
Exploring Castellaniella defragrans Linalool (De)hydratase-Isomerase for Enzymatic Hydration of Alkenes

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
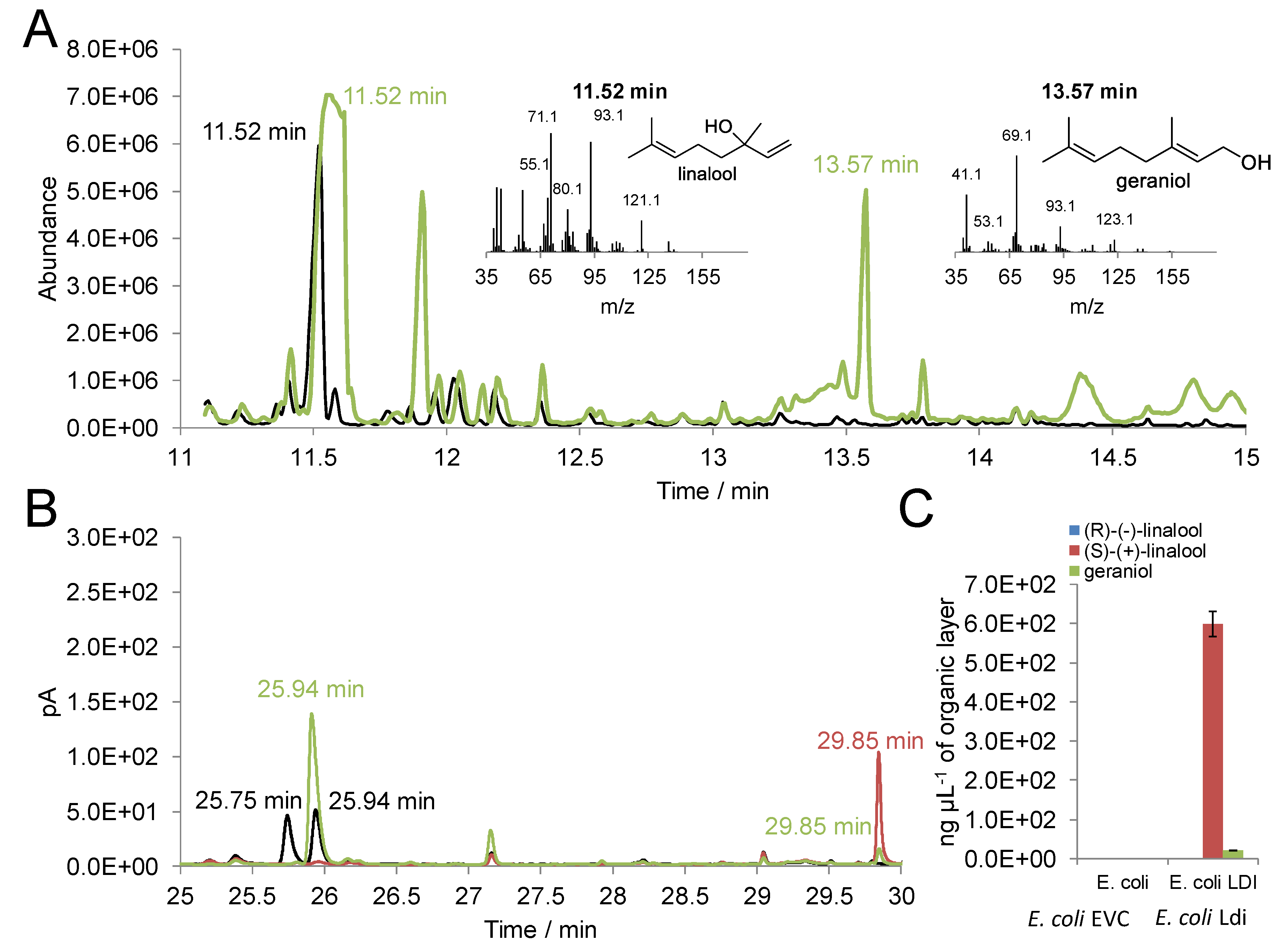
2.1. Activity of Recombinant Ldi in E. coli
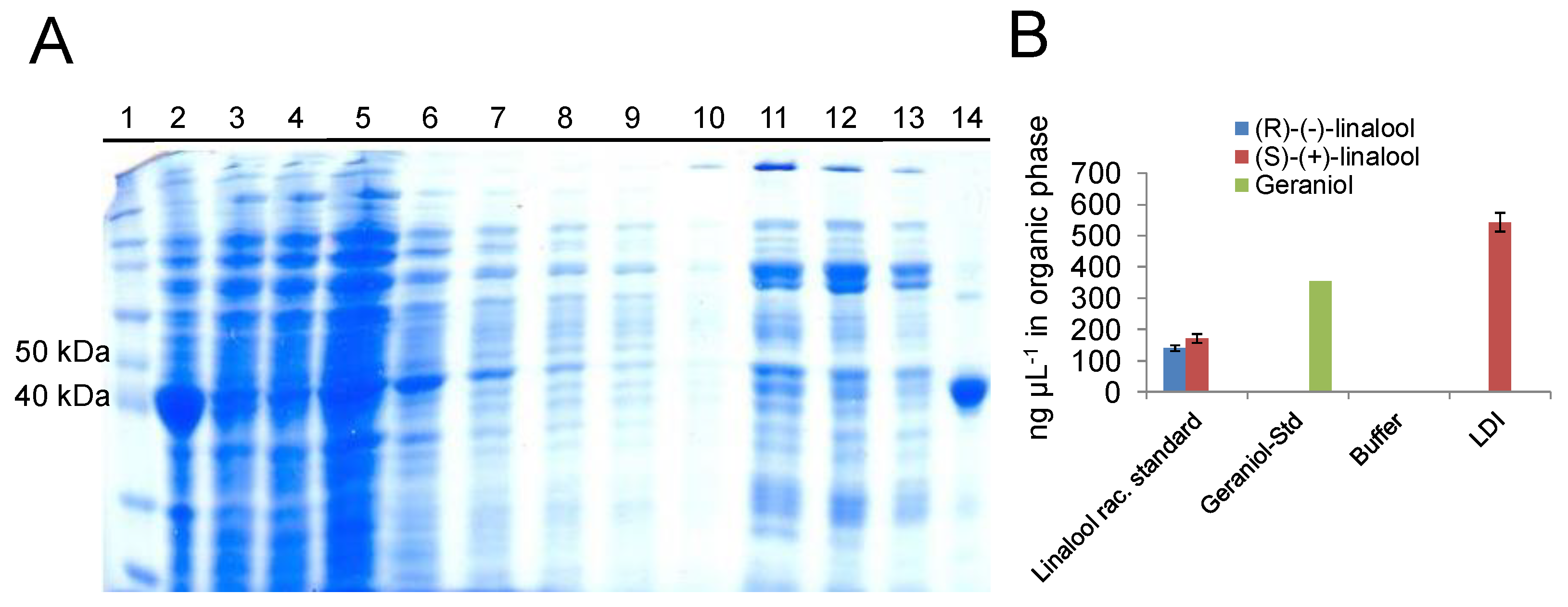
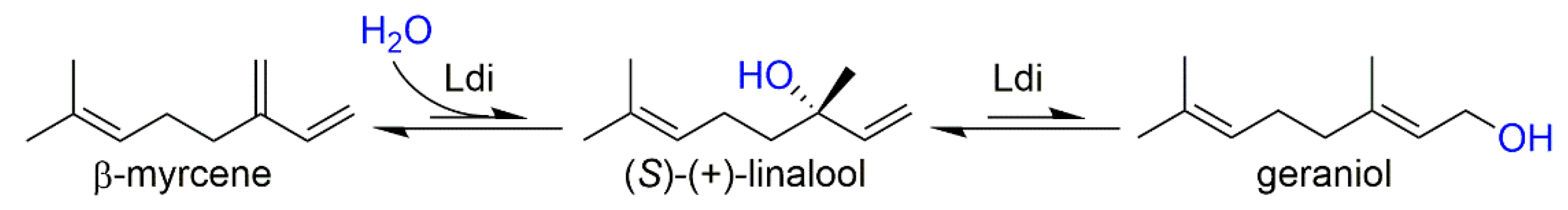
2.2. In Vitro Activity of Purified Ldi
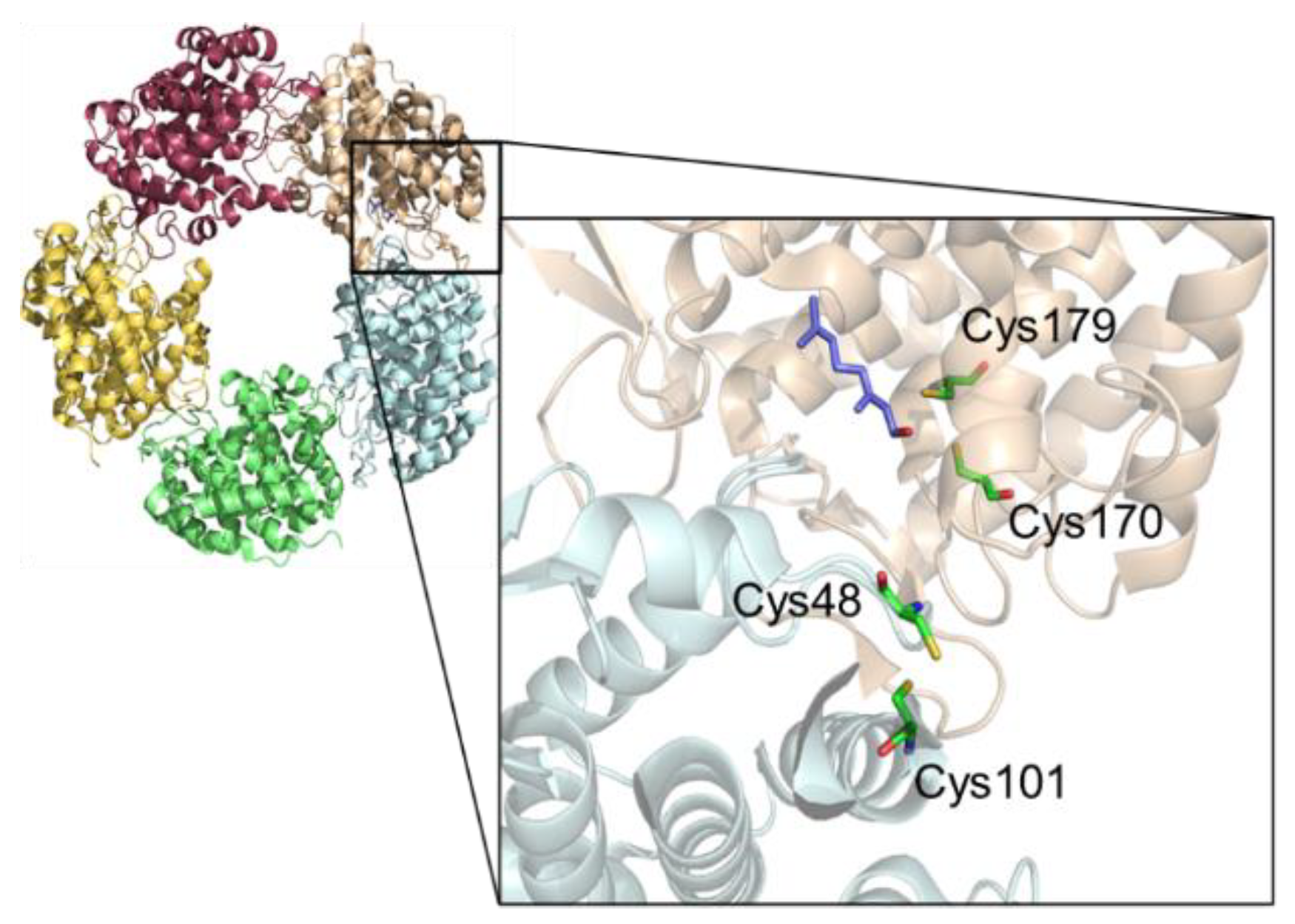
2.3. Impact of Cysteine Exchange on Ldi Activity
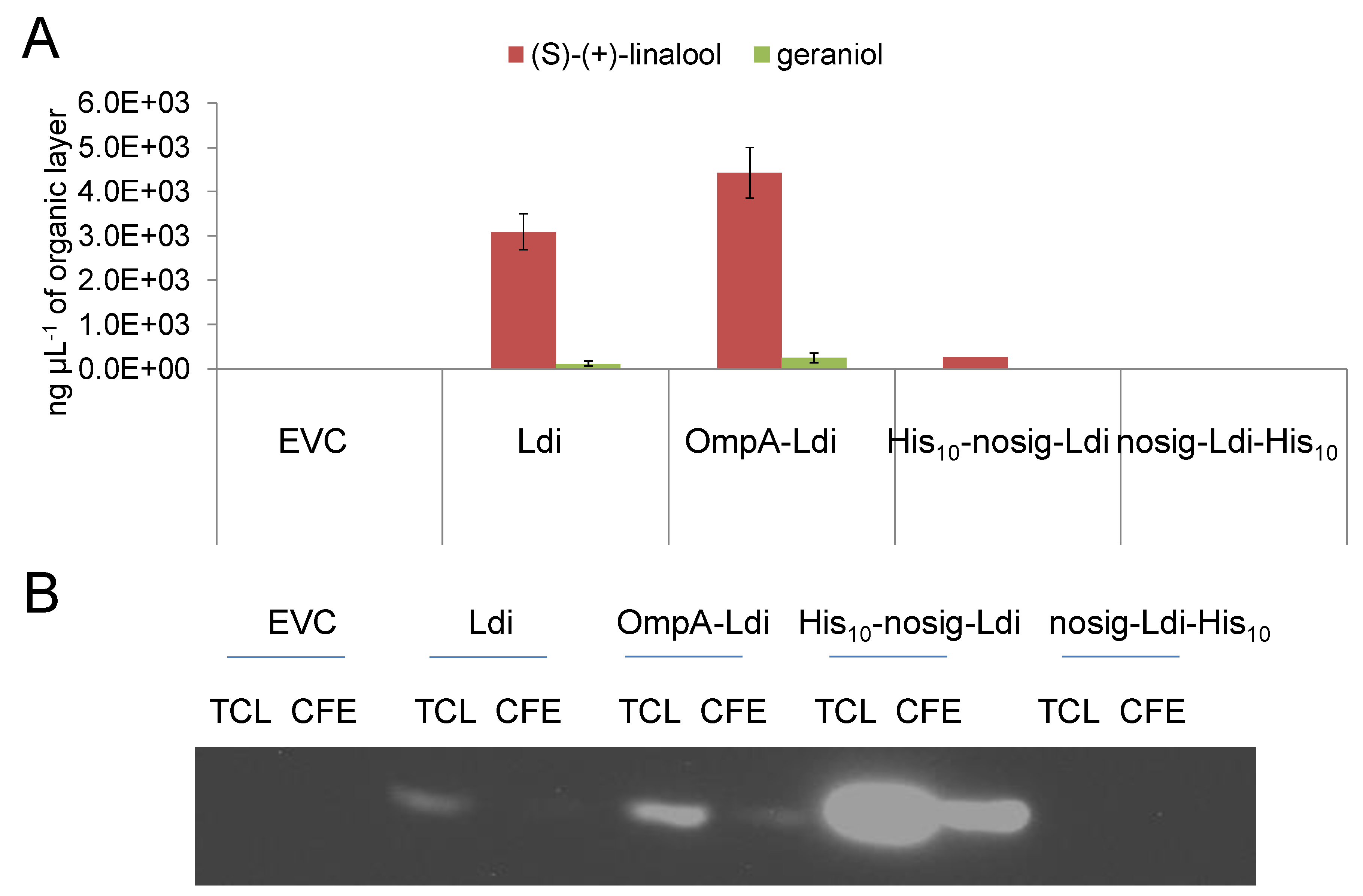
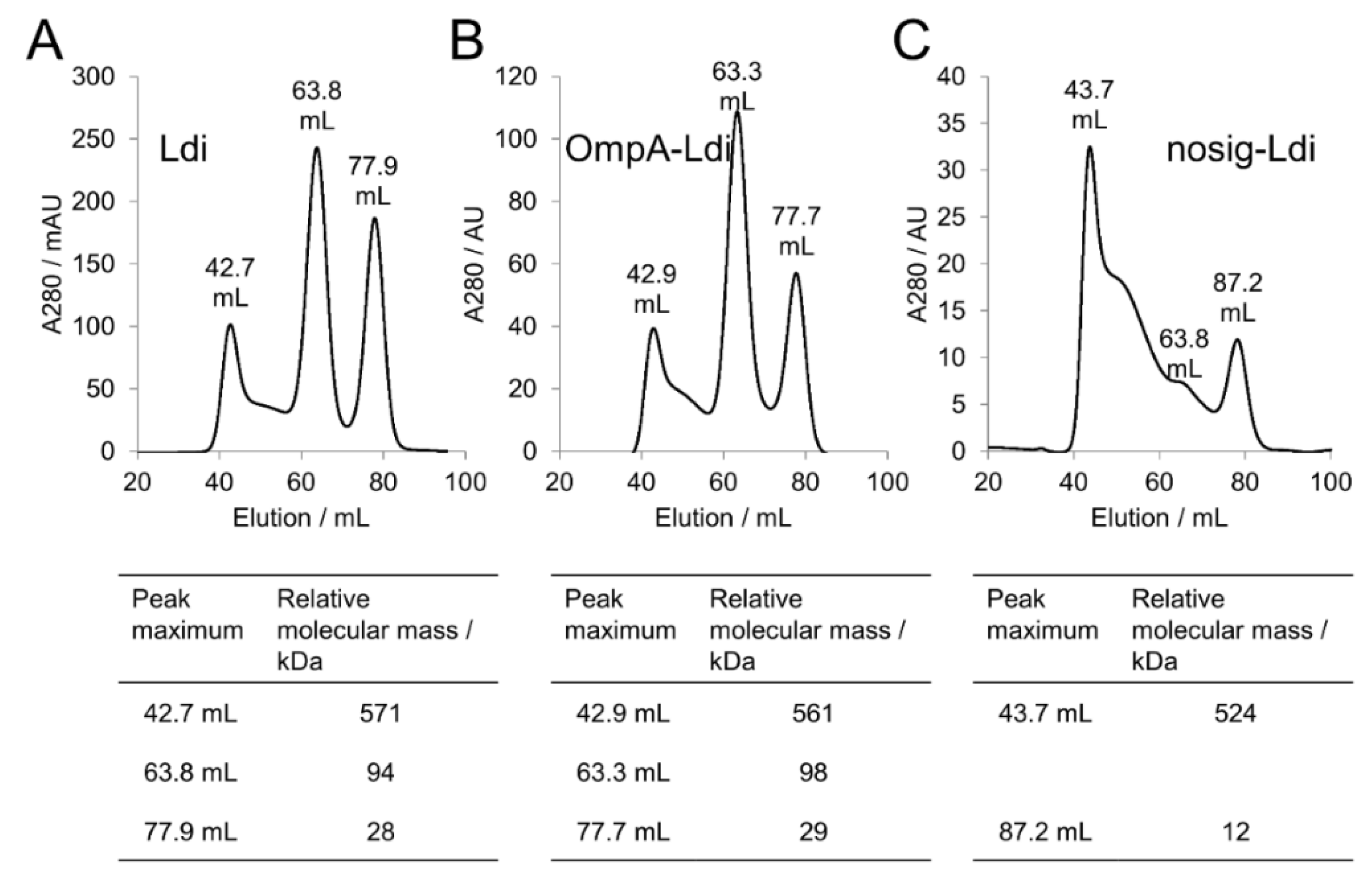
2.4. Periplasmic and Cytosolic Targeting of Ldi
2.5. Impact of Cellular Localization on Ldi Assembly and Activity
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Media
4.2. Cloning and Site-Directed Mutagenesis
4.3. Expression of Recombinant Ldi in E. coli
4.4. Whole-Cell Biotransformation of β-myrcene with E. coli
4.5. Cell Lysis and Purification of Ldi
4.6. In Vitro β-myrcene Conversion
4.7. Analysis of Monoterpenoids by GC-MS and GC-FID
4.8. Protein Analysis and Immunoblotting
4.9. Isolation of Recombinant Ldi from E. coli Periplasm and Cytosol
4.10. Circular Dichroism Spectroscopy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bicas, J.L.; Dionísio, A.P.; Pastore, G.M. Bio-oxidation of terpenes: An approach for the flavor Industry. Chem. Rev. 2009, 109, 4518–4531. [Google Scholar] [CrossRef] [PubMed]
- Brodkorb, D.; Gottschall, M.; Marmulla, R.; Lüddeke, F.; Harder, J. Linalool dehydratase-isomerase, a bifunctional enzyme in the anaerobic degradation of monoterpenes. J. Biol. Chem. 2010, 285, 30436–30442. [Google Scholar] [CrossRef]
- Demming, R.M.; Fischer, M.-P.; Schmid, J.; Hauer, B. (De)hydratases-recent developments and future perspectives. Curr. Opin. Chem. Biol. 2018, 43, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Lüddeke, F.; Harder, J. Enantiospecific (S)-(+)-Linalool Formation from β-Myrcene by Linalool Dehydratase-Isomerase. Z. Naturforsch. C 2011, 66, 409–412. [Google Scholar]
- Marmulla, R.; Šafarić, B.; Markert, S.; Schweder, T.; Harder, J. Linalool isomerase, a membrane-anchored enzyme in the anaerobic monoterpene degradation in Thauera linaloolentis 47Lol. BMC Biochem. 2016, 17, 1–11. [Google Scholar] [CrossRef]
- Weidenweber, S.; Marmulla, R.; Ermler, U.; Harder, J. X-ray structure of linalool dehydratase/isomerase from Castellaniella defragrans reveals enzymatic alkene synthesis. FEBS Lett. 2016, 590, 1375–1383. [Google Scholar] [CrossRef]
- De Marco, A. Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb. Cell Fact. 2009, 8, 1–18. [Google Scholar] [CrossRef]
- Nestl, B.M.; Geinitz, C.; Popa, S.; Rizek, S.; Haselbeck, R.J.; Stephen, R.; Noble, M.A.; Fischer, M.P.; Ralph, E.C.; Hau, H.T.; et al. Structural and functional insights into asymmetric enzymatic dehydration of alkenols. Nat. Chem. Biol. 2017, 13, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Ling, B.; Wang, X.; Su, H.; Liu, R.; Liu, Y. Protonation state and fine structure of the active site determine the reactivity of dehydratase: Hydration and isomerization of β-myrcene catalyzed by linalool dehydratase/isomerase from Castellaniella defragrans. Phys. Chem. Chem. Phys. 2018, 20, 17342–17352. [Google Scholar] [CrossRef]
- Oliver, J.L.; Marín, A. A relationship between GC content and coding-sequence length. J. Mol. Evol. 1996, 43, 216–223. [Google Scholar] [CrossRef]
- Petasch, J.; Disch, E.M.; Markert, S.; Becher, D.; Schweder, T.; Hüttel, B.; Reinhardt, R.; Harder, J. The oxygen-independent metabolism of cyclic monoterpenes in Castellaniella defragrans 65Phen. BMC Microbiol. 2014, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fisch, F.; Fleites, C.M.; Delenne, M.; Baudendistel, N.; Hauer, B.; Turkenburg, J.P.; Hart, S.; Bruce, N.C.; Grogan, G. A covalent succinylcysteine-like intermediate in the enzyme-catalyzed transformation of maleate to fumarate by maleate isomerase. J. Am. Chem. Soc. 2010, 132, 11455–11457. [Google Scholar] [CrossRef] [PubMed]
- Glavas, S.; Tanner, M.E. Active site residues of glutamate racemase. Biochemistry 2001, 40, 6199–6204. [Google Scholar] [CrossRef]
- Movva, N.R.; Nakamura, K.; Inouye, M. Amino acid sequence of the signal peptide of ompA protein, a major outer membrane protein of Escherichia coli. J. Biol. Chem. 1980, 255, 27–29. [Google Scholar]
- Gräslund, S.; Nordlund, P.; Weigelt, J.; Hallberg, B.M.; Bray, J.; Gileadi, O.; Knapp, S.; Oppermann, U.; Arrowsmith, C.; Hui, R.; et al. Protein production and purification. Nat. Methods 2008, 5, 135–146. [Google Scholar]
- Booth, W.T.; Schlachter, C.R.; Pote, S.; Ussin, N.; Mank, N.J.; Klapper, V.; Offermann, L.R.; Tang, C.; Hurlburt, B.K.; Chruszcz, M. Impact of an N-terminal Polyhistidine Tag on Protein Thermal Stability. ACS Omega 2018, 3, 760–768. [Google Scholar] [CrossRef]
- Tsirigotaki, A.; De Geyter, J.; Šoštarić, N.; Economou, A.; Karamanou, S. Protein export through the bacterial Sec pathway. Nat. Rev. Microbiol. 2017, 15, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, H.; Bardwell, J.C.A. Catalysis of disulfide bond formation and isomerization in the Escherichia coli periplasm. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1694, 111–119. [Google Scholar] [CrossRef]
- Mamathambika, B.S.; Bardwell, J.C. Disulfide-Linked Protein Folding Pathways. Annu. Rev. Cell Dev. Biol. 2008, 24, 211–235. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, S.; Rujas, E.; Ytterberg, A.J.; Zubarev, R.A.; Luirink, J.; de Gier, J.W. Optimizing heterologous protein production in the periplasm of E. coli by regulating gene expression levels. Microb. Cell. Fact. 2013, 12, 1–11. [Google Scholar] [CrossRef]
- Engleder, M.; Pichler, H. On the current role of hydratases in biocatalysis. Appl. Microbiol. Biotechnol. 2018, 102, 5841–5858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Naismith, J.H. A simple and efficient expression and purification system using two newly constructed vectors. Protein Expr. Purif. 2009, 63, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balzer, D.; Ziegelin, G.; Pansegrau, W.; Kruft, V.; Lanka, E. KorB protein of promiscuous plasmid RP4 recognizes inverted sequence repetitions in regions essential for conjugative plasmid transfer. Nucleic Acids Res. 1992, 20, 1851–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ausubel, M.; Brent, R.; Kingston, R.E.; Moore, D.D.; Seidman, J.G.; Smith, J.A.; Struhl, K. Current Protocols in Molecular Biology; Wiley-Liss, Inc.: New York, NY, USA, 1987. [Google Scholar]
- Ueguchi, C.; Ito, K. Escherichia coli sec mutants accumulate a processed immature form of maltose-binding protein (MBP), a late-phase intermediate in MBP export. J. Bacteriol. 1990, 172, 5643–5649. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ldi Construct | Pooled Fraction | Relative Molecular Mass/kDa | Assembly in Solution | Relative Activity for β-myrcene Hydration [%] |
|---|---|---|---|---|
| Ldi | 37–46 mL | 930–430 | Multimer (>8 subunits) | 4 |
| 57–70 mL | 168–55 | Oligomer (2 to 4 subunints) | 100 | |
| 72–84 mL | 47–17 | Monomer | 28 | |
| OmpA-Ldi | 38–47 mL | 854–395 | Multimer (>8 subunits) | ia |
| 58–70 mL | 154–55 | Oligomer (2 to 4 subunints) | 100 | |
| 73–83 mL | 43–18 | Monomer | 25 | |
| nosig-Ldi | 40–48 mL | 720–363 | Multimer (>8 subunits) | ia |
| 60–70 mL | 130–55 | Inconclusive | ia | |
| 74–83 mL | 39–18 | Inconclusive | ia |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Engleder, M.; Müller, M.; Kaluzna, I.; Mink, D.; Schürmann, M.; Leitner, E.; Pichler, H.; Emmerstorfer-Augustin, A. Exploring Castellaniella defragrans Linalool (De)hydratase-Isomerase for Enzymatic Hydration of Alkenes. Molecules 2019, 24, 2092. https://doi.org/10.3390/molecules24112092
Engleder M, Müller M, Kaluzna I, Mink D, Schürmann M, Leitner E, Pichler H, Emmerstorfer-Augustin A. Exploring Castellaniella defragrans Linalool (De)hydratase-Isomerase for Enzymatic Hydration of Alkenes. Molecules. 2019; 24(11):2092. https://doi.org/10.3390/molecules24112092
Chicago/Turabian StyleEngleder, Matthias, Monika Müller, Iwona Kaluzna, Daniel Mink, Martin Schürmann, Erich Leitner, Harald Pichler, and Anita Emmerstorfer-Augustin. 2019. "Exploring Castellaniella defragrans Linalool (De)hydratase-Isomerase for Enzymatic Hydration of Alkenes" Molecules 24, no. 11: 2092. https://doi.org/10.3390/molecules24112092
APA StyleEngleder, M., Müller, M., Kaluzna, I., Mink, D., Schürmann, M., Leitner, E., Pichler, H., & Emmerstorfer-Augustin, A. (2019). Exploring Castellaniella defragrans Linalool (De)hydratase-Isomerase for Enzymatic Hydration of Alkenes. Molecules, 24(11), 2092. https://doi.org/10.3390/molecules24112092