4.2. Synthesis of BTO-3-Alkanoic Esters (4b–7b) and Acids (4a–7a)
4.2.1. Stille Coupling of BTO Iodides (13a–13d) with Allyl Alcohol
3-(3-Oxopropyl)benzo[e][1,2,4]triazine 1-oxide (14a). Pd(OAc)
2 (447 mg, 1.99 mmol) was added to a degassed mixture of 3-iodobenzo[
e][1,2,4]triazine 1-oxide (
13a) [
78] (5.44 g, 19.9 mmol), NaHCO
3 (3.68 g, 43.8 mmol), tetrabutylammonium chloride (5.53 g, 19.9 mmol), and allyl alcohol (6.80 mL, 99.6 mmol) in acetonitrile (200 mL), and the mixture was stirred at reflux temperature for 2 h. The mixture was cooled and the solvent was evaporated. The residue was purified by chromatography, eluting with a gradient (30–50%) of EtOAc/pet. ether, to give aldehyde
14a (2.53 g, 62%) as a tan powder: mp (EtOAc/pet. ether) 86–88 °C;
1H-NMR δ 9.45 (s, 1 H, CHO), 8.45 (br d,
J = 8.7 Hz, 1 H, H-8), 7.73–7.98 (m, 2 H, H-5, H-7), 7.70 (ddd,
J = 8.5, 6.8 Hz, 1 H, H-6), 3.38 (dd,
J = 6.9, 6.7 Hz, 2 H, CH
2), 3.14 (dd,
J = 6.9, 6.7 Hz, 2 H, CH
2); MS
m/z 204.1 (MH
+, 100%). Anal. calcd for C
10H
9N
3O
2: C, 59.11, H, 4.46; N, 20.68. Found: C, 59.11; H, 4.42; N, 20.65%.
7-Methyl-3-(3-oxopropyl)benzo[e][1,2,4]triazine 1-oxide (14b). Similarly, the reaction of 3-iodo-7-methylbenzo[
e][1,2,4]triazine 1-oxide (
13b) [
78] (1.85 g, 6.44 mmol) with Pd(Oac)
2 (145 mg, 0.64 mmol), NaHCO
3 (1.19 g, 14.2 mmol), tetrabutylammonium chloride (1.79 g, 6.44 mmol) and allyl alcohol (2.20 mL, 32.3 mmol) gave aldehyde
14b (955 mg, 47%) as an grey solid: mp (EtOAc/pet. ether) 69–71 °C;
1H NMR δ 9.94 (s, 1 H, CHO), 8.23 (br s, 1 H, H-8), 7.85 (d,
J = 8.6, 1.8 Hz, 1 H, H-5), 7.75 (dd,
J = 8.6, 1.8 Hz, 1 H, H-6), 3.37 (t,
J = 6.9 Hz, 2 H, CH
2), 3.12 (t,
J = 6.9 Hz, 2 H, CH
2), 2.58 (s, 3 H, 7-CH
3); MS
m/z 218.1 (MH
+, 100%). Anal. calcd for C
11H
11N
3O
2: C, 60.82, H, 5.10; N, 19.34. Found: C, 60.72; H, 5.01; N, 19.12%.
6-Methyl-3-(3-oxopropyl)benzo[e][1,2,4]triazine 1-Oxide (14c). Similarly, reaction of 3-iodo-6-methylbenzo[
e][1,2,4]triazine 1-oxide (
13c) [
78] (3.00 g, 10.5 mmol) with Pd(OAc)
2 (234 mg, 1.05 mmol), NaHCO
3 (1.93 g, 23.0 mmol), tetrabutylammonium chloride (2.89 g, 10.5 mmol) and allyl alcohol (3.56 mL, 52.3 mmol) gave aldehyde
14c (1.40 g, 44%) as an cream powder: mp (EtOAc/pet. ether) 71–72 °C;
1H NMR δ 9.94 (s, 1 H, CHO), 8.32 (d,
J = 8.8 Hz, 1 H, H-8), 7.72 (br s, 1 H, H-5), 7.50 (dd,
J = 8.8, 1.7 Hz, 1 H, H-7), 3.36 (t,
J = 6.9 Hz, 2 H, CH
2), 3.12 (t,
J = 7.1 Hz, 2 H, CH
2), 2.59 (s, 3 H, 6-CH
3); MS
m/z 218.1 (MH
+, 100%). Anal. calcd for C
11H
11N
3O
2: C, 60.82, H, 5.10; N, 19.34. Found: C, 60.94; H, 5.06; N, 19.59%.
3-(1-Oxido-7,8-dihydro-6H-indeno[5,6-e][1,2,4]triazin-3-yl)propanal (14d). Similarly, the reaction of 3-iodo-7,8-dihydro-6H-indeno[5,6-e][1,2,4]triazine 1-oxide
13d [78] (7.15 g, 22.8 mmol) with Pd(OAc)
2 (516 mg, 2.28 mmol), NaHCO
3 (4.23 g, 50.0 mmol), tetrabutylammonium chloride (6.35 g, 22.8 mmol) and allyl alcohol (7.77 mL, 114 mmol) gave aldehyde
14d (3.54 g, 64%) as an off white solid: mp (EtOAc/pet. ether) 72–74 °C;
1H NMR δ 9.93 (t,
J = 0.9 Hz, 1 H, CHO), 8.25 (s, 1 H, H-9), 7.73 (s, 1 H, H-5), 3.35 (t,
J = 7.0 Hz, 2 H, CH
2), 3.07–3.14 (m, 6 H, H-6, H-8, CH
2), 2.21 (p,
J = 7.5 Hz, 2 H, H-7);
13C NMR δ 200.4, 163.9, 154.8, 149.1, 147.2, 132.3, 122.7, 114.2, 40.5, 33.1, 32.8, 29.4, 25.7; MS
m/z 244.2 (MH
+, 100%); HRMS (CI, CH
3OH) calcd for C
13H
14N
3O
2 (MH
+)
m/z 244.1086, found 244.1088. Anal. calcd for C
13H
13N
3O
2: C, 64.2; H, 5.4; N, 17.3. Found: C, 63.9; H, 5.5; N, 17.0%.
4.2.2. Oxidation of Aldehydes (14a–14d).
3-(2-Carboxyethyl)benzo[e][1,2,4]triazine 1-Oxide (15a). A solution of NaClO2 (1.59 g, 14.1 mmol) in water (20 mL) was added dropwise to a stirred mixture of aldehyde 14a (1.91 g, 9.39 mmol) in MeCN (80 mL) and KH2PO4 (319 mg, 2.35 mmol) in water (10 mL) and 35% H2O2 (1.15 mL, 11.3 mmol) at 5 °C and the mixture was stirred at 5 °C for 3 h. The mixture was acidified with aqueous HCl (1 M) and extracted with EtOAc (3 × 50 mL). The combined organic fraction was extracted with an aqueous NaOH solution (0.1 M, 3 × 50 mL). The aqueous fraction was acidified with an aqueous HCl solution (1 M) and extracted with EtOAc (3 × 50 mL). The organic fraction was dried and the solvent was evaporated. The residue was purified by chromatography, eluting with a gradient (50–100%) of EtOAc/pet. ether, to give acid 15a (1.82 g, 88%) as a cream powder: mp 152–154 °C (dec.); 1H NMR [(CD3)2SO] δ 12.52 (br s, 1 H, CO2H), 8.37 (dd, J = 8.7, 0.8 Hz, 1 H, H-8), 8.08 (ddd, J = 8.7, 6.8, 1.4 Hz, 1 H, H-7), 8.03 (dd, J = 8.4, 1.4 Hz, 1 H, H-5), 7.89 (ddd, J = 8.4, 6.8, 1.4 Hz, 1 H, H-6), 3.19 (t, J = 7.1 Hz, 2 H, CH2), 2.83 (t, J = 7.1 Hz, 2 H, CH2); MS m/z 204.1 (MH+, 100%). Anal. calcd for C10H9N3O3: C, 54.79; H, 4.14; N, 19.17. Found: C, 55.06; H, 4.00; N, 19.31%.
3-(2-Carboxyethyl)-7-methylbenzo[e][1,2,4]triazine 1-Oxide (15b). Similarly, the reaction of NaClO2 (0.70 g, 6.19 mmol) with aldehyde 14b (0.90 g, 4.12 mmol), KH2PO4 (140 mg, 1.03 mmol) and 30% H2O2 (0.51 mL, 4.94 mmol) gave acid 15b (771 mg, 80%) as white needles: mp 191–193 °C (dec.); 1H NMR [(CD3)2SO] δ 12.24 (br s, 1 H, CO2H), 8.18 (br s, 1 H, H-8), 7.90–7.94 (m, 2 H, H-5, H-6), 3.17 (br t, J = 7.1 Hz, 2 H, CH2), 2.82 (br t, J = 7.1 Hz, 2 H, CH2), 2.55 (s, 3 H, 7-CH3); MS m/z 234.1 (MH+, 100%). Anal. calcd for C11H11N3O3: C, 56.65; H, 4.75; N, 18.02. Found: C, 56.73; H, 4.80; N, 18.16%.
3-(2-Carboxyethyl)-6-methylbenzo[e][1,2,4]triazine 1-Oxide (15c). Similarly, the reaction of NaClO2 (1.00 g, 8.84 mmol) with aldehyde 14c (1.28 g, 5.89 mmol), KH2PO4 (200 mg, 1.47 mmol) and 30% H2O2 (0.72 mL, 7.07 mmol) gave acid 15c (1.32 g, 82%) as a white powder: mp 170–171 °C; 1H NMR [(CD3)2SO] δ 12.24 (br s, 1 H, CO2H), 8.32 (d, J = 8.8 Hz, 1 H, H-8), 7.74 (br s, 1 H, H-5), 7.51 (dd, J = 8.8, 1.7 Hz, 1 H, H-7), 3.35 (t, J = 7.1 Hz, 2 H, CH2), 3.02 (t, J = 7.1 Hz, 2 H, CH2), 2.59 (s, 3 H, 6-CH3); MS m/z 234.1 (MH+, 100%). Anal. calcd for C11H11N3O3: C, 56.65; H, 4.75; N, 18.02. Found: C, 56.92; H, 4.69; N, 18.25%.
3-(2-Carboxyethyl)-7,8-dihydro-6H-indeno[5,6-e][1,2,4]triazine 1-Oxide (15d). Similarly, the reaction of NaClO2 (222 mg, 1.96 mmol) with 3-(1-oxido-7,8-dihydro-6H-indeno[5,6-e][1,2,4] triazin-3-yl)-1-propanal 14d (341 mg, 1.40 mmol) gave acid 15d (261 mg, 72%) as a white powder: mp 215 °C (dec.); 1H NMR δ 8.24 (s, 1 H, H-9), 7.74 (s, 1 H, H-5), 3.34 (br t, J = 7.0 Hz, 2 H, CH2), 3.08–3.14 (m, 4 H, H-6, H-8), 3.01 (t, J = 7.0 Hz, 2 H, CH2), 2.22 (p, J = 7.4 Hz, 2 H, H-7), OH not observed; 13C NMR δ 176.4, 164.0, 155.2, 149.4, 147.2, 132.6, 122.8, 114.6, 33.3, 33.0, 31.4, 31.0, 25.9; MS m/z 260.3 (MH+, 100%); Anal. calcd for C13H13N3O3: C, 60.22; H, 5.05; N, 16.21. Found: C, 60.46; H, 5.08; N, 16.34%.
4.2.3. Esterification of Acids (15a–15d).
3-(3-Methoxy-3-oxopropyl)benzo[e][1,2,4]triazine 1-Oxide (16a). cH2SO4 (cat., 0.3 mL) was added to a stirred suspension of acid 15a (2.28 g, 10.4 mmol) in dry MeOH (100 mL) and the mixture was stirred at reflux temperature for 6 h. The mixture was cooled to 20 °C and the solvent was evaporated. The residue was partitioned between EtOAc (100 mL) and water (100 mL). The organic fraction was washed with an aqueous NaOH solution (0.1 M, 50 mL), then washed with water (25 mL), then washed with brine (30 mL), then dried, after which the solvent was evaporated. The residue was purified by chromatography, eluting with 50% EtOAc/pet. ether, to give ester 16a (2.267 g, 94%) as a white powder: mp (EtOAc/pet. ether) 45–47 °C; 1H NMR δ 8.44 (dd, J = 8.6, 1.2 Hz, 1 H, H-8), 7.97 (dd, J = 8.7, 1.4 Hz, 1 H, H-5), 7.93 (ddd, J = 8.6, 6.9, 1.3 Hz, 1 H, H-7), 7.70 (ddd, J = 8.4, 6.9, 1.4 Hz, 1 H, H-6), 3.71 (s, 3 H, OCH3), 3.37 (t, J = 7.2 Hz, 2 H, CH2), 2.99 (t, J = 7.2 Hz, 2 H, CH2); MS m/z 234.2 (MH+, 100%). Anal. calcd for C11H11N3O3: C, 56.65; H, 4.75; N, 18.02. Found: C, 56.86; H, 4.56; N, 18.21%.
3-(3-Methoxy-3-oxopropyl)-7-methylbenzo[e][1,2,4]triazine 1-Oxide (16b). Similarly, the reaction of cH2SO4 (cat., 0.2 mL) with acid 15b (730 mg, 3.13 mmol) in dry MeOH (50 mL) gave ester 16b (740 mg, 96%) as a white powder: mp (EtOAc/pet. ether) 130–132 °C; 1H NMR δ 8.24 (br s, 1 H, H-8), 7.86 (d, J = 8.6 Hz, 1 H, H-5), 7.74 (dd, J = 8.6, 1.8 Hz, 1 H, H-6), 3.70 (s, 3 H, OCH3), 3.35 (t, J = 7.2 Hz, 2 H, CH2), 2.98 (t, J = 7.2 Hz, 2 H, CH2), 2.58 (s, 3 H, 7-CH3); MS m/z 248.2 (MH+, 100%). Anal. calcd for C12H13N3O3: C, 58.29; H, 5.30; N, 16.99. Found: C, 58.18; H, 5.31; N, 17.10%.
3-(3-Methoxy-3-oxopropyl)-6-methylbenzo[e][1,2,4]triazine 1-Oxide (16c). Similarly, the reaction of cH2SO4 (cat., 0.3 mL) with acid 15c (1.09 g, 4.67 mmol) in dry MeOH (50 mL) gave ester 16c (1.12 g, 97%) as white needles: mp (EtOAc/pet. ether) 91–93 °C; 1H NMR δ 8.32 (d, J = 8.8 Hz, 1 H, H-8), 7.73 (br s, 1 H, H-5), 7.50 (dd, J = 8.8, 1.7 Hz, 1 H, H-7), 3.70 (s, 3 H, OCH3), 3.35 (t, J = 7.2 Hz, 2 H, CH2), 2.97 (t, J = 7.2 Hz, 2 H, CH2), 2.59 (s, 3 H, 6-CH3); MS m/z 248.2 (MH+, 100%). Anal. calcd for C12H13N3O3: C, 58.29; H, 5.30; N, 16.99. Found: C, 58.18; H, 5.38; N, 17.18%.
3-(3-Methoxy-3-oxopropyl)-7,8-dihydro-6H-indeno[5,6-e][1,2,4]triazine 1-Oxide (16d). Similarly, the reaction of cH2SO4 (cat., 3 drops) with acid 15d (330 mg, 1.27 mmol) in dry MeOH (30 mL) gave ester 16d (277 mg, 80%) as a white powder: mp (EtOAc/pet. ether) 119–120 °C; 1H NMR δ 8.25 (s, 1 H, H-9), 7.74 (s, 1 H, H-5), 3.70 (s, 3 H, OCH3), 3.34 (t, J = 7.3 Hz, 2 H, CH2), 3.08–3.14 (m, 4 H, H-6, H-8), 2.96 (t, J = 7.3 Hz, 2 H, CH2CO), 2.21 (p, J = 7.5 Hz, 2 H, H-7); MS m/z 274.3 (MH+, 100%). Anal. calcd for C14H15N3O3: C, 61.53; H, 5.53; N, 15.38. Found: C, 61.64; H, 5.48; N, 15.44%.
4.2.4. Oxidation of Benzotriazine 1-Oxides (16a–16d)
3-(3-Methoxy-3-oxopropyl)benzo[e][1,2,4]triazine 1,4-Dioxide (4b). H2O2 (30%, 9.8 mL, 95.1 mmol) was added dropwise to a stirred solution of TFAA (14.5 mL, 10.5 mmol) in DCM (30 mL) at 0–5 °C and the mixture was stirred at 20 °C for 1 h. This mixture was added dropwise to a stirred solution of ester 16a (2.218 g, 9.51 mmol) in DCM (30 mL) at 0 °C and the mixture was stirred at 20 °C for 24 h. The mixture was cooled to 0 °C and DMSO (6.75 mL, 95.1 mmol) was added and the mixture was stirred for 5 min. The mixture was carefully neutralized with a concentrated NH3 solution and the mixture was washed with water (3 × 30 mL), then dried, after which the solvent evaporated was. The residue was purified by chromatography, eluting with a gradient (50–100%) of EtOAc/pet. ether, to give di-oxide 4b (1.60 g, 68%) as a yellow powder: mp (EtOAc/pet. ether) 89–91 °C; 1H NMR δ 8.53 (dd, J = 8.7, 0.8 Hz, 1 H, H-8), 8.47 (dd, J = 8.7, 0.8 Hz, 1 H, H-5), 8.02 (ddd, J = 8.7, 7.1, 1.3 Hz, 1 H, H-7), 7.86 (ddd, J = 8.7, 7.1, 1.3 Hz, 1 H, H-6), 3.71 (s, 3 H, OCH3), 3.49 (t, J = 7.1 Hz, 2 H, CH2), 2.98 (t, J = 7.1 Hz, 2 H, CH2); 13C NMR [(CD3)2SO] δ 172.5, 154.1, 139.8, 135.7, 134.9, 132.1, 121.8, 119.7, 52.2, 28.8, 25.7; MS m/z 250.2 (MH+, 100%). Anal. calcd for C11H11N3O4: C, 53.01; H, 4.45; N, 16.86. Found: C, 53.17; H, 4.38; N, 16.93%.
3-(3-Methoxy-3-oxopropyl)-7-methylbenzo[e][1,2,4]triazine 1,4-Dioxide (5b). Similarly, the reaction of H2O2 (30%, 3.0 mL, 28.6 mmol) and TFAA (4.37 mL, 31.4 mmol) with ester 16b (706 mg, 2.86 mmol) gave starting material 16b (44 mg, 6%), spectroscopically identical to above, and di-oxide 5b (586 mg, 78%) as a yellow powder: mp (EtOAc/pet. ether) 165–167 °C; 1H NMR δ 8.40 (d, J = 8.8 Hz, 1 H, H-5), 8.25 (br s, 1 H, H-8), 7.82 (dd, J = 8.8, 1.5 Hz, 1 H, H-6), 3.71 (s, 3 H, OCH3), 3.48 (t, J = 7.1 Hz, 2 H, CH2), 2.97 (t, J = 7.1 Hz, 2 H, CH2), 2.63 (s, 3 H, 7-CH3); 13C NMR δ 172.5, 153.4, 143.8, 138.2, 137.7, 134.7, 120.5, 119.5, 52.2, 28.8, 25.6, 22.0; MS m/z 264.1 (MH+, 100%). Anal. calcd for C12H13N3O4: C, 54.75; H, 4.98; N, 15.96. Found: C, 54.83; H, 4.93; N, 16.23%.
3-(3-Methoxy-3-oxopropyl)-6-methylbenzo[e][1,2,4]triazine 1,4-Dioxide (6b). Similarly, the reaction of H2O2 (30%, 4.0 mL, 38.4 mmol) and TFAA (5.88 mL, 42.3 mmol) with ester 16c (950 mg, 3.84 mmol) gave starting material 16c (81 mg, 9%), spectroscopically identical to above, and di-oxide 6b (812 mg, 80%) as a yellow powder: mp (EtOAc/pet. ether) 166–168 °C; 1H NMR [(CD3)2SO] δ 8.26 (d, J = 8.9 Hz, 1 H, H-8), 8.18 (br s, 1 H, H-5), 7.78 (dd, J = 8.9, 1.6 Hz, 1 H, H-7), 3.63 (s, 3 H, OCH3), 3.27 (dt, J = 7.4, 7.1 Hz, 2 H, CH2), 2.97 (dt, J = 7.4, 7.1 Hz, 2 H, CH2), 2.61 (s, 3 H, 6-CH3); 13C NMR [(CD3)2SO] δ 172.2, 153.3, 147.4, 139.1, 133.9, 132.8, 120.8, 117.5, 51.6, 28.2, 25.2, 21.6; MS m/z 264.1 (MH+, 100%). Anal. calcd for C12H13N3O4: C, 54.75; H, 4.98; N, 15.96. Found: C, 54.51; H, 5.05; N, 16.22%.
3-(3-Methoxy-3-oxopropyl)-7,8-dihydro-6H-indeno[5,6-e][1,2,4]triazine 1,4-Dioxide (7b). Similarly, the reaction of H2O2 (30%, 1.25 mL, 10.0 mmol) and TFAA (1.26 mL, 9.1 mmol) with ester 16d (249 mg, 0.91 mmol) gave di-oxide 7b (200 mg, 76%) as a yellow powder: mp (EtOAc/pet. ether) 170–171 °C; 1H NMR δ 8.31 (s, 1 H, H-9), 8.25 (s, 1 H, H-5), 3.71 (s, 3 H, OCH3), 3.48 (t, J = 7.3 Hz, 2 H, CH2), 3.12–3.20 (m, 4 H, H-6, H-8), 2.97 (t, J = 7.2 Hz, 2 H, CH2CO), 2.26 (p, J = 7.5 Hz, 2 H, H-7); 13C NMR δ 172.6, 155.4, 153.2, 151.0, 139.2, 134.1, 116.1, 114.0, 52.2, 33.6, 33.1, 28.9, 25.8, 25.7; MS m/z 290.3 (MH+, 100%). Anal. calcd for C14H15N3O4: C, 58.13; H, 5.23; N, 14.53. Found: C, 57.91; H, 5.20; N, 14.44%.
4.2.5. Hydrolysis of Esters (4b–7b)
3-(2-Carboxyethyl)benzo[e][1,2,4]triazine 1,4-Dioxide (4a). A solution of HCl in dioxane (4 M, 52 mL, 209 mmol) was added dropwise to a stirred suspension of ester 4b (1.30 g, 5.22 mmol) in dioxane (30 mL) and water (1 mL) and the mixture was stirred at 20 °C for 5 d. The mixture was diluted with water (200 mL), extracted with CHCl3 (3 × 100 mL), then the combined organic fraction was dried and the solvent was evaporated. The residue was purified by chromatography, eluting with a gradient (0–5%) of MeOH/DCM, to give starting ester 4b (390 mg, 30%), spectroscopically identical to above, and acid 4a (728 mg, 59%) as a yellow powder: mp (EtOAc) 157 °C (dec.); 1H NMR [(CD3)2SO] δ 12.35 (br s, 1 H, CO2H), 8.35–8.40 (m, 2 H, H-5, H-8), 8.12 (br ddd, J = 8.6, 7.1, 1.4 Hz, 1 H, H-7), 7.96 (br ddd, J = 8.6, 7.1, 1.4 Hz, 1 H, H-6), 3.23 (br t, J = 7.2 Hz, 2 H, CH2), 2.77 (br t, J = 7.2 Hz, 2 H, CH2); 13C NMR [(CD3)2SO] δ 173.2, 153.5, 139.3, 135.7, 134.4, 132.1, 121.1, 118.8, 28.4, 25.3; MS m/z 276.3 (MH+, 100%). Anal. calcd for C10H9N3O4: C, 51.07; H, 3.86; N, 17.87. Found: C, 51.10; H, 3.80; N, 17.58%. HPLC purity 95.1%.
3-(2-Carboxyethyl)-7-methylbenzo[e][1,2,4]triazine 1,4-Dioxide (5a). Similarly, the reaction of HCl in dioxane (4 M, 25 mL, 100 mmol) with ester 5b (522 mg, 1.98 mmol) gave starting ester 5b (249 mg, 48%), spectroscopically identical to above, and acid 5a (200 mg, 40%) as a yellow powder: mp (EtOAc/MeOH) 181–183 °C (dec.); 1H NMR [(CD3)2SO] δ 12.34 (br s, 1 H, CO2H), 8.26 (d, J = 8.8 Hz, 1 H, H-5), 8.18 (br s, 1 H, H-8), 7.95 (dd, J = 8.8, 1.6 Hz, 1 H, H-6), 3.22 (t, J = 7.1 Hz, 2 H, CH2), 2.77 (t, J = 7.2 Hz, 2 H, CH2), 2.57 (s, 3 H, 7-CH3); 13C NMR [(CD3)2SO] δ 173.2, 152.9, 143.1, 137.7, 137.5, 134.2, 119.6, 118.6, 28.4, 25.2, 21.1; MS m/z 250.1 (MH+, 100%). Anal. calcd for C11H11N3O4·¼CH3OH: C, 52.07; H, 4.94; N, 15.84. Found: C, 52.10; H, 4.80; N, 16.19%. HPLC purity 99.1%.
3-(2-Carboxyethyl)-6-methylbenzo[e][1,2,4]triazine 1,4-Dioxide (6a). Similarly, the reaction of HCl in dioxane (4 M, 34 mL, 136 mmol) with ester 6b (714 mg, 2.71 mmol) gave starting ester 6b (289 mg, 40%), spectroscopically identical to above, and acid 6a (385 mg, 57%) as a yellow powder: mp (EtOAc) 173–175 °C (dec.); 1H NMR [(CD3)2SO] δ 12.35 (br s, 1 H, CO2H), 8.25 (d, J = 8.8 Hz, 1 H, H-8), 8.18 (br s, 1 H, H-5), 7.79 (dd, J = 8.8, 1.6 Hz, 1 H, H-7), 3.23 (t, J = 7.2 Hz, 2 H, CH2), 2.76 (t, J = 7.2 Hz, 2 H, CH2), 2.60 (s, 3 H, 6-CH3); 13C NMR [(CD3)2SO] δ 173.2, 153.6, 147.4, 139.1, 133.8, 132.8, 120.8, 117.5, 28.4, 25.3, 21.6; MS m/z 250.1 (MH+, 100%). Anal. calcd for C11H11N3O4: C, 53.01; H, 4.45; N, 16.86. Found: C, 52.94; H, 4.47; N, 16.80%. HPLC purity 99.6%.
3-(2-Carboxyethyl)-7,8-dihydro-6H-indeno[5,6-e][1,2,4]triazine 1,4-Dioxide (7a). Similarly, the reaction of HCl in dioxane (4 M, 4.32 mL, 17.3 mmol) with ester 7b (100 mg, 0.34 mmol) gave starting ester 7b (13 mg, 13%), spectroscopically identical to above, and acid 7a (60 mg, 64%) as a yellow powder: mp (EtOAc) 159 °C (dec.); 1H NMR δ 12.34 (br s, 1 H, CO2H), 8.18 (s, 1 H, H-9), 8.17 (s, 1 H, H-5), 3.22 (t, J = 7.2 Hz, 2 H, CH2), 3.07–3.16 (m, 4 H, H-6, H-8), 2.76 (t, J = 7.2 Hz, 2 H, CH2CO), 2.13 (p, J = 7.5 Hz, 2 H, H-7); 13C NMR δ 173.2, 154.6, 152.7, 150.2, 138.7, 133.5, 115.2, 113.1, 32.8, 32.3, 28.5, 25.2 (2); MS m/z 276.3 (MH+, 100%). Anal. calcd for C13H13N3O4: C, 56.72; H, 4.76; N, 15.27. Found: C, 57.00; H, 4.73; N, 15.08%. HPLC purity 99.7%.
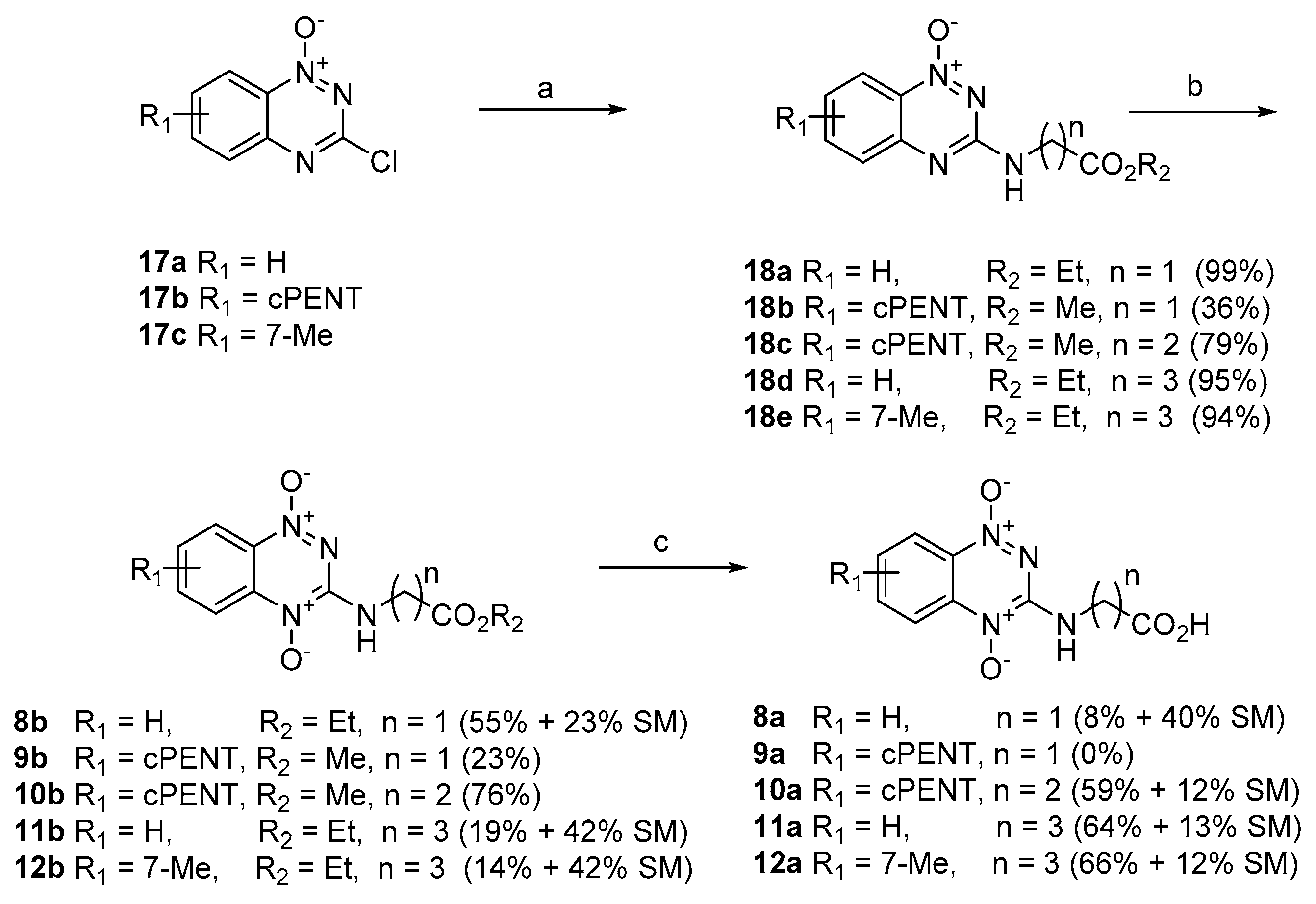
4.2.6. Synthesis of BTO-3-aminoalkanoic Esters (8b–12b) and Acids (8a–12a)
Ethyl [(1-Oxido-1,2,4-benzotriazin-3-yl)amino]acetate (18a). A mixture of 3-chloro-1,2,4-benzotriazine 1-oxide (
17a)[
78] ( 2.02 g, 11.1 mmol), glycine ethyl ester hydrochloride (2.33 g, 16.7 mmol), and Et
3N (4.2 mL, 30 mmol) in DME (100 mL) was heated at reflux temperature for 6 h. The solvent was evaporated and the residue was partitioned between DCM/water (200 mL), then the aqueous fraction extracted with DCM (2 × 50 mL), then the combined organic fraction was dried and the solvent was evaporated. The residue was purified by chromatography, eluting with 10% EtOAc/DCM, to give the ester
18a (2.75 g, 99%) as a yellow solid: mp (EtOAc/DCM) 136–138 °C;
1H NMR δ 8.27 (dd,
J = 8.6, 1.0 Hz, 1 H, H 8), 7.72 (ddd,
J = 8.5, 7.0, 1.4 Hz, 1 H, H 6), 7.62 (dd,
J = 8.5, 1.0 Hz, 1 H, H 5), 7.34 (ddd,
J = 8.6, 7.0, 1.0 Hz, 1 H, H 7), 5.87 (br s, 1 H, NH), 4.30 (d,
J = 5.7 Hz, 2 H, CH
2N), 4.26 (q,
J = 7.2 Hz, 2 H, CH
2O), 1.31 (t,
J = 7.2 Hz, 3 H, CH
3);
13C NMR δ 169.9, 158.4, 148.5, 135.6, 131.2, 126.7, 125.5, 120.4, 61.6, 43.2, 14.2; MS
m/z 249.2 (MH
+, 100%). Anal. calcd for C
11H
12N
4O
3: C, 53.2; H, 4.9; N, 22.6. Found: C, 53.4; H, 5.0; N, 22.6%.
3-((2-methoxy-2-oxoethyl)amino)-7,8-dihydro-6H-indeno[5,6-e][1,2,4]triazine 1-oxide (18b). Similarly, the reaction of 3-chloride 17b (0.84 g, 3.79 mmol), glycine methyl ester hydrochloride (0.95 g, 7.59 mmol) and iPr2NEt (2.5 mL, 15.19 mmol) in DME (100 mL) gave ester 18b (0.38 g, 36%) as a yellow powder: mp (MeOH) 182–184 °C; 1H NMR δ 8.10 (s, 1 H, H-9), 7.43 (s, 1 H, H-5), 5.65 (t, J = 5.1 Hz, 1 H, NH), 4.29 (d, J = 5.6 Hz, 2 H, CH2CO), 3.80 (s, 3 H, OCH3), 2.94–3.05 (m, 4 H, H-6, H-8), 2.15 (t, J = 7.4 Hz, 2 H, H-7), 2.15 (pent, J = 7.4 Hz, 2 H, H-7); MS m/z 275.2 (MH+, 100%). Anal. calcd for C13H14N4O3·0.2 CH3OH: C, 56.48; H, 5.31; N, 19.96. Found: C, 56.24; H, 5.03; N, 20.20%.
3-((3-Methoxy-3-oxopropyl)amino)-7,8-dihydro-6H-indeno[5,6-e][1,2,4]triazine 1-Oxide (18c). Similarly the reaction of 3-chloro-7,8-dihydro-6
H-indeno[5,6-
e][1,2,4]triazine 1-oxide (
17b) [
79] (1.02 g, 4.60 mmol), β-alanine methyl ester (1.28 g, 9.20 mmol) and iPr
2NEt (3.80 mL, 23.0 mmol) in DME (100 mL) gave the ester
18c (1.46 g, 79%) as a yellow solid: mp (EtOAc/DCM) 160–161 °C;
1H NMR δ 8.08 (s, 1 H, H-9), 7.41 (s, 1 H, H-5), 5.56 (t,
J = 6.1 Hz, 1 H, NH), 3.81 (dt,
J = 6.3, 6.1 Hz, 2 H, CH
2N), 3.71 (s, 3 H, OCH
3), 2.98–3.06 (m, 4 H, H-6, H-8), 2.71 (t,
J = 6.1 Hz, 2 H, CH
2O), 2.15 (pent,
J = 7.4 Hz, 2 H, H-7); MS
m/z 298.2 (MH
+, 100%). Anal. calcd for C
14H
16N
4O
3: C, 58.32; H, 5.59; N, 19.43. Found: C, 58.41; H, 5.51; N, 19.39%.
3-((4-Ethoxy-4-oxobutyl)amino)benzo[e][1,2,4]triazine 1-Oxide (18d). Similarly, the reaction of chloride 17a (1.26 g, 6.94 mmol), ethyl 4-aminobutanoate hydrochloride (1.51 g, 9.00 mmol) and Et3N (2.90 mL, 20.8 mmol) in DME (100 mL) gave ester 18d (1.83 g, 95%) as a yellow powder: mp (EtOAc/pet. ether) 130–131 °C; 1H NMR δ 8.26 (dd, J = 8.7, 1.4 Hz, 1 H, H-8), 7.70 (ddd, J = 8.7, 6.9, 1.4 Hz, 1 H, H-7), 7.60 (br d, J = 8.4 Hz, 1 H, H-5), 7.29 (ddd, J = 8.4, 6.9, 1.4 Hz, 1 H, H-6), 5.42 (br s, 1 H, NH), 4.15 (q, J = 7.1 Hz, 2 H, CH2O), 3.58 (dd, J = 6.8, 6.2 Hz, 2 H, CH2N), 2.44 (t, J = 7.2 Hz, 2 H, CH2), 2.02 (pent, J = 7.0 Hz, 2 H, CH2), 1.26 (t, J = 7.1 Hz, 3 H, CH3); MS m/z 277.1 (MH+, 100%). Anal. calcd for C13H16N4O3: C, 56.51, H, 5.84; N, 20.28. Found: C, 56.78; H, 5.89; N, 20.47%.
3-((4-Ethoxy-4-oxobutyl)amino)-7-methylbenzo[e][1,2,4]triazine 1-Oxide (18e). Similarly, the reaction of 3-chloro-7-methylbenzo[
e][1,2,4]triazine 1-oxide (
17b) [
78] (2.65 g, 13.5 mmol), ethyl 4-aminobutanoate hydrochloride (3.41 g, 20.3 mmol) and Et
3N (5.64 mL, 40.5 mmol) gave ester
18e (3.68 g, 94%) as a yellow powder: mp (EtOAc) 158–159 °C;
1H NMR δ 8.06 (br s, 1 H, H-8), 7.54 (dd,
J = 8.7, 1.8 Hz, 1 H, H-6), 7.50 (d,
J = 8.7 Hz, 1 H, H-5), 5.34 (br s, 1 H, NH), 4.15 (q,
J = 7.1 Hz, 2 H, CH
2O), 3.57 (dd,
J = 6.8, 6.2 Hz, 2 H, CH
2N), 2.46 (s, 3 H, 7-CH
3), 2.44 (t,
J = 7.2 Hz, 2 H, CH
2), 2.02 (pent,
J = 7.0 Hz, 2 H, CH
2), 1.24 (t,
J = 7.1 Hz, 3 H, CH
3); MS
m/z 290.1 (MH
+, 100%). Anal. calcd for C
14H
18N
4O
3: C, 57.92, H, 6.25; N, 19.30. Found: C, 57.75; H, 6.34; N, 19.38%.
4.2.7. Oxidation of BTO-3-aminoalkanoic Esters (18a–18e)
Ethyl [(1,4-Dioxido-1,2,4-benzotriazin-3-yl)amino]acetate (8b). H2O2 (30%, 10.9 mL, 106 mmol) was added dropwise to a stirred solution of TFAA (16.2 mL, 117 mmol) in DCM (50 mL) at 0–5 °C and the mixture was stirred at 20 °C for 1 h. The mixture was added dropwise to a stirred solution of ester 18a (2.63 g, 10.6 mmol) in DCM (50 mL) at 0 °C and the mixture was stirred at 20 °C for 5 days. The mixture was cooled to 0 °C and DMSO (7.5 mL, 106 mmol) was added. The mixture was carefully neutralized with a concentrated NH3 solution (ca. 15 mL) and the mixture was washed with water (3 × 30 mL), dried and the solvent was evaporated. The residue was purified by chromatography, eluting with a gradient (0–10%) of MeOH/EtOAc, to give (i) starting material 18a (593 mg, 23%), spectroscopically identical with the sample above; and (ii) di-oxide 8b (1.535 g, 55%) as a red powder: mp (EtOAc/pet. ether) 156–158 °C; 1H NMR δ 8.34 (d, J = 8.7 Hz, 2 H, H 5, H 8), 7.91 (br s, 1 H, NH), 7.50–7.61 (m, 2 H, H 6, H 7), 4.37 (br s, 2 H, CH2), 4.26 (q, J = 7.1 Hz, 2 H, CH2), 1.30 (t, J = 7.1 Hz, 3 H, CH3); 13C NMR δ 168.5, 149.7, 138.4, 136.0, 131.0, 129.6, 121.7, 117.6, 61.9, 42.8, 14.1; MS (EI+) m/z 264 (M+, 30%), 284 (40), 175 (100); HRMS (EI+) calc. for C11H12N4O4 (M+) m/z 264.0859, found 264.0852. Anal. calcd for C11H12N4O4: C, 50.0; H, 4.6; N, 21.2. Found: C, 50.8; H, 4.5; N, 21.1%.
3-((2-Methoxy-2-oxoethyl)amino)-7,8-dihydro-6H-indeno[5,6-e][1,2,4]triazine 1,4-dioxide (9b). Similarly, the reaction of H2O2 (30%, 1.4 mL, 14.1 mmol) and TFAA (1.8 mL, 12.8 mmol) with ester 18b (350 mg, 1.28 mmol) gave di-oxide 9b (86 mg, 23%) as a red powder: mp (MeOH/EtOAc) 171–178 °C; 1H NMR δ 8.14 (s, 1 H, H-9), 8.13 (s, 1 H, H-5), 7.40 (br s, 1 H, NH), 4.36 (d, J = 5.9 Hz, 2 H, CH2N), 3.80 (s, 3 H, OCH3), 3.13 (t, J = 7.2 Hz, 2 H, H-8), 3.07 (dt, J = 7.4, 1.0 Hz, 2 H, H-6), 2.12 (p, J = 7.5 Hz, 2 H, H-7); 13C NMR δ 169.3, 156.1, 149.3, 146.9, 138.2, 130.6, 116.0, 112.0, 52.9, 43.0, 33.7, 32.6, 25.8; MS m/z 291.2 (MH+, 100%). Anal. calcd for C13H14N4O4: C, 53.79; H, 4.86; N, 19.30. Found: C, 53.84; H, 4.79; N, 19.41%. HPLC purity 96.4%.
3-((3-Methoxy-3-oxopropyl)amino)-7,8-dihydro-6H-indeno[5,6-e][1,2,4]triazine 1,4-Dioxide (10b). Similarly, the reaction of H2O2 (30%, 1.25 mL, 10.0 mmol) and TFAA (1.26 mL, 9.1 mmol) with ester 18c (249 mg, 0.91 mmol) gave di-oxide 10b (200 mg, 76%) as a yellow powder: mp (EtOAc/pet. ether) 170–171 °C; 1H NMR δ 8.31 (s, 1 H, H-9), 8.25 (s, 1 H, H-5), 3.71 (s, 3 H, OCH3), 3.48 (t, J = 7.3 Hz, 2 H, CH2), 3.12–3.20 (m, 4 H, H-6, H-8), 2.97 (t, J = 7.2 Hz, 2 H, CH2CO), 2.26 (p, J = 7.5 Hz, 2 H, H-7); 13C NMR δ 172.6, 155.4, 153.2, 151.0, 139.2, 134.1, 116.1, 114.0, 52.2, 33.6, 33.1, 28.9, 25.8, 25.7; MS m/z 290.3 (MH+, 100%). Anal. calcd for C14H15N3O4: C, 58.13; H, 5.23; N, 14.53. Found: C, 57.91; H, 5.20; N, 14.44%. HPLC purity 99.3%.
3-((4-Ethoxy-4-oxobutyl)amino)benzo[e][1,2,4]triazine 1,4-Dioxide (11b). Similarly, the reaction of H2O2 (30%, 6.6 mL, 64.4 mmol) and TFAA (9.6 mL, 70.9 mmol) in DCM (40 mL) with ester 18d (1.78 g, 6.44 mmol) gave starting ester 18d (751 mg, 42%), spectroscopically identical to above, and di-oxide 11b (355 mg, 19%) as a red powder: mp (EtOAc) 163–165 °C; 1H NMR δ 8.34 (dd, J = 8.7, 0.8 Hz, 1 H, H-8), 8.30 (dd, J = 8.7, 1.3 Hz, 1 H, H-5), 7.88 (ddd, J = 8.7, 7.0, 1.3 Hz, 1 H, H-7), 7.52 (ddd, J = 8.7, 7.0, 1.0 Hz, 1 H, H-6), 7.30 (br s, 1 H, NH), 4.16 (q, J = 7.1 Hz, 2 H, CH2O), 3.67 (dd, J = 6.8, 6.6 Hz, 2 H, CH2N), 2.46 (t, J = 7.2 Hz, 2 H, CH2), 2.08 (pent, J = 7.1 Hz, 2 H, CH2), 1.26 (t, J = 7.1 Hz, 3 H, CH3); 13C NMR [(CD3)2SO] δ 172.9, 150.0, 138.4, 136.0, 130.7, 127.4, 121.8, 117.5, 60.8, 41.0, 31.5, 24.7, 14.3; MS m/z 293.2 (MH+, 100%). Anal. calcd for C13H16N4O4: C, 53.42; H, 5.52; N, 19.17. Found: C, 53.41; H, 5.49; N, 19.40%. HPLC purity 99.9%.
3-((4-Ethoxy-4-oxobutyl)amino)-7-methylbenzo[e][1,2,4]triazine 1,4-Dioxide (12b). Similarly, the reaction of H2O2 (30%, 5.0 mL, 48.2 mmol) and TFAA (7.4 mL, 53.0 mmol) with ester 18e (1.40 g, 4.82 mmol) gave starting ester 18b (586 mg, 42%), spectroscopically identical to above, and di-oxide 12b (202 mg, 14%) as a red powder: mp (EtOAc) 191–192 °C; 1H NMR δ 8.19 (d, J = 8.9 Hz, 1 H, H-5), 8.13 (br s, 1 H, H-8), 7.70 (dd, J = 8.9, 1.6 Hz, 1 H, H-6), 7.13 (br s, 1 H, NH), 4.16 (q, J = 7.1 Hz, 2 H, CH2O), 3.65 (dd, J = 7.0, 5.7 Hz, 2 H, CH2N), 2.53 (s, 3 H, 7-CH3), 2.45 (t, J = 7.2 Hz, 2 H, CH2), 2.06 (pent, J = 7.0 Hz, 2 H, CH2), 1.26 (t, J = 7.1 Hz, 3 H, CH3); 13C NMR [(CD3)2SO] δ 173.0, 149.7, 138.6, 138.3, 137.0, 130.6, 120.4, 117.3, 60.9, 41.0, 31.6, 24.7, 21.6, 14.4; MS m/z 307.2 (MH+, 100%). Anal. calcd for C14H18N4O4: C, 54.89; H, 5.92; N, 18.29. Found: C, 55.03; H, 5.77; N, 18.50%. HPLC purity 99.9%.
4.2.8. Hydrolysis of Esters (8b–12b)
3-((Carboxymethyl)amino)benzo[e][1,2,4]triazine 1,4-dioxide (8a). A solution of HCl in dioxane (4 M, 69 mL, 277 mmol) was added dropwise to a stirred suspension of ester 8b (1.44 g, 5.55 mmol) in dioxane (40 mL) and water (5 drops), and the mixture was stirred at 20 °C for 6 d. The mixture was diluted with water (50 mL) and extracted with CHCl3 (3 × 60 mL), then the combined organic fraction was dried and the solvent was evaporated. The residue was purified by chromatography, eluting with a gradient (0–5%) of MeOH/DCM, to give (i) starting ester 8b (570 mg, 40%), spectroscopically identical to above; and (ii) acid 8a (99 mg, 8%) as a red powder, mp (H2O) 190–191 °C; 1H NMR [(CD3)2SO] δ 8.45 (br s, 1 H, NH), 8.23 (d, J = 8.7 Hz, 1 H, H 8), 8.19 (d, J = 8.5 Hz, 1 H, H 5), 7.99 (ddd, J = 8.5, 7.0, 1.3 Hz, 1 H, H 6), 7.63 (ddd, J = 8.7, 7.0, 1.3 Hz, 1 H, H 7), 4.09 (d, J = 5.4 Hz, 2 H, CH2), OH not observed; 13C NMR [(CD3)2SO] δ 170.5, 149.8, 138.2, 135.8, 130.3, 127.6, 121.1, 117.0, 42.3. Anal calcd for C9H8N4O4·¼H2O; C, 44.9; H, 3.6; N, 23.3. Found: C, 44.5; H, 3.4; H, 23.4%. HPLC purity 98.4%.
3-((3-Carboxypropyl)amino)benzo[e][1,2,4]triazine 1,4-Dioxide (10a). Similarly, the reaction of HCl in dioxane (4 M, 13 mL, 52 mmol) with ester 10b (298 mg, 1.02 mmol) in dioxane (10 mL) and water (0.5 mL) gave starting ester 10b (36 mg, 12%), spectroscopically identical to above, and acid 10a (158 mg, 59%) as an orange powder: mp (EtOAc) 190–193 °C; 1H NMR [(CD3)2SO] δ 12.06 (br s, 1 H, CO2H), 8.35 (t, J = 6.2 Hz, 1 H, NH), 8.19 (dd, J = 8.7, 0.7 Hz, 1 H, H-8), 8.12 (dd, J = 8.7, 0.7 Hz, 1 H, H-5), 7.92 (ddd, J = 8.7, 6.9, 1.2 Hz, 1 H, H-7), 7.55 (ddd, J = 8.7, 6.9, 1.2 Hz, 1 H, H-6), 3.43 (q, J = 6.4 Hz, 2 H, CH2N), 2.30 (t, J = 7.4 Hz, 2 H, CH2), 1.84 (pent, J = 7.1 Hz, 2 H, CH2); 13C NMR [(CD3)2SO] δ 174.2, 149.8, 138.2, 135.4, 129.9, 126.9, 121.1, 116.9, 40.1, 31.0, 24.1; MS m/z 265.2 (MH+, 100%). Anal. calcd for C11H12N4O4: C, 50.00; H, 4.58; N, 21.20. Found: C, 50.31; H, 4.54; N, 20.82%. HPLC purity 98.6%.
3-((2-Carboxyethyl)amino)-7,8-dihydro-6H-indeno[5,6-e][1,2,4]triazine 1,4-Dioxide (11a). Similarly the reaction of HCl in dioxane (4 M, 4.32 mL, 17.3 mmol) with ester 11b (100 mg, 0.34 mmol) gave starting ester 11b (13 mg, 13%), spectroscopically identical to above, and acid 11a (60 mg, 64%) as a yellow powder: mp (EtOAc) 159 °C (dec.); 1H NMR δ 12.34 (br s, 1 H, CO2H), 8.18 (s, 1 H, H-9), 8.17 (s, 1 H, H-5), 3.22 (t, J = 7.2 Hz, 2 H, CH2), 3.07–3.16 (m, 4 H, H-6, H-8), 2.76 (t, J = 7.2 Hz, 2 H, CH2CO), 2.13 (p, J = 7.5 Hz, 2 H, H-7); 13C NMR δ 173.2, 154.6, 152.7, 150.2, 138.7, 133.5, 115.2, 113.1, 32.8, 32.3, 28.5, 25.2 (2); MS m/z 276.3 (MH+, 100%). Anal. calcd for C13H13N3O4: C, 56.72; H, 4.76; N, 15.27. Found: C, 57.00; H, 4.73; N, 15.08%. HPLC purity 99.7%.
3-((3-Carboxypropyl)amino)-7-methylbenzo[e][1,2,4]triazine 1,4-Dioxide (12a). Similarly, the reaction of HCl in dioxane (4 M, 3.5 mL, 13.8 mmol) with ester 12b (85 mg, 0.27 mmol) gave starting ester 12b (36 mg, 12%), spectroscopically identical to above, and acid 12a (50 mg, 66%) as a red powder: mp (MeOH/H2O) 185–188 °C; 1H NMR [(CD3)2SO] δ 12.06 (br s, 1 H, CO2H), 8.26 (t, J = 6.1 Hz, 1 H, NH), 8.04 (d, J = 8.9 Hz, 1 H, H-5), 8.01 (br s, 1 H, H-8), 7.77 (dd, J = 8.9, 1.5 Hz, 1 H, H-6), 3.40 (q, J = 6.7 Hz, 2 H, CH2N), 2.47 (s, 3 H, 7-CH3), 2.30 (t, J = 7.4 Hz, 2 H, CH2), 1.85 (pent, J = 7.1 Hz, 2 H, CH2); 13C NMR [(CD3)2SO] δ 174.3, 149.5, 137.5, 137.4, 136.8, 129.7, 119.5, 116.8, 40.2, 31.1, 24.1, 20.8; MS m/z 278.2 (MH+, 100%). Anal. calcd for C12H14N4O4·¼H2O: C, 50.97; H, 5.17; N, 19.81. Found: C, 50.73; H, 5.04; N, 19.59%. HPLC purity 98.5%.

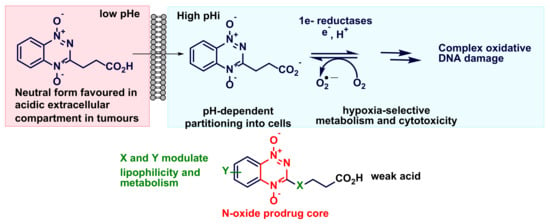
 ,
, 












{kind=link}
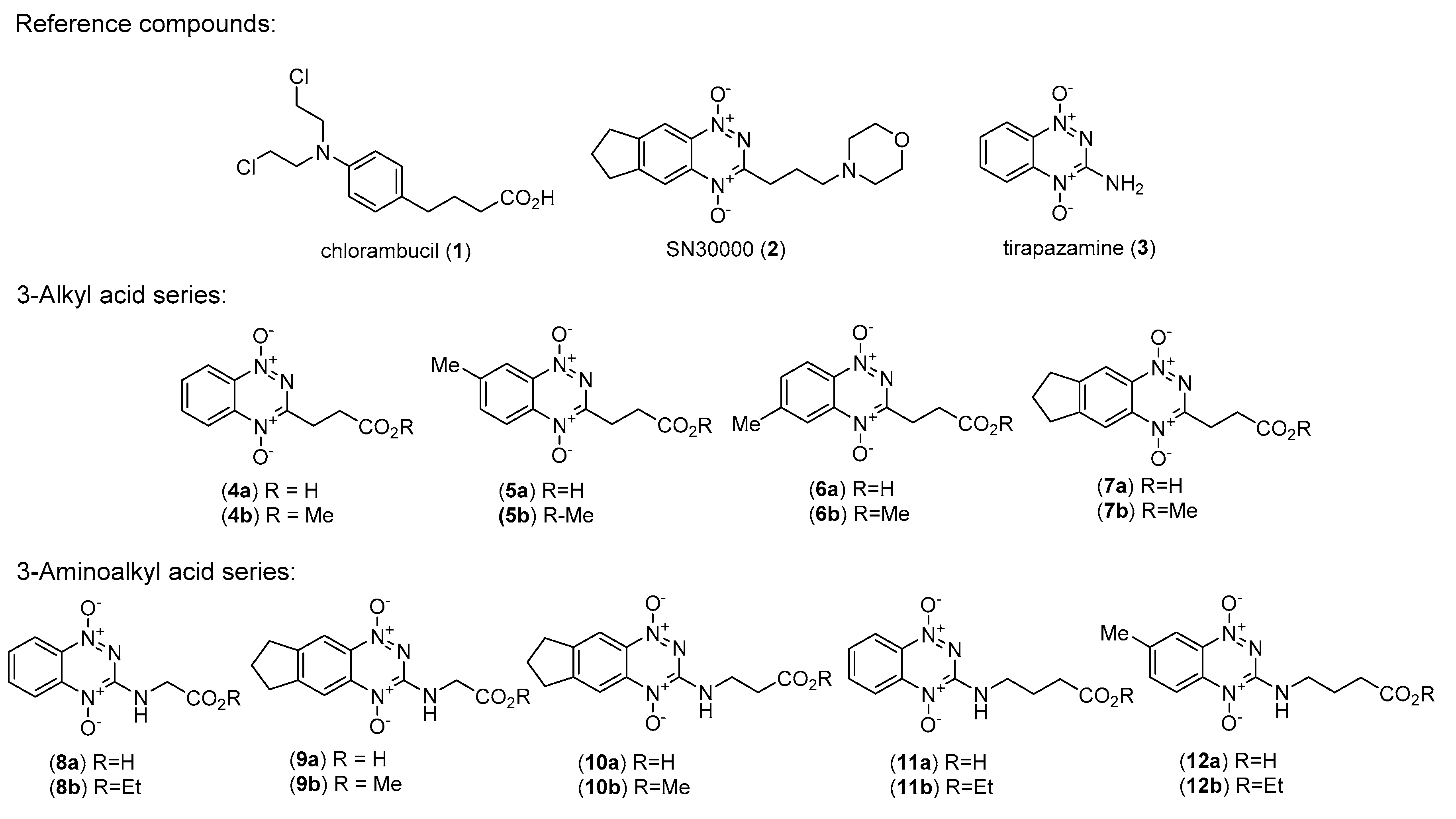{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
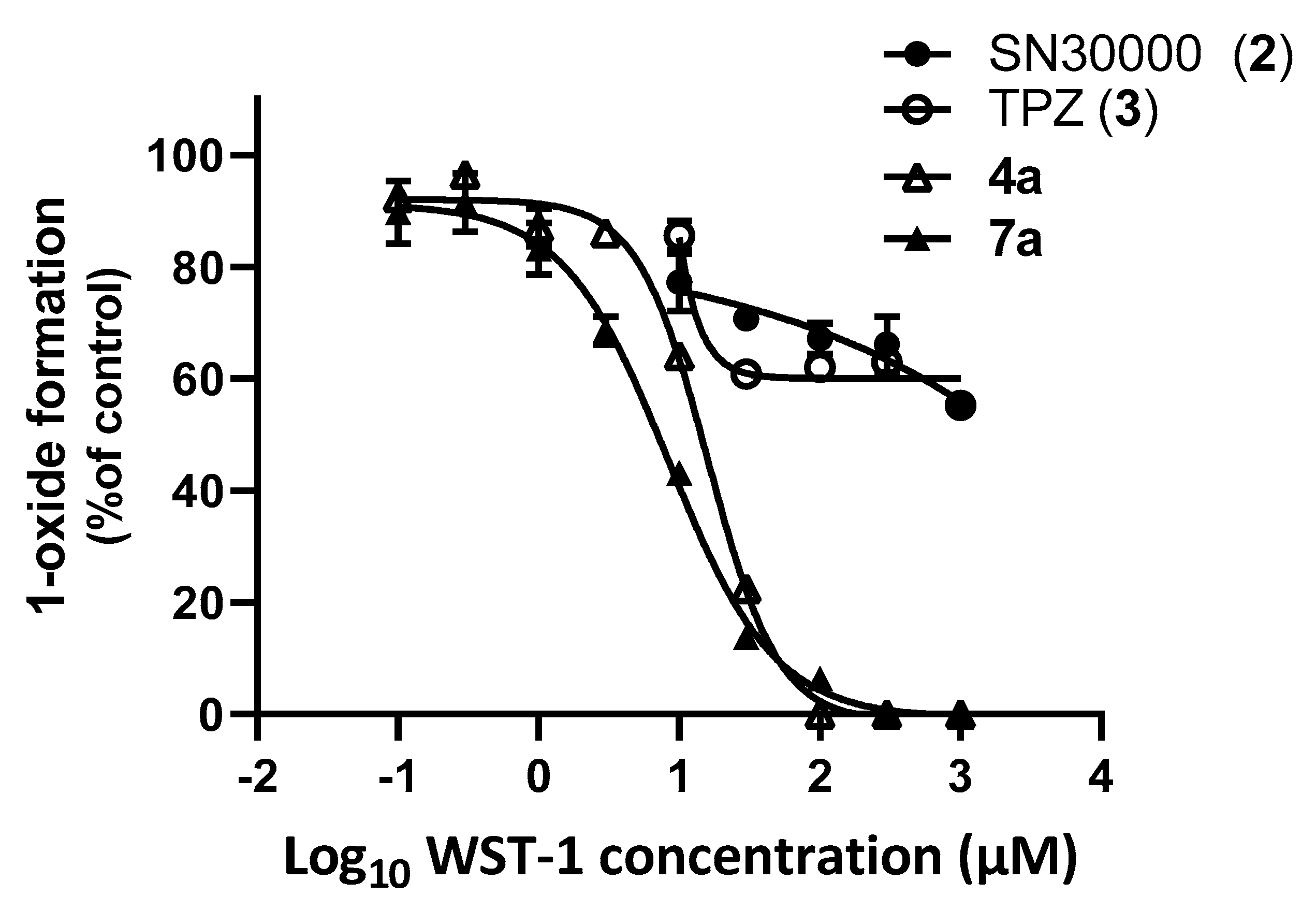{kind=link}
{kind=link}
{kind=link}
{kind=link}