Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–5

 ,
,  ,
,  ,
,  ,
, 
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  , ,
, ,  , ,
, ,  ,
,  ,
,  ,
,  ,
,  , ,
, ,  ,
,  ,
,  and
and  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Inhibition of 5-Lipoxygenase-Activating Protein: A New Therapeutic Strategy to Combat Residual Inflammatory Risk in Patients with Ischaemic Heart Disease?
3. Repurposing Ticagrelor: When an Antiplatelet Drug Could Emerge as a Potent Antibacterial Agent
4. Anticancer Effective Tantalum Organo-Complexes
5. Design, Synthesis, and Biological Activities of New Pyrazole Derivatives Possessing Both Coxib and Combretastatins Pharmacophores
6. Novel Epoxyketone-Based Proteasome Inhibitors with Enhanced Metabolic Stability and Activity against PI-Resistant Models
7. Tyrosyl-DNA Phosphodiesterase 1 as a Novel Target to Enhance the Efficacy and Utility of Topotecan and Irinotecan
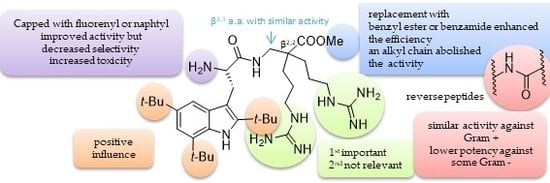
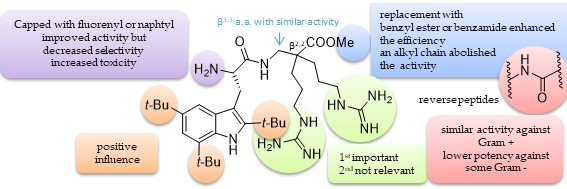
8. “KISS” Antimicrobial Peptides: “Keep It Simple and Stable” to Reach Selective New Antibiotics
9. Amino Acid-Based Prodrugs of a Fosmidomycin Surrogate as Antimalarial and Antitubercular Agents
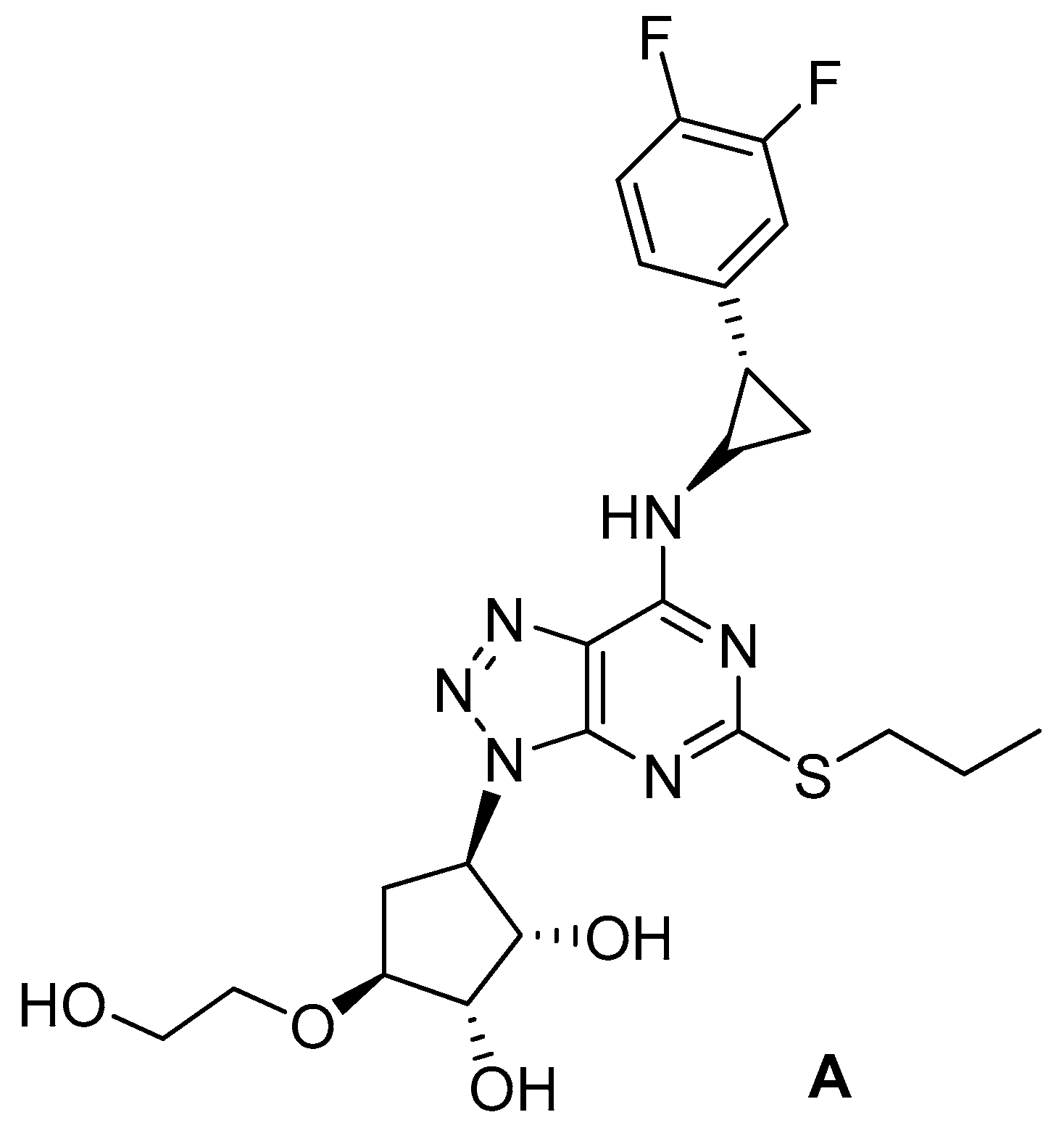
10. Horizon for a Prenatal Prevention of Cystic Fibrosis?
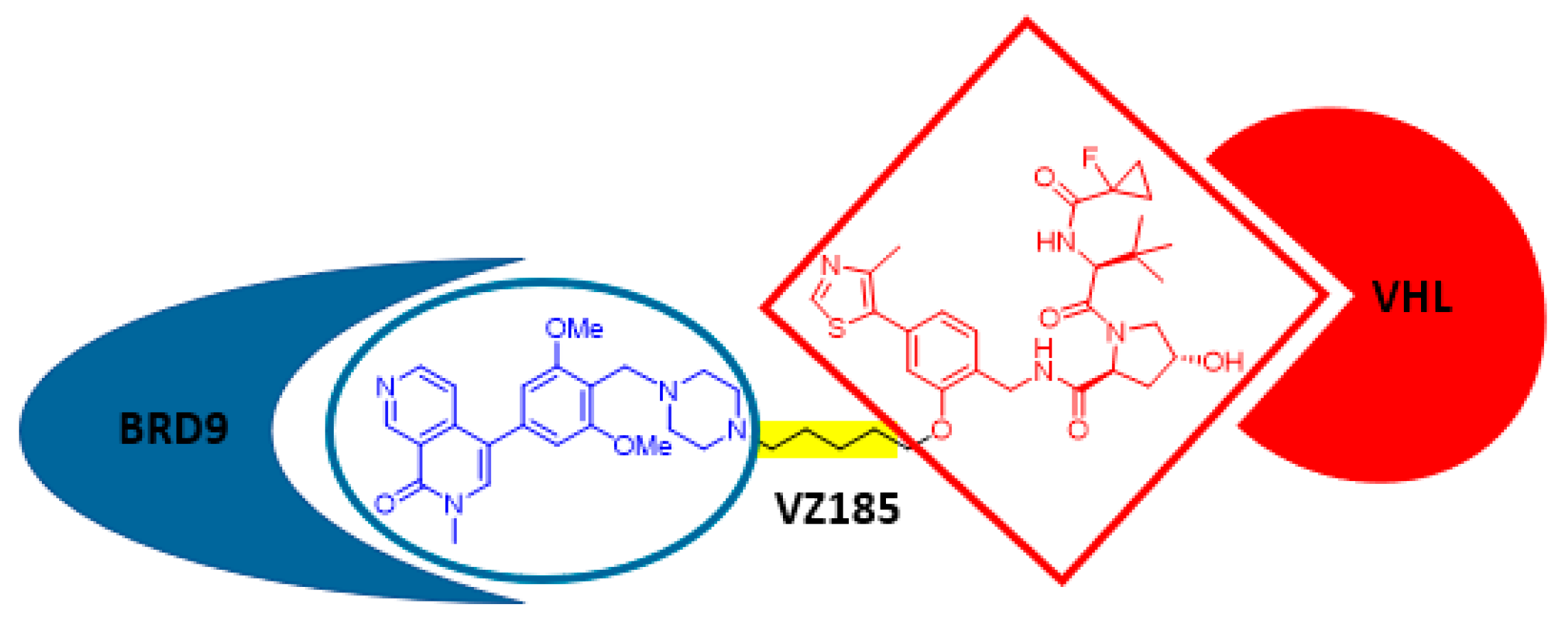
11. Small-Molecule PROTACs: A Promising Approach for the Development of Selective Drugs
12. Screen Using Huge Virtual Docking Library to Accelerate the Discovery of Candidate Drugs
13. Expanding the Range of Targeted Residues for the Design of Covalent Inhibitors
14. Amino Acid Versus Phosphate Ester Prodrugs for Improving the Oral Bioavailability of Atazanavir
15. A General Method to Quantify Ligand-Driven Oligomerization from Fluorescence-Based Images
16. Reactivation of PTEN Tumor Suppressor for Cancer Treatment Through Inhibition of a MYC–WWP1 Inhibitory Pathway
17. Derivatives of the Natural Alkaloid Matrine: New Anti-Fibrotic Tools in the Fight Against Idiopathic Pulmonary Fibrosis
18. Multi-Targeting Therapy for Glioblastoma: A Promising New Design
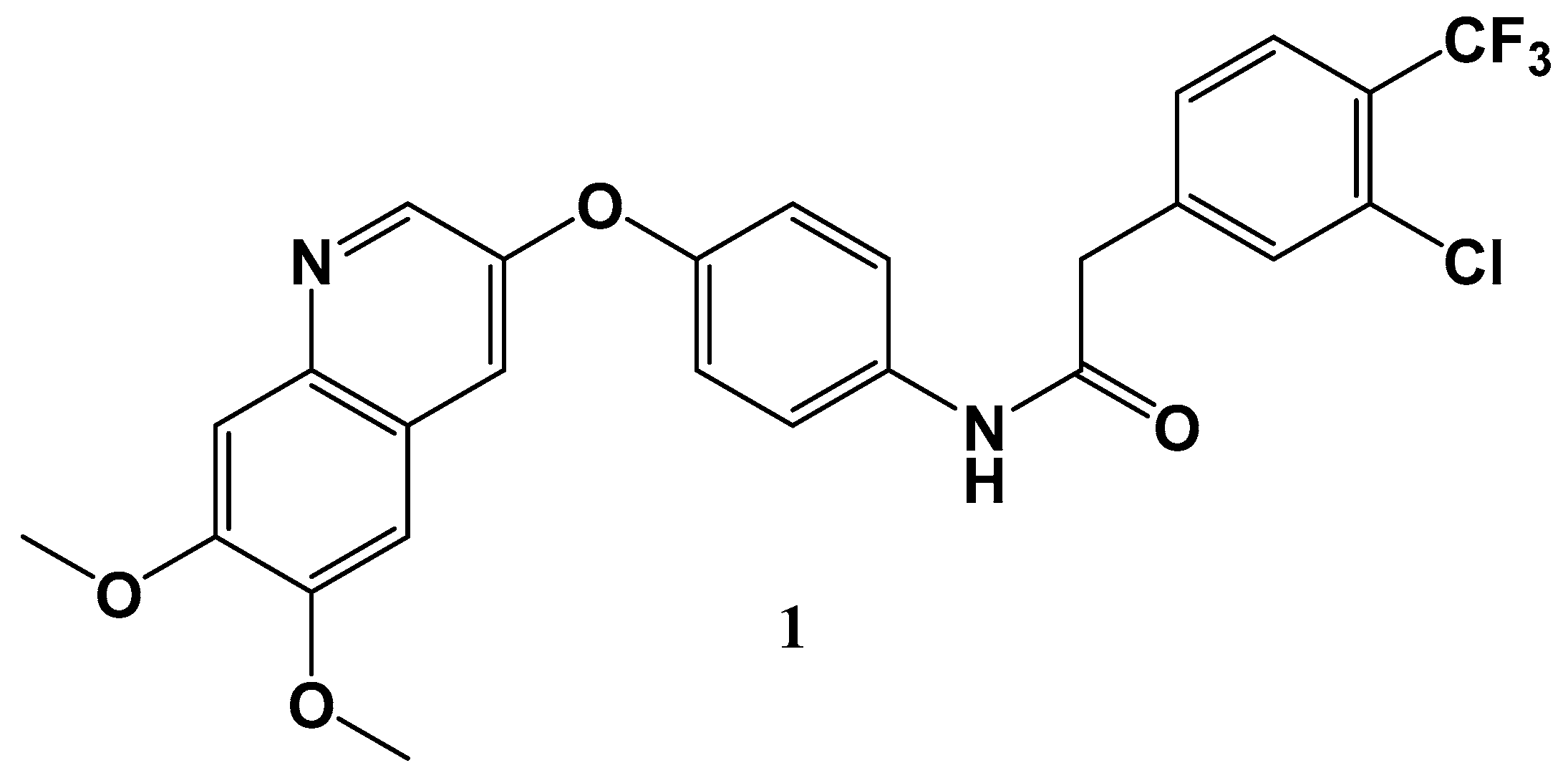
19. New Potential Therapeutics for Gastrointestinal Stromal Tumors
20. Directly Targeting Riboswitches with Small Molecules to Regulate Gene Expression: Identification of PreQ1 Riboswitches Ligands
21. Discovery of Protein–Protein Interaction Stabilizers by Site-Directed Fragment-Based Screening
22. The Short-Chain Fatty Acid Pentanoate as A Potent Immunomodulatory Molecule
23. Small-Molecular-Weight Synthetic Benzophenone Derivatives as New Potential Breast and Prostate Anticancer Drugs
24. Sex-Specific Leukotriene Formation is Causative for Sex Dimorphism in Murine Asthma-Like Features
25. New Asymmetric Pd-Catalyzed Synthesis of Nucleoside Analogs
26. Experimental Data Shed Light on the Molecular Basis of Cancer Resistance
27. PROTACing the UnPROTACable
Conflicts of Interest
References
- Kalkman, D.N.; Aquino, M.; Claessen, B.E.; Baber, U.; Guedeney, P.; Sorrentino, S.; Vogel, B.; de Winter, R.J.; Sweeny, J.; Kovacic, J.C.; et al. Residual inflammatory risk and the impact on clinical outcomes in patients after percutaneous coronary interventions. Eur. Heart J. 2018, 39, 4101–4108. [Google Scholar] [CrossRef] [PubMed]
- Hersberger, M. Potential role of the lipoxygenase derived lipid mediators in atherosclerosis: Leukotrienes, lipoxins and resolvins. Clin. Chem. Lab. Med. 2010, 48, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, D.; Broddefalk, J.; Emtenas, H.; Hayes, M.A.; Lemurell, M.; Swanson, M.; Ulander, J.; Whatling, C.; Amilon, C.; Ericsson, H.; et al. Discovery and early clinical development of an inhibitor of 5-lipoxygenase activating protein (AZD5718) for treatment of coronary artery disease. J. Med. Chem. 2019, 62, 4312–4324. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.R.; Zhang, G.; Macaulay, T.E.; Callahan, L.A.; Charnigo, R.; Vsevolozhskaya, A.; Li, Z.; Smyth, S. Ticagrelor reduces thromboinflammatory markers in patients with pneumonia. JACC Basic Transl. Sci. 2018, 3, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Lancellotti, P.; Musumeci, L.; Jacques, N.; Servais, L.; Goffin, E.; Pirotte, B.; Oury, C. Antibacterial activity of ticagrelor in conventional antiplatelet dosages against antibiotic-resistant Gram-positive bacteria. JAMA Cardiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal complexes in cancer therapy – an update from drug design perspective. Drug Des. Dev. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Starha, P.; Travnicek, Z.; Dvorak, Z. A cytotoxic tantalum(V) half-sandwich complex: A new challenge for metal-based anticancer agents. Chem. Commun. 2018, 54, 9533–9536. [Google Scholar] [CrossRef] [PubMed]
- Travnicek, Z.; Starha, P.; Cajan, M.; Dvorak, Z. A half-sandwich TaV dichlorido complex containing an O,N,O′-tridentate Schiff base ligand: Synthesis, crystal structure and in vitro cytotoxicity. Acta Cryst. 2019, C75, 248–254. [Google Scholar]
- Starha, P.; Travnicek, Z.; Dvorak, Z. Dichloro-complexes of tantalum, method of their preparation and their use in preparation of drugs for cancer treatment. CZ 307 696 B6, 9 January 2019. [Google Scholar]
- de Oliveira Pedrosa, M.; Duarte da Cruz, R.M.; de Oliveira Viana, J.; de Moura, R.O.; Ishiki, H.M.; Barbosa Filho, J.M.; Diniz, M.F.; Scotti, M.T.; Scotti, L.; Bezerra Mendonca, F.J. Hybrid Compounds as Direct Multitarget Ligands: A Review. Curr. Top. Med. Chem. 2017, 17, 1044–1079. [Google Scholar] [CrossRef]
- Thi, T.H.N.; Thi, Y.T.; Nguyen, L.A.; Vo, B.N.; Ngo, Q.A. Design, Synthesis and Biological Activities of New Pyrazole Derivatives Possessing Both Coxib and Combretastatins Pharmacophores. Chem. Biodivers. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Bhattarai, D.; Yoo, J.; Miller, Z.; Park, J.E.; Lee, S.; Lee, W.; Driscoll, J.J.; Kim, K.B. Development of novel epoxyketone-based proteasome inhibitors as a strategy to overcome cancer resistance to carfilzomib and bortezomib. J. Med. Chem. 2019, 62, 4444–4455. [Google Scholar] [CrossRef] [PubMed]
- Koldysheva, E.; Men’shchikova, A.; Lushnikova, E.; Popova, N.; Kaledin, V.; Nikolin, V.; Zakharenko, A.; Luzina, O.; Salakhutdinov, N.; Lavrik, O. Antimetastatic activity of combined topotecan and tyrosyl-DNA phosphodiesterase-1 inhibitor on modeled lewis lung carcinoma. Bull. Exp. Biol. Med. 2019, 166, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Strøm, M.B.; Haug, B.E.; Skar, M.L.; Stensen, W.; Stiberg, T.; Svendsen, J.S. The pharmacophore of short cationic antibacterial peptides. J. Med. Chem. 2003, 46, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Boullet, H.; Bentot, F.; Hequet, A.; Ganem-Elbaz, C.; Bechara, C.; Pacreau, E.; Launay, P.; Sagan, S.; Jolivalt, C.; Lacombe, C.; et al. Small antiMicrobial peptide with in vivo activity against sepsis. Molecules 2019, 24, 1702. [Google Scholar] [CrossRef] [PubMed]
- Courtens, C.; Risseeuw, M.; Caljon, G.; Maes, L.; Martin, A.; Van Calenbergh, S. Amino acid based prodrugs of a fosmidomycin surrogate as antimalarial and antitubercular agents. Bioorg. Med. Chem. 2019, 27, 729–747. [Google Scholar] [CrossRef] [PubMed]
- Jomaa, H.; Wiesner, J.; Sanderbrand, S.; Altincicek, B.; Weidemeyer, C.; Hintz, M.; Türbachova, I.; Eberl, M.; Zeidler, J.; Lichtenthaler, H.K.; et al. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 1999, 285, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Ruangweerayut, R.; Looareesuwan, S.; Hutchinson, D.; Chauemung, A.; Banmairuroi, V.; Na-Bangchang, K. Assessment of the pharmacokinetics and dynamics of two combination regimens of fosmidomycin-clindamycin in patients with acute uncomplicated falciparum malaria. Malaria J. 2008, 7, 225. [Google Scholar] [CrossRef]
- Su, X.; Yi, Y.; Yan, Z.; Rosen, B.H.; Liang, B.; Winter, M.C.; Evans, I.A.; Rotti, P.H.G.; Yu, Y.; Gray, J.S.; et al. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci. Trans. Med. 2019, eaau7531. [Google Scholar] [CrossRef]
- Smith, B.E.; Wang, S.L.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun. 2019, 10, 131. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, S.; Balius, T.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224. [Google Scholar] [CrossRef]
- Liu, R.; Yue, Z.; Tsai, C.-C.; Shen, J. Assessing lysine and cysteine reactivities for designing targeted covalent kinase inhibitors. J. Am. Chem. Soc. 2019, 141, 6553–6560. [Google Scholar] [CrossRef] [PubMed]
- Murugaiah, A.M.; Subbaiah, S.M.; Sridhar, D.; Thangeswaran, R.; Lakshumanan, S.; Mathiazhagan, A.; Salil, D.; Desai, S.S.; Susan, M.; Jenkins, M.R.; et al. Design, synthesis, and pharmacokinetic evaluation of phosphate and amino acid ester prodrugs for improving the oral bioavailability of the HIV-1 protease inhibitor atazanavir. J. Med. Chem. 2019, 62, 3553–3574. [Google Scholar]
- Stoneman, M.R.; Biener, G.; Ward, R.J.; Pediani, J.D.; Badu, D.; Eis, A.; Popa, I.; Milligan, G.; Raicu, V. A general method to quantify ligand-driven oligomerization from fluorescence-based images. Nat. Methods 2019. [Google Scholar] [CrossRef] [PubMed]
- Boosani, C.S.; Agrawal, D.K. PTEN modulators: A patent review. Expert Opin. Ther. Pat. 2013, 23, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.Y.; Fu, T.M.; Chen, H.; Ishikawa, T.; Chiang, S.Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364, eaau0159. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ma, L.; Wang, D.; Jia, H.; Yu, M.; Gu, Y.; Shang, H.; Zou, Z. Design and synthesis of matrine derivatives as novel anti-pulmonary fibrotic agents via repression of the TGFβ/Smad pathway. Molecules 2019, 24, 1108. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Chuan, D.; Hou, H.; Chen, H.; Han, B.; Zhang, X.; Zhou, L.; Tong, A.; Xu, J.; Guo, G. Development of a hybrid nanocarrier-recognizing tumor vasculature and penetrating the BBB for glioblastoma multi-targeting therapy. Nanoscale 2019. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Beilei, W.; Junjie, W.; Shuang, Q.; Fengming, Z.; Ziping, Q.; Feiyang, L.; Qingwang, L.; Cheng, C.; Chen, H.; et al. Discovery of 2-(4-Chloro-3-(trifluoromethyl)phenyl)-N-(4-((6,7-dimethoxyquinolin-4-yl)oxy)phenyl)acetamide (CHMFL-KIT-64) as a novel orally available potent inhibitor against broad-spectrum mutants of c-KIT kinase for gastrointestinal stromal tumors. J. Med. Chem. 2019. [Google Scholar] [CrossRef]
- Connelly, C.M.; Numata, T.; Boer, R.; Moon, M.H.; Sinniah, R.S.; Barchi, J.J.; Ferré-D’Amaré, A.R.; Schneekloth, J.S., Jr. Synthetic ligands for PreQ1 riboswitches provide structural and mechanistic insights into targeting RNA tertiary structure. Nat. Commun. 2019, 10, 1501. [Google Scholar] [CrossRef]
- Andrei, S.A.; Sijbesma, E.; Hann, M.; Davis, J.; O’Mahony, G.; Perry, M.W.; Karawajczyk, A.; Eickhoff, J.; Brunsveld, L.; Doveston, R.G.; et al. Stabilization of protein-protein interactions in drug discovery. Expert Opin. Drug Discov. 2017, 12, 925–940. [Google Scholar] [CrossRef] [Green Version]
- Sijbesma, E.; Hallenbeck, K.K.; Leysen, S.; de Vink, P.J.; Skóra, L.; Jahnke, W.; Brunsveld, L.; Arkin, M.R.; Ottmann, C. Site-directed fragment-based screening for the discovery of protein-protein interaction stabilizers. J. Am. Chem. Soc. 2019, 141, 3524–3531. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Luu, M.; Pautz, S.; Kohl, V.; Singh, R.; Romero, R.; Lucas, S.; Hofmann, J.; Raifer, H.; Vachharajani, N.; Carrascosa, L.C.; et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic epigenetic crosstalk in lymphocytes. Nat. Commun. 2019, 10, 760. [Google Scholar] [CrossRef] [PubMed]
- Saidi, L.; Rocha, D.H.A.; Talhi, O.; Bentarzi, Y.; Nedjar-Kolli, B.; Bachari, K.; Almeida Paz, F.A.; Helguero, L.A.; Silva, A.M.S. Synthesis of benzophenones and in vitro evaluation of their anticancer potential in breast and prostate cancer cells. ChemMedChem 2019, 14, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Roviezzo, F.; Sorrentino, R.; Riemma, M.A.; Cerqua, I.; Bilancia, R.; Spaziano, G.; Troisi, F.; Pace, S.; Pinto, A.; et al. Leukotriene-mediated sex dimorphism in murine asthma-like features duringallergensensitization. Pharmacol. Res. 2019, 139, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Pace, S.; Pergola, C.; Dehm, F.; Rossi, A.; Gerstmeier, J.; Troisi, F.; Pein, H.; Schaible, A.M.; Weinigel, C.; Rummler, S.; et al. Androgen-mediated sex bias impairs efficiency of leukotriene biosynthesis inhibitors in males. J. Clin. Investig. 2017, 127, 3167–3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, S.; Rossi, A.; Krauth, V.; Dehm, F.; Troisi, F.; Bilancia, R.; Weinigel, C.; Rummler, S.; Werz, O.; Sautebin, L. Sex differences in prostaglandin biosynthesis inneutrophils during acute inflammation. Sci. Rep. 2017, 7, 3759. [Google Scholar] [CrossRef]
- Trost, B.M.; Xu, S.; Sharif, E.U. New catalytic asymmetric formation of oxygen heterocycles bearing nucleoside bases at the anomeric carbon. J. Am. Chem. Soc. 2019. [Google Scholar] [CrossRef]
- Birkinshaw, R.W.; Gong, J.N.; Luo, C.S.; Lio, D.; White, C.A.; Anderson, M.A.; Blombery, P.; Lessene, G.; Majewski, I.J.; Thijssen, R.; et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat. Commun. 2019, 10, 2385. [Google Scholar] [CrossRef]
- Casara, P.; Davidson, J.; Claperon, A.; Le Toumelin-Braizat, G.; Vogler, M.; Bruno, A.; Chanrion, M.; Lysiak-Auvity, G.; Le Diguarher, T.; Starck, J.B.; et al. S55746 is a novel orally active BCL-2 selective and potent inhibitor that impairs hematological tumor growth. Oncotarget 2018, 9, 20075–20088. [Google Scholar] [CrossRef]
- Zoppi, V.; Hughes, S.J.; Maniaci, C.; Testa, A.; Gmaschitz, T.; Wieshofer, C.; Koegl, M.; Riching, K.M.; Daniels, D.L.; Spallarossa, A.; et al. Iterative design and optimization of initially inactive proteolysis targeting chimeras (PROTACs) identify VZ185 as a potent, fast, and selective von Hippel-Lindau (VHL) based dual degrader probe of BRD9 and BRD7. J. Med. Chem. 2019, 62, 699–726. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangoni, A.A.; Eynde, J.J.V.; Jampilek, J.; Hadjipavlou-Litina, D.; Liu, H.; Reynisson, J.; Sousa, M.E.; Gomes, P.A.C.; Prokai-Tatrai, K.; Tuccinardi, T.; et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–5. Molecules 2019, 24, 2415. https://doi.org/10.3390/molecules24132415
Mangoni AA, Eynde JJV, Jampilek J, Hadjipavlou-Litina D, Liu H, Reynisson J, Sousa ME, Gomes PAC, Prokai-Tatrai K, Tuccinardi T, et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–5. Molecules. 2019; 24(13):2415. https://doi.org/10.3390/molecules24132415
Chicago/Turabian StyleMangoni, Arduino A., Jean Jacques Vanden Eynde, Josef Jampilek, Dimitra Hadjipavlou-Litina, Hong Liu, Jóhannes Reynisson, Maria Emília Sousa, Paula A. C. Gomes, Katalin Prokai-Tatrai, Tiziano Tuccinardi, and et al. 2019. "Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–5" Molecules 24, no. 13: 2415. https://doi.org/10.3390/molecules24132415
APA StyleMangoni, A. A., Eynde, J. J. V., Jampilek, J., Hadjipavlou-Litina, D., Liu, H., Reynisson, J., Sousa, M. E., Gomes, P. A. C., Prokai-Tatrai, K., Tuccinardi, T., Sabatier, J. -M., Luque, F. J., Rautio, J., Karaman, R., Vasconcelos, M. H., Gemma, S., Galdiero, S., Hulme, C., Collina, S., ... Muñoz-Torrero, D. (2019). Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes–5. Molecules, 24(13), 2415. https://doi.org/10.3390/molecules24132415