J. Clust. Sci. 2003, 14, 505–526. [Google Scholar] [CrossRef]
- Wang, S.-S.; Yang, G.-Y. Recent advances in polyoxometalate-catalyzed reactions. Chem. Rev. 2015, 115, 4893–4962. [Google Scholar] [CrossRef]
- Müller, C.E.; Iqbal, J.; Baqi, Y.; Zimmermann, H.; Röllich, A.; Stephan, H. Polyoxometalates—a new class of potent ecto-nucleoside triphosphate diphosphohydrolase (NTPDase) inhibitors. Bioor. Med. Chem. Lett. 2006, 16, 5943–5947. [Google Scholar] [CrossRef]
- Proust, A.; Thouvenot, R.; Gouzerh, P. Functionalization of polyoxometalates: towards advanced applications in catalysis and materials science. Chem. Commun. 2008, 16, 1837–1852. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Shen, C.; He, L. Recent advances of polyoxometalate-catalyzed selective oxidation based on structural classification. Acta Crystallogr. Sect. C Struct. Chem. 2018, 74, 1182–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepankiewicz, S.H.; Ippolito, C.M.; Santora, B.P.; Van De Ven, T.J.; Ippolito, G.A.; Fronckowiak, L.; Wiatrowski, F.; Power, T.; Kozik, M. Interaction of carbon dioxide with transition-metal-substituted heteropolyanions in nonpolar solvents. spectroscopic evidence for complex formation. Inorg. Chem. 1998, 37, 4344–4352. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Li, F.; Xu, L.; Liu, X.; Yang, Y. CO2 coordination by inorganic polyoxoanion in water. J. Am. Chem. Soc. 2008, 130, 10838–10839. [Google Scholar] [CrossRef] [PubMed]
- He, L.-N.; Wang, M.-Y.; Song, Q.-W.; Ma, R.; Xie, J.-N. Efficient conversion of carbon dioxide at atmospheric pressure to 2-oxazolidinones promoted by bifunctional Cu(II)-substituted polyoxometalate-based ionic liquids. Green Chem. 2016, 18, 282–287. [Google Scholar]
- Cheng, W.; Xue, Y.-S.; Luo, X.-M.; Xu, Y.; Xue, Y. A rare three-dimensional POM-based inorganic metal polymer bonded by CO2 with high catalytic performance for CO2 cycloaddition. Chem. Commun. 2018, 54, 12808–12811. [Google Scholar] [CrossRef]
- Fang, X.; Anderson, T.M.; Neiwert, W.A.; Hill, C.L. Yttrium polyoxometalates. Synthesis and characterization of a carbonate-encapsulated sandwich-type complex. Inorg. Chem. 1998, 37, 4344–4352. [Google Scholar]
- Chen, B.; Neumann, R. Coordination of carbon dioxide to the Lewis acid site of a zinc-substituted polyoxometalate and formation of an adduct using a polyoxometalate-2,4,6-trimethylpyridine frustrated Lewis pair. Eur. J. Inorg. Chem. 2018, 2018, 791–794. [Google Scholar] [CrossRef]
- Garai, S.; Haupt, E.T.K.; Bögge, H.; Merca, A.; Müller, A. Picking up 30 CO2 molecules by a porous metal oxide capsule based on the same number of receptors. Angew. Chem. Int. Ed. 2012, 51, 10528–10531. [Google Scholar] [CrossRef]
- Selimkhanov, J.; Taylor, B.; Yao, J.; Pilko, A.; Albeck, J.; Hoffmann, A.; Tsimring, L.; Wollman, R. Accurate information transmission through dynamic biochemical signaling networks. Science 2014, 346, 1370–1373. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Peng, B.; Peng, T. Recent advances in heterogeneous photocatalytic CO2 conversion to solar fuels. ACS Catal. 2016, 6, 7485–7527. [Google Scholar] [CrossRef]
- Sohn, Y.; Huang, W.; Taghipour, F. Recent progress and perspectives in the photocatalytic CO2 reduction of Ti-oxide-based nanomaterials. Appl. Surf. Sci. 2017, 396, 1696–1711. [Google Scholar] [CrossRef]
- Inoue, T.; Fujishima, A.; Konishi, S.; Honda, K. Photoelectrocatalytic reduction of carbon dioxide in aqueous suspensions of semiconductor powders. Nature 1979, 277, 637–638. [Google Scholar] [CrossRef]
- Zeng, S.; Kar, P.; Thakur, U.K.; Shankar, K. A review on photocatalytic CO2 reduction using perovskite oxide nanomaterials. Nanotechnology 2018, 29, 052001. [Google Scholar] [CrossRef]
- Shi, R.; Waterhouse, G.I.; Zhang, T. Recent Progress in Photocatalytic CO2 reduction over perovskite oxides. Sol. RRL 2017, 1, 1700126. [Google Scholar] [CrossRef]
- Ye, S.; Wang, R.; Wu, M.-Z.; Yuan, Y.-P. A review on g-C3N4 for photocatalytic water splitting and CO2 reduction. Appl. Surf. Sci. 2015, 358, 15–27. [Google Scholar] [CrossRef]
- Li, R.; Zhang, W.; Zhou, K. Metal-organic-framework-based catalysts for photoreduction of CO2. Adv. Mater. 2018, 30, e1705512. [Google Scholar] [CrossRef]
- Sprick, R.S.; Jiang, J.-X.; Bonillo, B.; Ren, S.; Ratvijitvech, T.; Guiglion, P.; Zwijnenburg, M.A.; Adams, D.J.; Cooper, A.I. Tunable organic photocatalysts for visible-light-driven hydrogen evolution. J. Am. Chem. Soc. 2015, 137, 3265–3270. [Google Scholar] [CrossRef]
- Ghosh, S.; Kouamé, N.A.; Ramos, L.; Rémita, S.; Dazzi, A.; Deniset-Besseau, A.; Beaunier, P.; Goubard, F.; Aubert, P.-H.; Remita, H. Conducting polymer nanostructures for photocatalysis under visible light. Nat. Mater. 2015, 14, 505–511. [Google Scholar] [CrossRef]
- Papaconstantinou, E. Photochemistry of polyoxometallates of molybdenum and tungsten and/or vanadium. Chem. Soc. Rev. 1989, 18, 1. [Google Scholar] [CrossRef]
- Hill, C.L.; Prosser-McCartha, C.M. Photocatalytic and Photoredox Properties of Polyoxometalate Systems; Springer Nature: Dordrecht, Netherlands, 1993. [Google Scholar]
- Nemodruk, A.A.; Bezrogova, E.V. The use of photochemical reduction for the determination of silicon and phosphorus as their blue heteropoly acids. Zh. Anal. Khim. 1969, 24, 292. [Google Scholar]
- Morosanova, S.A.; Kolli, N.Y.; Kushnirenko, T.G. Photochemical reduction of 12-molybdoarsenic acid in aqueous-organic media. Zh. Anal. Khim. 1977, 32, 96. [Google Scholar]
- Streb, C. New trends in polyoxometalate photoredox chemistry: From photosensitisation to water oxidation catalysis. Dalton Trans. 2012, 41, 1651–1659. [Google Scholar] [CrossRef]
- Tucher, J.; Wu, Y.; Nye, L.C.; Ivanović-Burmazović, I.; Khusniyarov, M.M.; Streb, C. Metal substitution in a Lindqvist polyoxometalate leads to improved photocatalytic performance. Dalton Trans. 2012, 41, 9938. [Google Scholar] [CrossRef]
- Walsh, J.J.; Bond, A.M.; Forster, R.J.; Keyes, T.E. Hybrid polyoxometalate materials for photo(electro-) chemical applications. Coord. Chem. Rev. 2016, 306, 217–234. [Google Scholar] [CrossRef]
- Khenkin, A.M.; Efremenko, I.; Weiner, L.; Martin, J.M.L.; Neumann, R. Photochemical reduction of carbon dioxide catalyzed by a ruthenium-substituted polyoxometalate. Chem. Eur. J. 2010, 16, 1356–1364. [Google Scholar] [CrossRef]
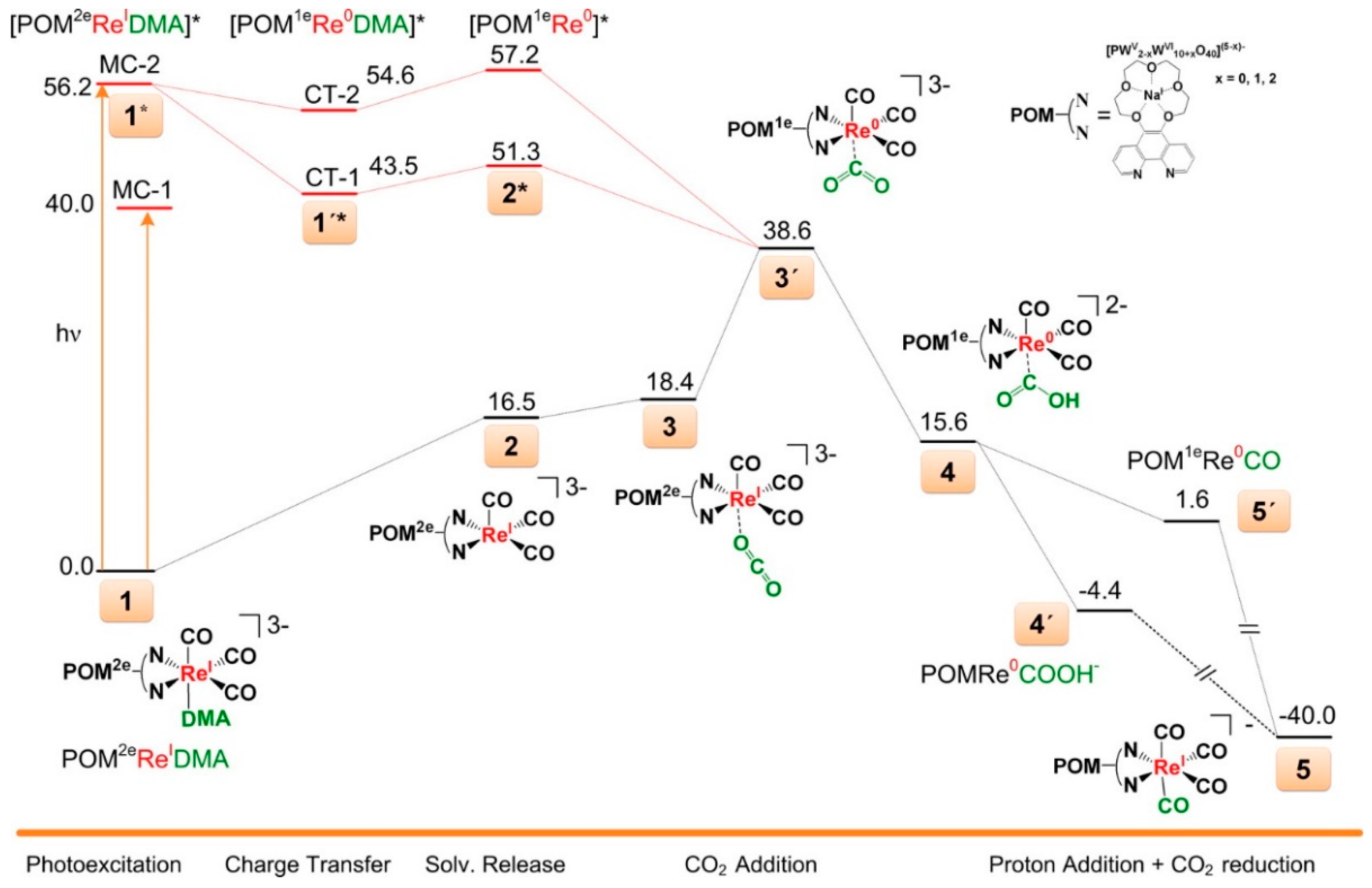
- Ettedgui, J.; Diskin-Posner, Y.; Weiner, L.; Neumann, R. Photoreduction of carbon dioxide to carbon monoxide with hydrogen catalyzed by a rhenium(I) phenanthroline−polyoxometalate hybrid complex. J. Am. Chem. Soc. 2011, 133, 188–190. [Google Scholar] [CrossRef]
- Poblet, J.M. The Photoreduction mechanism of CO2 to CO catalyzed by a rhenium(I)-polyoxometalate hybrid compound. ACS Catal. 2016, 6, 6422–6428. [Google Scholar]
- Haviv, E.; Shimon, L.J.W.; Neumann, R. Photochemical reduction of CO2 with visible light using a polyoxometalate as photoreductant. Chem. Eur. J. 2017, 23, 92–95. [Google Scholar] [CrossRef]
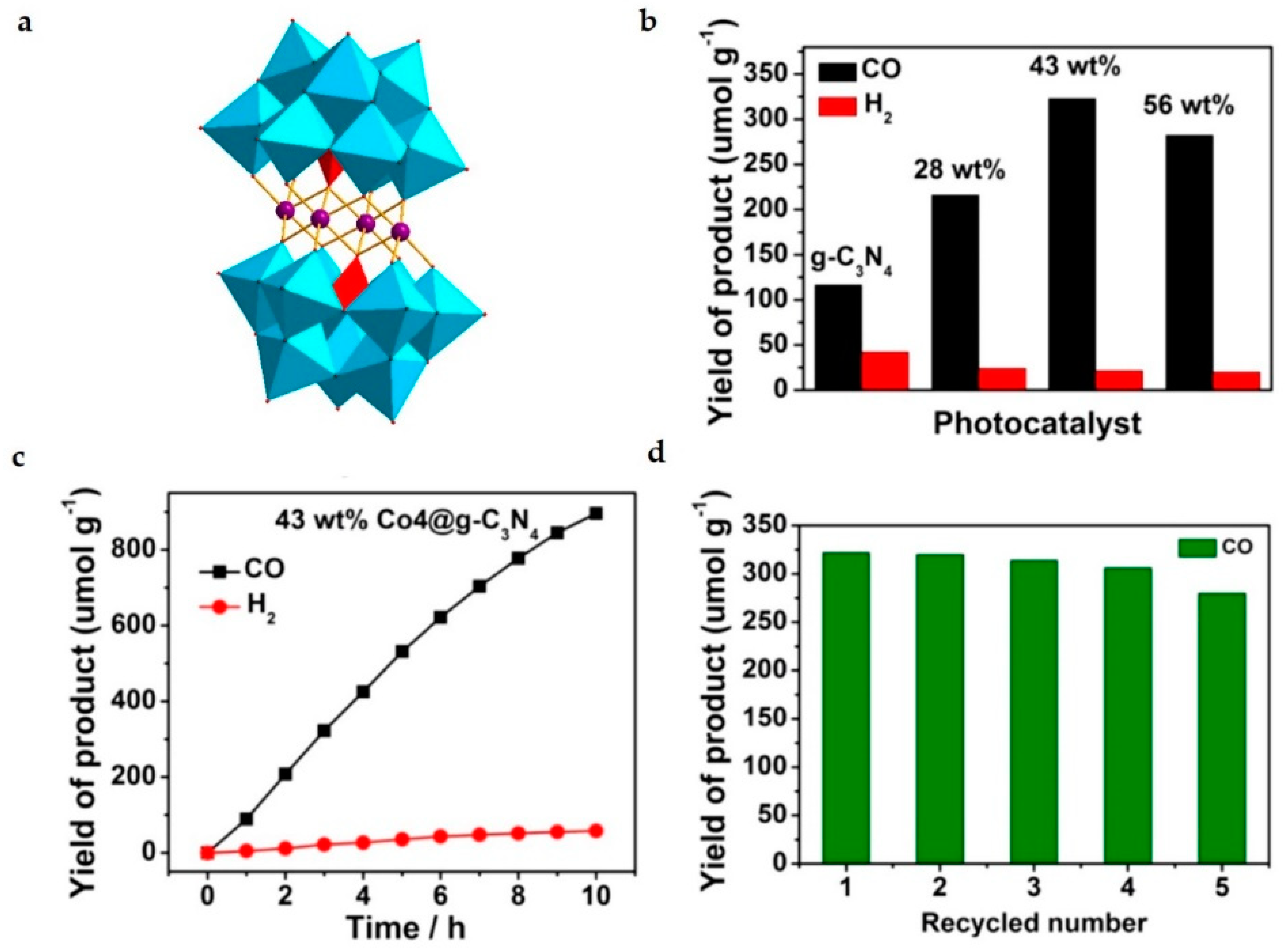
- Zhou, J.; Chen, W.; Sun, C.; Han, L.; Qin, C.; Chen, M.; Wang, X.; Wang, E.; Su, Z. Oxidative polyoxometalates modified graphitic carbon nitride for visible-light CO2 reduction. ACS Appl. Mater. Interfaces 2017, 9, 11689–11695. [Google Scholar] [CrossRef]
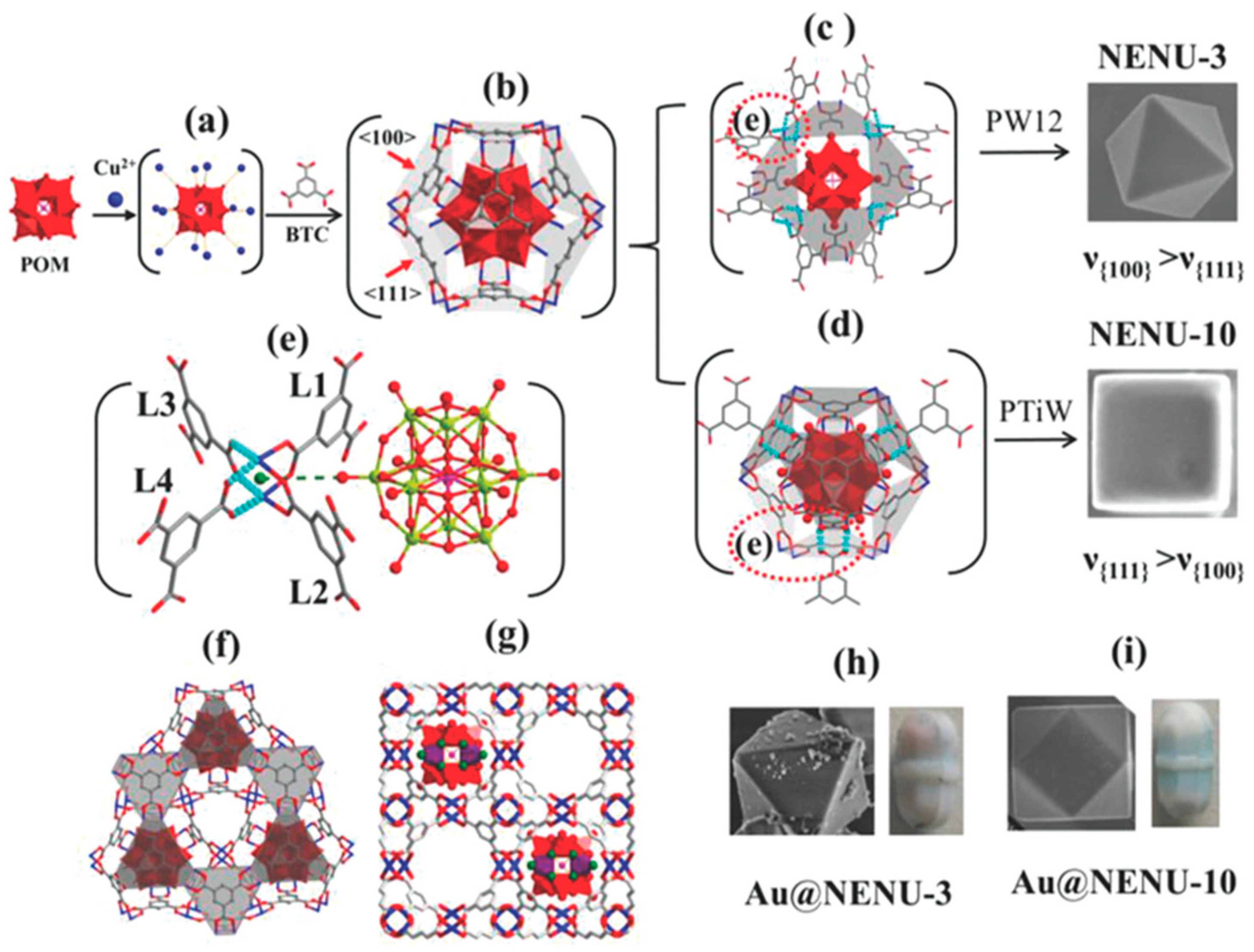
- Liu, S.-M.; Zhang, Z.; Li, X.; Jia, H.; Ren, M.; Liu, S. Ti-substituted Keggin-type polyoxotungstate as proton and electron reservoir encaged into metal-organic framework for carbon dioxide photoreduction. Adv. Mater. Interfaces 2018, 5, 1801062. [Google Scholar] [CrossRef]
- Das, S.; Biswas, S.; Balaraju, T.; Barman, S.; Pochamoni, R.; Roy, S. Photochemical reduction of carbon dioxide coupled with water oxidation using various soft-oxometalate (SOM) based catalytic systems. J. Mater. Chem. A 2016, 4, 8875–8887. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Kumar, S.; Garai, S.; Pochamoni, R.; Paul, S.; Roy, S. Softoxometalate [{K6.5Cu(OH)(8.5)(H2O)(7.5)}(0.5)@{K3PW12O40}](n) (n=1348-2024) as an efficient inorganic material for CO2 reduction with concomitant water oxidation. ACS Appl. Mater. Interfaces 2017, 9, 35086–35094. [Google Scholar] [CrossRef]
- Das, S.; Balaraju, T.; Barman, S.; Sreejith, S.S.; Pochamoni, R.; Roy, S. A Molecular CO2 reduction catalyst based on giant polyoxometalate {Mo-368}. Front. Chem. 2018, 6, 514. [Google Scholar] [CrossRef] [PubMed]
- Yamase, T.; Sugeta, M. Photoreduction of CO2 to CH4 in water using dititanodecatungstophosphate as multielectron transfer catalyst. Inorg. Chim. Acta. 1990, 172, 131–134. [Google Scholar] [CrossRef]
- Xie, S.-L.; Liu, J.; Dong, L.-Z.; Li, S.-L.; Lan, Y.-Q.; Su, Z.-M. Hetero-metallic active sites coupled with strongly reductive polyoxometalate for selective photocatalytic CO2-to-CH4 conversion in water. Chem. Sci. 2019, 10, 185–190. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef]
- Paul, J.; Page, P.; Sauers, P.; Ertel, K.; Pasternak, C.; Lin, W.; Kozik, M. Transition-Metal-Substituted Heteropoly Anions in Nonpolar Solvents-Structures and Interaction with Carbon Dioxide; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Girardi, M.; Blanchard, S.; Griveau, S.; Simon, P.; Fontecave, M.; Bedioui, F.; Proust, A. Electro-assisted reduction of CO2 to CO and formaldehyde by (TOA)6[α-SiW11O39Co(_)] polyoxometalate. Eur. J. Inorg. Chem. 2015, 2015, 3642–3648. [Google Scholar] [CrossRef]
- Girardi, M.; Platzer, D.; Griveau, S.; Bedioui, F.; Alves, S.; Proust, A.; Blanchard, S. Assessing the electrocatalytic properties of the {Cp*RhIII}2+-polyoxometalate derivative [H2PW11O39{(RhIIICp*(OH2)}]3− towards CO2 reduction. Eur. J. Inorg. Chem. 2019, 3–4, 387–393. [Google Scholar] [CrossRef]
- Szczęśniak, B.; Choma, J.; Jaroniec, M. Gas adsorption properties of hybrid graphene-MOF materials. J. Colloid Interface Sci. 2018, 514, 801–813. [Google Scholar] [CrossRef]
- Genovese, M.; Lian, K. Polyoxometalate modified inorganic–organic nanocomposite materials for energy storage applications: A review. Curr. Opin. Solid State Mater. Sci. 2015, 19, 126–137. [Google Scholar] [CrossRef]
- Fan, D.; Hao, J.; Wei, Q. Assembly of polyoxometalate-based composite materials. J. Inorg. Organomet. Polym. Mater. 2012, 22, 301–306. [Google Scholar] [CrossRef]
- Wang, Y.-R.; Huang, Q.; He, C.-T.; Chen, Y.; Liu, J.; Shen, F.-C.; Lan, Y.-Q. Oriented electron transmission in polyoxometalate-metalloporphyrin organic framework for highly selective electroreduction of CO2. Nat. Commun. 2018, 9, 4466. [Google Scholar] [CrossRef]
- Guo, S.-X.; MacFarlane, D.R.; Zhang, J. Bioinspired electrocatalytic CO2 reduction by bovine serum albumin-capped silver nanoclusters mediated by [α-SiW12O40]4−. Chemsuschem 2016, 9, 80–87. [Google Scholar] [CrossRef]
- Wang, Y.; Weinstock, I.A. Polyoxometalate-decorated nanoparticles. Chem. Soc. Rev. 2012, 41, 7479. [Google Scholar] [CrossRef] [PubMed]
- Jameel, U.; Zhu, M.; Chen, X.; Tong, Z. Recent progress of synthesis and applications in polyoxometalate and nanogold hybrid materials. J. Mater. Sci. 2016, 51, 2181–2198. [Google Scholar] [CrossRef]
- Guo, S.-X.; Li, F.; Chen, L.; Macfarlane, D.R.; Zhang, J. Polyoxometalate-promoted electrocatalytic CO2 reduction at nanostructured silver in dimethylformamide. ACS Appl. Mater. Interfaces 2018, 10, 12690–12697. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Opgenorth, P.H.; Wernick, D.G.; Rogers, S.; Wu, T.-Y.; Higashide, W.; Malati, P.; Huo, Y.-X.; Cho, K.M.; Liao, J.C. Integrated electromicrobial conversion of CO2 to higher alcohols. Science 2012, 335, 1596. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, A.S.; McTernan, P.M.; Lian, H.; Kelly, R.M.; Adams, M.W. Biological conversion of carbon dioxide and hydrogen into liquid fuels and industrial chemicals. Curr. Opin. Biotechnol. 2013, 24, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, A.; Fricke, W.F.; Reinecke, F.; Kusian, B.; Liesegang, H.; Cramm, R.; Eitinger, T.; Ewering, C.; Pötter, M.; Schwartz, E.; et al. Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat. Biotechnol. 2006, 24, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Freire, C.; Nunes, M.; Fernandes, D.M.; Abdelkader, V.K. POM&MOF-based electrocatalysts for energy-related reactions. ChemCatChem 2018, 10, 1703–1730. [Google Scholar]
- Wang, M.; Zhong, W.; Zhang, S.; Liu, R.; Xing, J.; Zhang, G. An overall water-splitting polyoxometalate catalyst for the electromicrobial conversion of CO2 in neutral water. J. Mater. Chem. A 2018, 6, 9915–9921. [Google Scholar] [CrossRef]
- Yuan, G.; Qi, C.; Wu, W.; Jiang, H. Recent advances in organic synthesis with CO2 as C1 synthon. Green Sustain. Chem. 2017, 3, 22–27. [Google Scholar]
- Yu, B.; Zou, B.; Hu, C.-W. Recent applications of polyoxometalates in CO2 capture and transformation. J. CO2 Util. 2018, 26, 314–322. [Google Scholar] [CrossRef]
- La, K.W.; Youn, M.H.; Chung, J.S.; Baeck, S.H.; Song, I.K. Synthesis of dimethyl carbonate from methanol and carbon dioxide by heteropolyacid/metal oxide catalysts. Solid State Phenom. 2007, 119, 287–290. [Google Scholar] [CrossRef]
- Aouissi, A.; Al-Othman, Z.A.; Al-Amro, A. Gas-phase synthesis of dimethyl carbonate from methanol and carbon dioxide over Co1.5PW12O40 Keggin-type heteropolyanion. Int. J. Mol. Sci. 2010, 11, 1343–1351. [Google Scholar] [CrossRef]
- Lee, H.J.; Park, S.; Jung, J.C.; Song, I.K. Direct synthesis of dimethyl carbonate from methanol and carbon dioxide over H3PW12O40/CeXZr1-XO2 catalysts: Effect of acidity of the catalysts. Korean J. Chem. Eng. 2011, 28, 1518–1522. [Google Scholar] [CrossRef]
- La, K.W.; Jung, J.C.; Kim, H.; Baeck, S.-H.; Song, I.K. Effect of acid–base properties of H3PW12O40/CexTi1−xO2 catalysts on the direct synthesis of dimethyl carbonate from methanol and carbon dioxide: A TPD study of H3PW12O40/CexTi1−xO2 catalysts. J. Mol. Catal. A: Chem. 2007, 269, 41–45. [Google Scholar] [CrossRef]
- Kimura, T.; Kamata, K.; Mizuno, N. A bifunctional tungstate catalyst for chemical fixation of CO2 at atmospheric pressure. Angew. Chem. Int. Ed. 2012, 51, 6700–6703. [Google Scholar] [CrossRef] [PubMed]
- Sunaba, H.; Kimura, T.; Kamata, K.; Mizuno, N. Efficient [WO4]2− -catalyzed chemical fixation of carbon dioxide with 2-aminobenzonitriles to quinazoline-2,4(1H,3H)-diones. Inorg. Chem. 2012, 51, 13001–13008. [Google Scholar]
- Wang, D.; Zhong, S. Study on CuPMo/TiO2 catalyst for direct synthesis of methacrylic acid from propylene and carbon dioxide. Chin. J. Catal. 2003, 24, 705–710. [Google Scholar]
- Wang, D.-W.; Zhong, S.-H. Study on CuPW/TiO2 catalyst for direct synthesis of MAA from propylene and carbon dioxide. J. Fuel Chem. Technol. 2004, 32, 219–224, (In Chinese with English abstract). [Google Scholar]
- Wang, M.-Y.; Ma, R.; He, L.-N. Polyoxometalate-based ionic liquids-promoted CO2 conversion. Sci. China Chem. 2016, 59, 507–516. [Google Scholar] [CrossRef]
- Kamata, K.; Sugahara, K. Base catalysis by mono- and polyoxometalates. Catalysts 2017, 7, 345. [Google Scholar] [CrossRef]
- Guo, L.; Jin, X.; Wang, X.; Yin, L.; Wang, Y.; Yang, Y.-W. Immobilizing polyether imidazole ionic liquids on zsm-5 zeolite for the catalytic synthesis of propylene carbonate from carbon dioxide. Molecules 2018, 23, 2710. [Google Scholar] [CrossRef]
- Han, Q.; Qi, B.; Ren, W.; He, C.; Niu, J.; Duan, C. Polyoxometalate-based homochiral metal-organic frameworks for tandem asymmetric transformation of cyclic carbonates from olefins. Nat. Commun. 2015, 6, 10007. [Google Scholar] [CrossRef]
- Jia, J.; Niu, Y.; Zhang, P.; Zhang, D.; Ma, P.; Zhang, C.; Niu, J.; Wang, J. A Monomeric tricobalt(II)-substituted dawson-type polyoxometalate decorated by a metal carbonyl group: [P2W15O56Co3(H2O)3(OH)3Mn(CO)3]8−. Inorg. Chem. 2017, 56, 10131–10134. [Google Scholar] [CrossRef]
- Lu, J.; Ma, X.; Singh, V.; Zhang, Y.; Wang, P.; Feng, J.; Ma, P.; Niu, J.; Wang, J. Facile CO2 cycloaddition to epoxides by using a tetracarbonyl metal selenotungstate derivate [{Mn(CO)3}4(Se2W11O43)]8−. Inorg. Chem. 2018, 57, 14632–14643. [Google Scholar] [CrossRef]
- Ge, W.; Wang, X.; Zhang, L.; Du, L.; Zhou, Y.; Wang, J. Fully-occupied Keggin type polyoxometalate as solid base for catalyzing CO2 cycloaddition and Knoevenagel condensation. Catal. Sci. Technol. 2016, 6, 460–467. [Google Scholar] [CrossRef]
- Chen, A.; Chen, C.; Xiu, Y.; Liu, X.; Chen, J.; Guo, L.; Zhang, R.; Hou, Z. Niobate salts of organic base catalyzed chemical fixation of carbon dioxide with epoxides to form cyclic carbonates. Green Chem. 2015, 17, 1842–1852. [Google Scholar] [CrossRef]
- Hayashi, S.; Yamazoe, S.; Koyasu, K.; Tsukuda, T. Lewis base catalytic properties of [Nb10O28]6− for CO2 fixation to epoxide: kinetic and theoretical studies. Chem. – Asian J. 2017, 12, 1635–1640. [Google Scholar] [CrossRef]
- Al-Garni, T.; Al-Jallal, N.; Aouissi, A. Synthesis of propylene carbonate from epoxide and CO2 catalyzed by carbon nanotubes supported Fe1.5PMo12O40. J. Chem. 2017, 2017, 1–9. [Google Scholar] [CrossRef]
- Gómez-Romer, P. Polyoxometalates as photoelectrochemical models for quantum-sized colloidal semiconducting oxides. Solid State Ionics 1997, 101, 243–248. [Google Scholar]
- Vickers, J.W.; Lv, H.; Sumliner, J.M.; Zhu, G.; Luo, Z.; Musaev, D.G.; Geletii, Y.V.; Hill, C.L. Differentiating homogeneous and heterogeneous water oxidation catalysis: confirmation that [Co4(H2O)2(α-PW9O34)2]10− is a molecular water oxidation catalyst. J. Am. Chem. Soc. 2013, 135, 14110–14118. [Google Scholar] [CrossRef]
- Rozes, L.; Sanchez, C. Titanium oxo-clusters: Precursors for a Lego-like construction of nanostructured hybrid materials. Chem. Soc. Rev. 2011, 40, 1006–1030. [Google Scholar] [CrossRef]
- Izarova, N.V.; Pope, M.T.; Kortz, U. Noble metals in polyoxometalates. Angew. Chem., Int. Ed. 2012, 51, 9492–9510. [Google Scholar] [CrossRef]
- Dan-Hardi, M.; Serre, C.; Frot, T.; Rozes, L.; Maurin, G.; Sanchez, C.; Férey, G. A new photoactive crystalline highly porous titanium(IV) dicarboxylate. J. Am. Chem. Soc. 2009, 131, 10857–10859. [Google Scholar] [CrossRef]
- Habisreutinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K.; Schmidt-Mende, L.; Schmidt-Mende, L. Photocatalytic reduction of CO2 on TiO2 and other semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef]
- Abdullah, H.; Khan, M.M.R.; Ong, H.R.; Yaakob, Z. Modified TiO2 photocatalyst for CO2 photocatalytic reduction: an overview. J. CO2 Util. 2017, 22, 15–32. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Jaroniec, M.; Chen, X. Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem. Rev. 2019, 119, 3962–4179. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Ayass, W.W.; Taffa, D.H.; Schneemann, A.; Semrau, A.L.; Wannapaiboon, S.; Altmann, P.J.; Pöthig, A.; Nisar, T.; Balster, T.; et al. Discovery of polyoxo-noble-metalate-based metal–organic frameworks. J. Am. Chem. Soc. 2019, 141, 3385–3389. [Google Scholar] [CrossRef] [PubMed]
- Pley, M.; Wickleder, M.S. The cluster ion [Pt12O8(SO4)12]4−. Angew. Chem. Int. Ed. 2004, 43, 4168–4170. [Google Scholar] [CrossRef]
- Goloboy, J.C.; Klemperer, W.G. Are particulate noble-metal catalysts metals, metal oxides, or something in-between? Angew. Chem. Int. Ed. 2009, 48, 3562–3564. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wang, J.; Xu, H.; Xiong, Y. Coordination chemistry in the design of heterogeneous photocatalysts. Chem. Soc. Rev. 2017, 46, 2799–2823. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.D.; Katsoukis, G.; Frei, H. Photoinduced electron transfer from ZrOCo binuclear light absorber to pyridine elucidated by transient optical and infrared spectroscopy. J. Phys. Chem. C 2018, 122, 20176–20185. [Google Scholar] [CrossRef]
- Kim, W.; Frei, H. Directed assembly of cuprous oxide nanocatalyst for CO2 reduction coupled to heterobinuclear ZrOCoII light absorber in mesoporous silica. ACS Catal. 2015, 5, 5627–5635. [Google Scholar] [CrossRef]
- Kim, W.; Yuan, G.; McClure, B.A.; Frei, H. Light induced carbon dioxide reduction by water at binuclear ZrOCoII unit coupled to Ir oxide nanocluster catalyst. J. Am. Chem. Soc. 2014, 136, 11034–11042. [Google Scholar] [CrossRef]
- Macnaughtan, M.L.; Soo, H.S.; Frei, H. Binuclear ZrOCo metal-to-metal charge-transfer unit in mesoporous silica for light-driven CO2 reduction to CO and formate. J. Phys. Chem. C 2014, 118, 7874–7885. [Google Scholar] [CrossRef]
- Lin, W.; Frei, H. Photochemical CO2 splitting by metal-to-metal charge-transfer excitation in mesoporous ZrCu(I)-MCM-41 silicate sieve. J. Am. Chem. Soc. 2005, 127, 1610–1611. [Google Scholar] [CrossRef]
- McClure, B.A.; Frei, H. Excited state electron transfer of all-inorganic heterobinuclear TiOMn2+ chromophore anchored on silica nanoparticle surface. J. Phys. Chem. C 2014, 118, 11601–11611. [Google Scholar] [CrossRef]
- Takashima, T.; Yamaguchi, A.; Hashimoto, K.; Nakamura, R. Multielectron-transfer reactions at single Cu(II) centers embedded in polyoxotungstates driven by photo-induced metal-to-metal charge transfer from anchored Ce(III) to framework W(VI). Chem. Commun. 2012, 48, 2964. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Takashima, T.; Hashimoto, K.; Nakamura, R. Design of metal-to-metal charge-transfer chromophores for visible-light activation of oxygen-evolving Mn oxide catalysts in a polymer film. Chem. Mater. 2017, 29, 7234–7242. [Google Scholar] [CrossRef]
- Glass, E.N.; Fielden, J.; Kaledin, A.L.; Musaev, D.G.; Lian, T.; Hill, C.L. Extending metal-to-polyoxometalate charge transfer lifetimes: the effect of heterometal location. Chem. Eur. J. 2014, 20, 4297–4307. [Google Scholar] [CrossRef]
- Glass, E.N.; Musaev, D.G.; Lian, T.; Hill, C.L.; Fielden, J.; Huang, Z.; Xiang, X. Transition metal substitution effects on metal-to-polyoxometalate charge transfer. Inorg. Chem. 2016, 55, 4308–4319. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Takashima, T.; Hashimoto, K.; Nakamura, R. Preparation of polyoxometalate-based photo-responsive membranes for the photo-activation of manganese oxide catalysts. J. Vis. Exp. 2018, 138, e58072. [Google Scholar] [CrossRef]
- Meng, Y.-S.; Sato, O.; Liu, T. Manipulating metal-to-metal charge transfer for materials with switchable functionality. Angew. Chem. Int. Ed. 2018, 57, 12216–12226. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, D.; Xu, X.; Sheng, Y.; Xu, J.; Han, Y.-F. Application of operando spectroscopy on catalytic reactions. Curr. Opin. Chem. Eng. 2016, 12, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Freund, T.; Gomes, W.P. Electrochemical methods for investigating catalysis by semiconductors. Catal. Rev. 1970, 3, 1–36. [Google Scholar] [CrossRef]
- Lopez, X.; Carbó, J.J.; Bo, C.; Poblet, J.M. Structure, properties and reactivity of polyoxometalates: A theoretical perspective. Chem. Soc. Rev. 2012, 41, 7537. [Google Scholar] [CrossRef]
- Lyon, D.K.; Miller, W.K.; Novet, T.; Domaille, P.J.; Evitt, E.; Johnson, D.C.; Finke, R.G. Highly oxidation resistant inorganic-porphyrin analog polyoxometalate oxidation catalysts. 1. The synthesis and characterization of aqueous-soluble potassium salts of α2-P2W17O61(Mn+·OH2)(n-10) and organic solvent soluble tetra-n-butylammonium salts of α2-P2W17O61(Mn+·Br)(n-11) (M = Mn3+, Fe3+, Co2+, Ni2+, Cu2+). J. Am. Chem. Soc. 1991, 113, 7209–7221. [Google Scholar]
- Randell, W.J.; Weakley, T.J.R.; Finke, R.G. Oxidation resistant inorganic porphyrin analog polyoxometalates. 3. The synthesis and X-ray crystallographic characterization of a new heteropolyoxoanion structural type, the diruthenium-oxo-bridged “bimetallic unorganic-polyphyrin analog KLi15[O{RuIVCl(α2-P2W17O61)}2]·2KCl·60H2O”. Inorg. Chem. 1993, 32, 1068–1071. [Google Scholar]
- Neumann, R.; Dahan, M. A ruthenium-substituted polyoxometalate as an inorganic dioxygenase for activation of molecular oxygen. Nature 1997, 388, 353–355. [Google Scholar] [CrossRef]
- Corbin, N.; Zeng, J.; Williams, K.; Manthiram, K. Heterogeneous molecular catalysts for electrocatalytic CO2 reduction. Nano Res. 2019, 1–33. [Google Scholar] [CrossRef]
- Shih, C.F.; Zhang, T.; Li, J.; Bai, C. Powering the future with liquid sunshine. Joule 2018, 2, 1925–1949. [Google Scholar] [CrossRef]
- Xu, Q.; Niu, Y.; Wang, G.; Li, Y.; Zhao, Y.; Singh, V.; Niu, J.; Wang, J. Polyoxoniobates as a superior Lewis base efficiently catalyzed Knoevenagel condensation. Mol. Catal. 2018, 453, 93–99. [Google Scholar] [CrossRef]
Sample Availability: No available |





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Products | Products Reaction E0 (V) | E0 (V) pH = 7 |
|---|---|---|
| - | HO + 2 h+ → 1/2O2 + 2H+ | +0.82 |
| - | CO2 + e− → | −1.9 |
| HCOOH | CO2 + 2 H+ + 2 e− → HCOOH | −0.61 |
| CO | CO2 + 2 H+ + 2 e− → CO + H2O | −0.53 |
| HCHO | CO2 + 2 H+ + 4 e− → HCHO + H2O | −0.48 |
| CH3OH | CO2 + 6 H+ + 6 e− → CH3OH + H2O | −0.38 |
| CH4 | CO2 + 8 H+ + 8 e− → CH4 + 2H2O | −0.24 |
| - | 2 H+ + 2 e− → H2 | –0.41 |
| Entry | Catalysts | Products (μmol g−1 h−1) | ||
|---|---|---|---|---|
| CO | CH4 | H2 | ||
| 1 | Au@NENU-10 | 12.8 | 2.1 | 2.6 |
| 2 | Au@NENU-3 | 0.5 | - | 0.15 |
| 3 | Au/Na3PW12 O40) | - | - | 0.45 |
| 4 | Au/K7(PTi2W10O40) | 2.1 | 0.35 | 0.29 |
| 5 | NENU-10 | - | - | - |
| 6 | NENU-3 | - | - | - |
| 7 | HKUST-1 | - | - | - |
| Catalyst | HCOOH Yield (μmol) | TON | TOF (s−1) |
|---|---|---|---|
| {Mo154}1165 | 116.7 | 778 | 377 |
| {Mn6P3W24}931 | 40.6 | 270 | 56 |
| {Mo132}1064@RGO | 205 | 1366 | 610 |
| Entry | Catalyst | CO (nmol/g) | CH4 (nmol/g) | CH4-TON (10−3) | CH4-TOF (10−3 h−1) | All-TON (10−3) | All-TOF (10−3 h−1) |
|---|---|---|---|---|---|---|---|
| 1 | NENU-605 | 52 | 170 | 104.1 | 5.5 | 135.9 | 7.2 |
| 2 | NENU-606 | 68 | 402 | 241.4 | 10.5 | 282.2 | 12.3 |
| 3 | NENU-607 | 47 | 70 | 15.2 | 0.75 | 25.4 | 1.3 |
| 4 b | NENU-606 | n.d. | n.d. | - | - | - | - |
| 5 c | NENU-606 | n.d. | n.d. | - | - | - | - |
| 6 d | NENU-606 | n.d. | n.d. | - | - | - | - |
| 7 | blank | n.d. | n.d. | - | - | - | - |
| Products | Half-Electrochemical Thermodynamic Reactions | V vs. SHE |
|---|---|---|
| C | CO2(g) + 4 H+ + 4 e− = C(s) + 2 H2O(l) | −0.210 |
| C | CO2(g) + 2 H2O(l) + 4 e− = C(s) + 4 OH− | −0.627 |
| HCOOH | CO2(g) + 2 H+ + 2e− = HCOOH(l) | −0.250 |
| HCOO− | CO2(g) + 2 H2O(l) + 2 e− = HCOO− (aq) + OH− | −1.078 |
| CO | CO2(g) + 2 H+ + 2 e− = CO(g) + H2O(l) | −0.106 |
| CO | CO2(g) + 2 H2O(l) + 2 e− = CO(g) + 2 OH− | −0.934 |
| HCHO | CO2(g) + 4 H+ + 4e− = HCHO(l) + H2O(l) | −0.070 |
| HCHO | CO2(g) + 3 H2O(l) + 4 e− = HCHO(l) + 4 OH− | −0.898 |
| CH3OH | CO2(g) + 6 H+ + 6 e− = CH3OH(l) + H2O(l) | 0.016 |
| CH3OH | CO2(g) + 5 H2O(l) + 6 e− = CH3OH(l) + 6 OH− | −0.812 |
| CH4 | CO2(g) + 8 H+ + 8 e− = CH4(g) + 2 H2O (l) | 0.169 |
| CH4 | CO2(g) + 6 H2O(l) + 8 e− = CH4(g) + 8 OH− | −0.659 |
| oxalic acid | 2 CO2(g) + 2 H+ + 2 e− = (COOH)2(aq) | −0.500 |
| oxalate | 2 CO2(g) + 2 e− = C2(aq) | −0.590 |
| ethylene | 2 CO2(g) + 12 H+ + 12 e− = CH2CH2(g) + 4 H2O(l) | 0.064 |
| ethylene | 2 CO2(g) + 8 H2O(l) + 12 e− = CH2CH2(g) + 12 OH− | −0.764 |
| ethanol | 2 CO2(g) + 12 H+ + 12 e− = CH3CH2OH(l) + 3 H2O(l) | 0.084 |
| ethanol | 2 CO2(g) + 9 H2O(l) + 12 e− = CH3CH2OH(l) + 12 OH− | −0.744 |
| Catalysts | E V vs. SHE | Onset Potential V | jCO mA cm−2 | Tafel Slope (mV dec−1) | ECSA mF cm−2 | EIS Ω | FEco % | TOF H−1 |
|---|---|---|---|---|---|---|---|---|
| Co-PMOF | −0.8 | −0.35 | 18.08 | 98 | 12.17 | 9.83 | 98.7 | 1656 |
| Fe-PMOF | −0.7 | −0.53 | 0.47 | 211 | 10.26 | 10.26 | 28.8 | 17.45 |
| Ni-PMOF | −0.8 | −0.58 | 0.27 | 675 | 10.16 | 10.70 | 18.5 | 8.11 |
| Zn-PMOF | −0.9 | −0.60 | 0.02 | 206 | 9.83 | 12.17 | 0.95 | 0.005 |
| Co-TMCP | −0.9 | −0.53 | NA b | 151 | NA | NA | 40 | NA |
| TMCP | −0.6 | −0.67 | NA | 552 | NA | NA | 0.77 | NA |
| NNU-12 | −0.6 | −0.6 | NA | 413 | NA | NA | 1.8 | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.; Chen, Q.; Shen, C.; He, L. Polyoxometalate-Based Catalysts for CO2 Conversion. Molecules 2019, 24, 2069. https://doi.org/10.3390/molecules24112069
Cao Y, Chen Q, Shen C, He L. Polyoxometalate-Based Catalysts for CO2 Conversion. Molecules. 2019; 24(11):2069. https://doi.org/10.3390/molecules24112069
Chicago/Turabian StyleCao, Yanwei, Qiongyao Chen, Chaoren Shen, and Lin He. 2019. "Polyoxometalate-Based Catalysts for CO2 Conversion" Molecules 24, no. 11: 2069. https://doi.org/10.3390/molecules24112069
APA StyleCao, Y., Chen, Q., Shen, C., & He, L. (2019). Polyoxometalate-Based Catalysts for CO2 Conversion. Molecules, 24(11), 2069. https://doi.org/10.3390/molecules24112069