Revisiting the Allosteric Regulation of Sodium Cation on the Binding of Adenosine at the Human A2A Adenosine Receptor: Insights from Supervised Molecular Dynamics (SuMD) Simulations

 , ,
, ,  and
and 
Abstract
:
1. Introduction
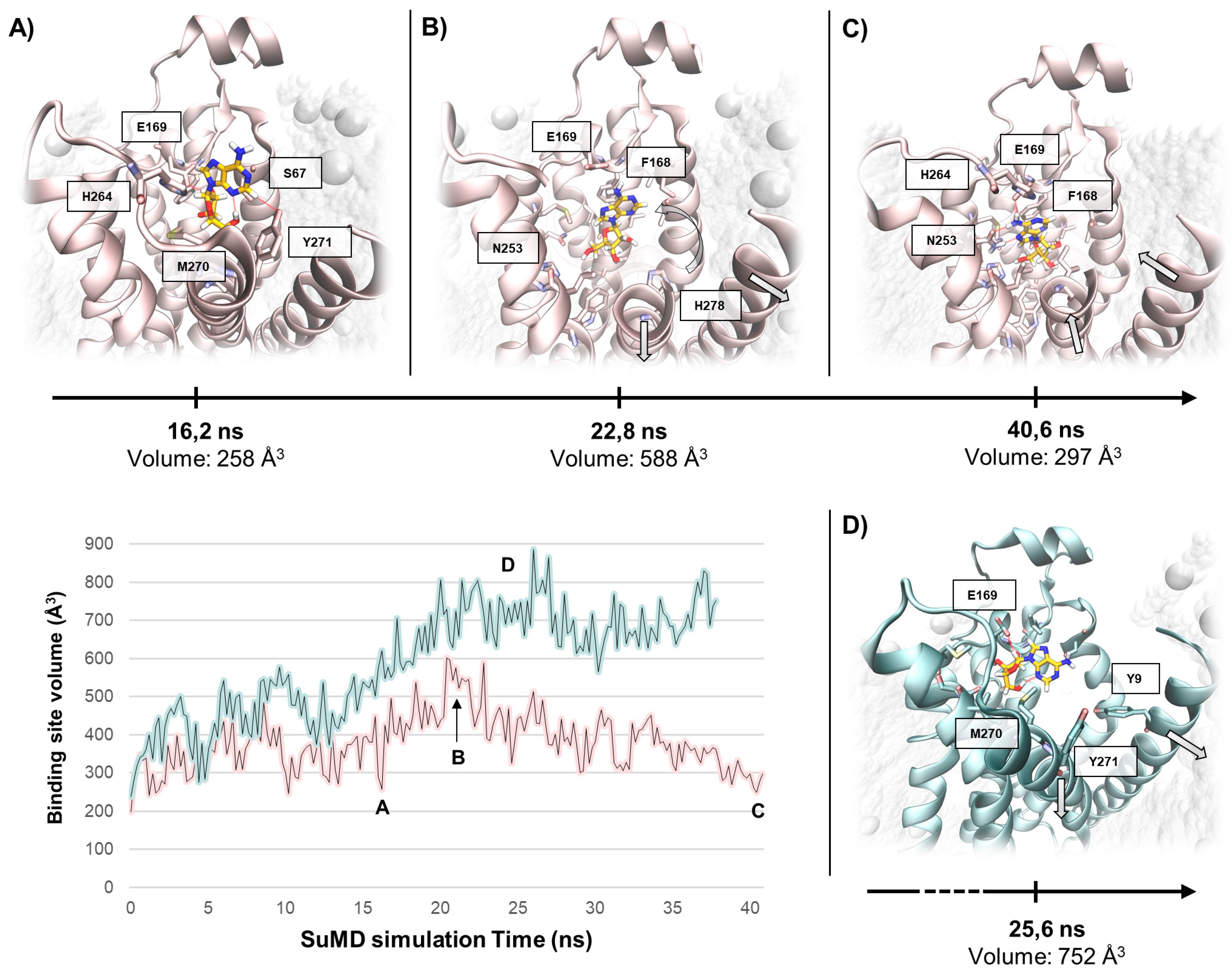
2. Results and Discussion
2.1. SuMD Simulations of the Sodium Ion on the A2A AR
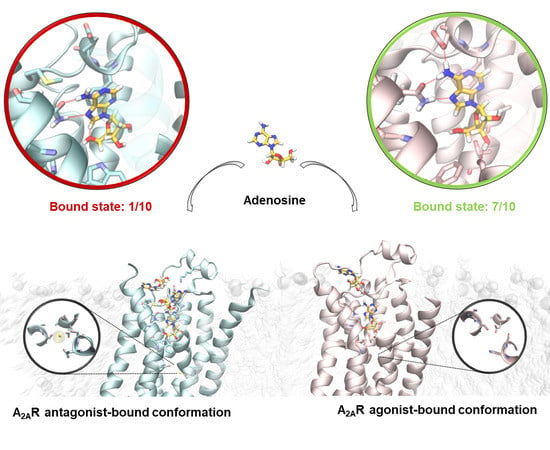
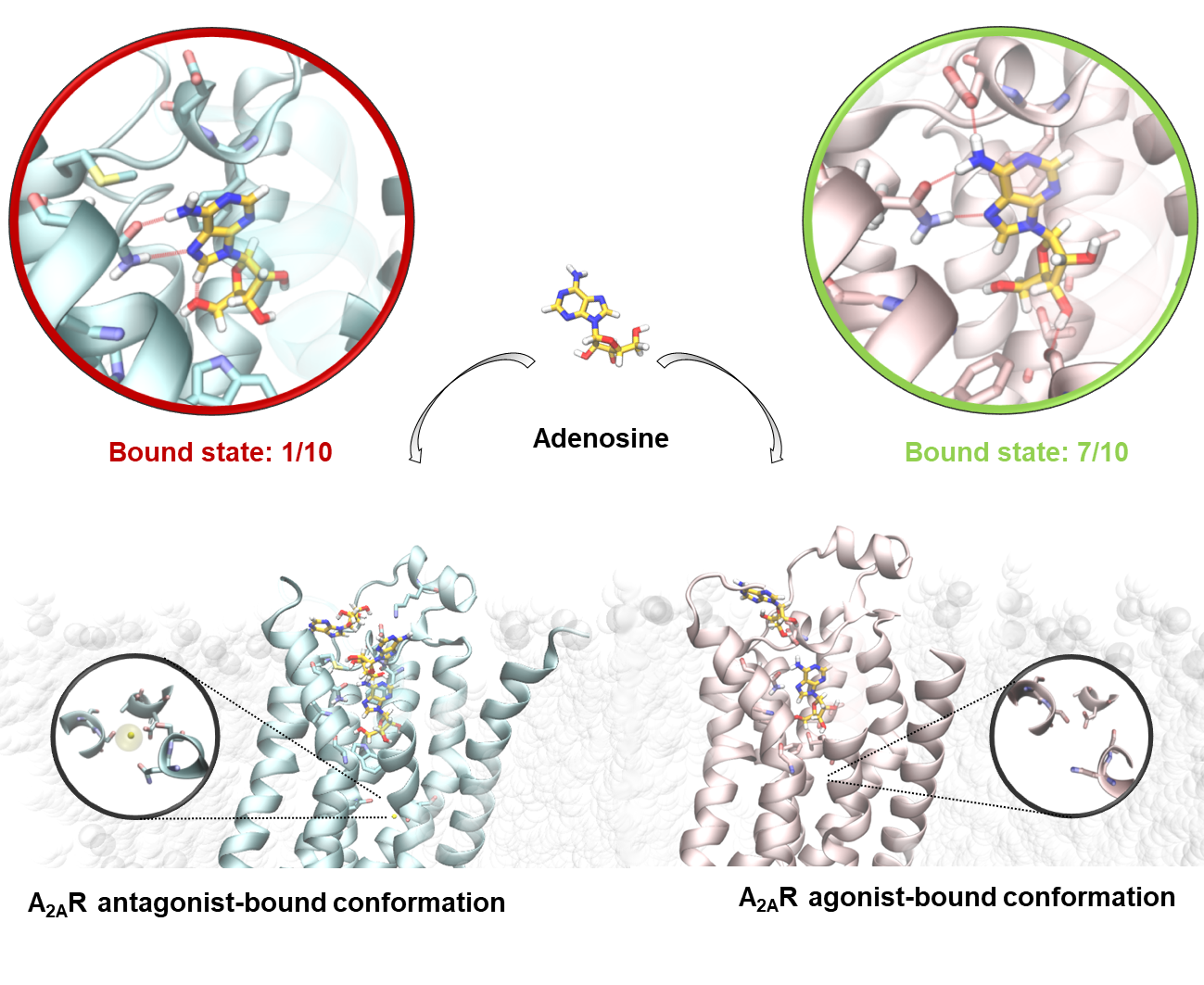
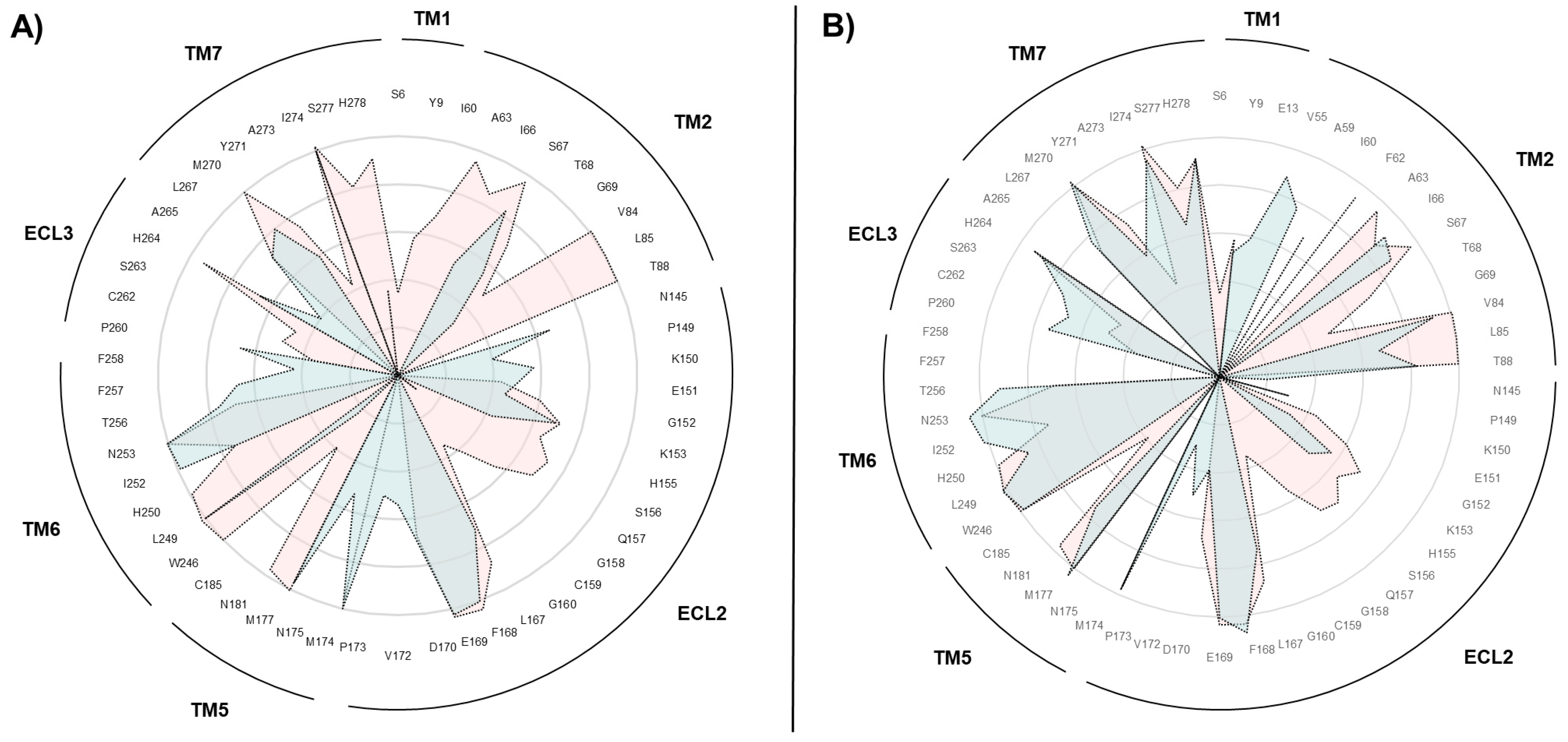
2.2. SuMD Simulations of the Adenosine on the Intermediate-Active, Sodium-Free, A2A AR Conformation
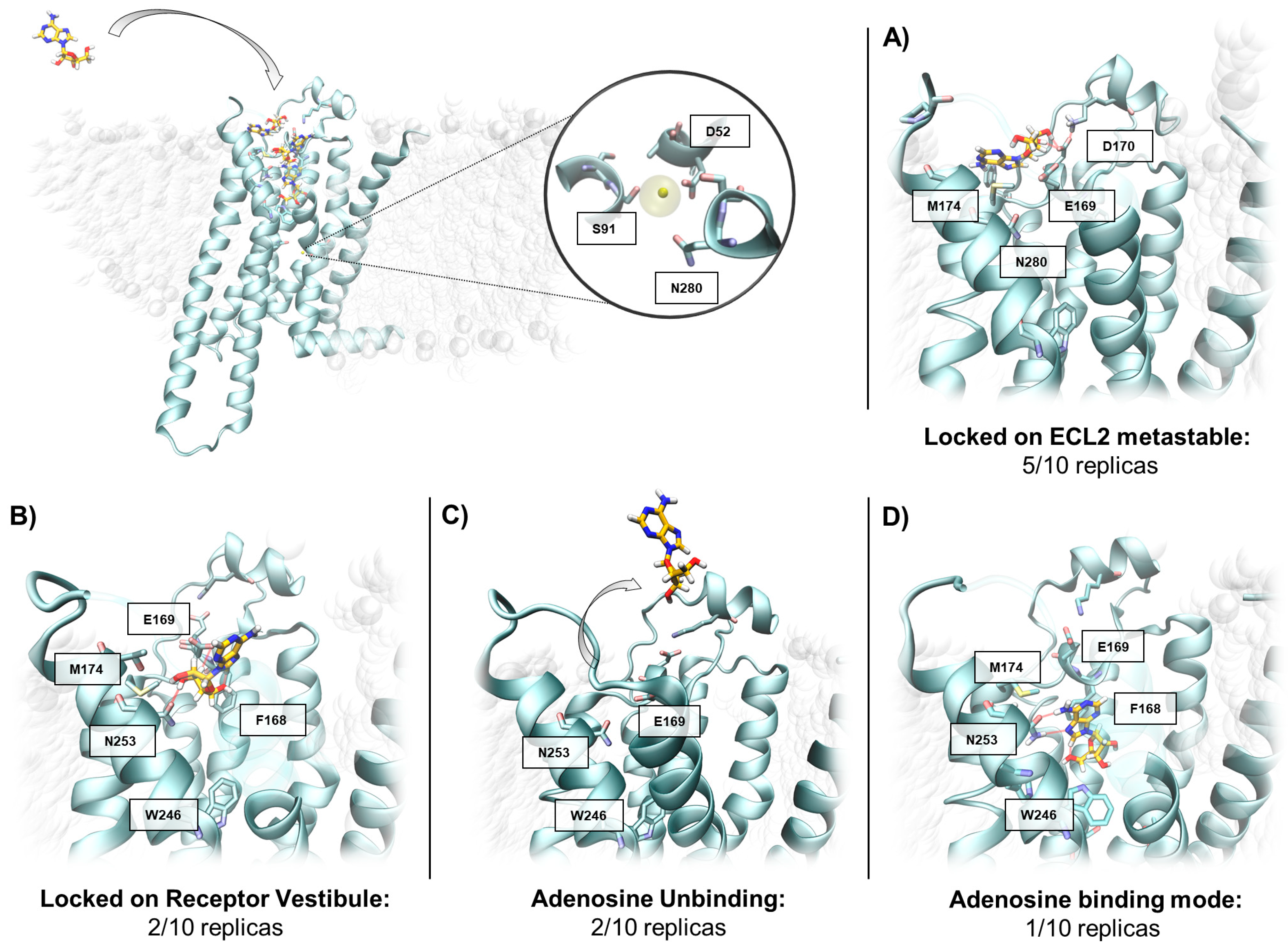
2.3. SuMD Simulations of the Adenosine on the Inactive, Sodium-Bound, A2A AR Conformation
2.4. Insight on the Role of the Sodium Ion in the Recognition of A2A AR Agonists
3. Materials and Methods
3.1. General
3.2. Systems Preparation
3.3. Solvated System Setup and Equilibration
3.4. Supervised Molecular Dynamics (SuMD) Simulations
3.5. SuMD Trajectory Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SuMD | Supervised Molecular Dynamics |
| GPCR | G protein-coupled receptor |
| A2A AR | A2A Adenosine Receptor |
| TM | Transmembrane |
| EC | Extracellular |
| PDB | Protein Data Bank |
| MD | Molecular Dynamics |
| RMSD | Root-mean-square deviation |
| TMD | Transmembrane domain |
| ECL | Extracellular loop |
| POPC | 1-palmitoyl-2oleyl-sn-glycerol-3-phospho-choline |
| OPM | Orientations of Proteins in Membrane |
| NPT | Isothermal–isobaric ensemble |
References
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchio, E.A.; Baltos, J.-A.; Nguyen, A.T.N.; Christopoulos, A.; White, P.J.; May, L.T. New paradigms in adenosine receptor pharmacology: Allostery, oligomerization and biased agonism. Br. J. Pharmacol. 2018, 175, 4036–4046. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Lebon, G. Human Adenosine A2A Receptor: Molecular Mechanism of Ligand Binding and Activation. Front. Pharmacol. 2017, 8, 898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Pert, C.B.; Pasternak, G.; Snyder, S.H. Opiate agonists and antagonists discriminated by receptor binding in brain. Science 1973, 182, 1359–1361. [Google Scholar] [CrossRef]
- Pert, C.B.; Snyder, S.H. Opiate Receptor Binding of Agonists and Antagonists Affected Differentially by Sodium. Mol. Pharmacol. 1974, 10, 868–879. [Google Scholar]
- Katritch, V.; Fenalti, G.; Abola, E.E.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 2014, 39, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Massink, A.; Gutiérrez-De-Terán, H.; Lenselink, E.B.; Zacarías, N.V.O.; Xia, L.; Heitman, L.H.; Katritch, V.; Stevens, R.C.; Ijzerman, A.P. Sodium Ion Binding Pocket Mutations and Adenosine A2A Receptor Functions. Mol. Pharmacol. 2015, 87, 305–313. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Ijzerman, A.P. Allosteric modulation of A2A adenosine receptors by amiloride analogues and sodiumions. Biochem. Pharmacol. 2000, 60, 669–676. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef]
- Margiotta, E.; Deganutti, G.; Moro, S. Could the presence of sodium ion influence the accuracy and precision of the ligand-posing in the human A2A adenosine receptor orthosteric binding site using a molecular docking approach? Insights from Dockbench. J. Comput. Aided Mol. Des. 2018, 32, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Selvam, B.; Shamsi, Z.; Shukla, D. Universality of the Sodium Ion Binding Mechanism in Class AG-Protein-Coupled Receptors. Angew. Chem. 2018, 130, 3102–3107. [Google Scholar] [CrossRef]
- Gutiérrez-de-Terán, H.; Massink, A.; Rodríguez, D.; Liu, W.; Han, G.W.; Joseph, J.S.; Katritch, I.; Heitman, L.H.; Xia, L.; IJzerman, A.P.; et al. The Role of a Sodium Ion Binding Site in the Allosteric Modulation of the A2A Adenosine G Protein-Coupled Receptor. Structure 2013, 21, 2175–2185. [Google Scholar] [CrossRef] [Green Version]
- Vickery, O.N.; Carvalheda, C.A.; Zaidi, S.A.; Pisliakov, A.V.; Katritch, V.; Zachariae, U. Intracellular Transfer of Na+ in an Active-State G-Protein-Coupled Receptor. Structure 2018, 26, 171–180. [Google Scholar] [CrossRef]
- Hu, X.; Wang, Y.; Hunkele, A.; Provasi, D.; Pasternak, G.W.; Filizola, M. Kinetic and thermodynamic insights into sodium ion translocation through the μ-opioid receptor from molecular dynamics and machine learning analysis. PLoS Comput. Biol. 2019, 15, e1006689. [Google Scholar] [CrossRef]
- Shang, Y.; LeRouzic, V.; Schneider, S.; Bisignano, P.; Pasternak, G.W.; Filizola, M. Mechanistic Insights into the Allosteric Modulation of Opioid Receptors by Sodium Ions. Biochemistry 2014, 53, 5140–5149. [Google Scholar] [CrossRef]
- Fleetwood, O.; Matricon, P.; Carlsson, J.; Delemotte, L. Energy landscapes reveal agonist’s control of GPCR activation via microswitches. BioRxiv 2019, 627026. [Google Scholar] [CrossRef]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR—Ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef]
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. Exploring Protein-Peptide Recognition Pathways Using a Supervised Molecular Dynamics Approach. Structure 2017, 25, 655–662. [Google Scholar] [CrossRef]
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the complexity of ligand—Protein recognition pathways using supervised molecular dynamics (SuMD) simulations. J. Chem. Inf. Model. 2016, 56, 687–705. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Proceedings of the 2nd International Conference on Knowledge Discovery and Data Mining, Portland, OR, USA, 2–4 August 1996; AAAI press: Portland, OR, USA, 1996. [Google Scholar]
- Segala, E.; Guo, D.; Cheng, R.K.Y.; Bortolato, A.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Heitman, L.H.; IJzerman, A.P.; Marshall, F.H.; et al. Controlling the Dissociation of Ligands from the Adenosine A2A Receptor through Modulation of Salt Bridge Strength. J. Med. Chem. 2016, 59, 6470–6479. [Google Scholar] [CrossRef]
- Pang, X.; Yang, M.; Han, K. Antagonist binding and induced conformational dynamics of GPCR A2A adenosine receptor. Proteins 2013, 81, 1399–1410. [Google Scholar] [CrossRef]
- Yuan, S.; Hu, Z.; Filipek, S.; Vogel, H. W2466.48 opens a gate for a continuous intrinsic water pathway during activation of the adenosine A2A receptor. Angew. Chem. Int. Ed. 2015, 54, 556–559. [Google Scholar]
- Igonet, S.; Raingeval, C.; Cecon, E.; Pučić-Baković, M.; Lauc, G.; Cala, O.; Baranowski, M.; Perez, J.; Jockers, R.; Krimm, I.; et al. Enabling STD-NMR fragment screening using stabilized native GPCR: A case study of adenosine receptor. Sci. Rep. 2018, 8, 8142. [Google Scholar] [CrossRef]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.W.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef] [Green Version]
- Sabbadin, D.; Ciancetta, A.; Deganutti, G.; Cuzzolin, A.; Moro, S. Exploring the recognition pathway at the human A2A adenosine receptor of the endogenous agonist adenosine using supervised molecular dynamics simulations. Medchemcomm 2015, 6, 1081–1085. [Google Scholar] [CrossRef]
- Deganutti, G.; Welihinda, A.; Moro, S. Comparison of the Human A2A Adenosine Receptor Recognition by Adenosine and Inosine: New Insight from Supervised Molecular Dynamics Simulations. ChemMedChem 2017, 12, 1319–1326. [Google Scholar] [CrossRef]
- Lee, S.; Nivedha, A.K.; Tate, C.G.; Vaidehi, N. Dynamic Role of the G Protein in Stabilizing the Active State of the Adenosine A2A Receptor. Structure 2019, 27, 703–712.e3. [Google Scholar] [CrossRef] [Green Version]
- Chemical Computing Group (CCG) Inc. Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, QC, Canada, 2019. [Google Scholar]
- Harvey, M.J.; Giupponi, G.; Fabritiis, G. De ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [Green Version]
- Labute, P. Protonate 3D: Assignment of Macromolecular Protonation State and Geometry; Chem. Comput. Group Inc: Montreal, QC, Canada, 2007. [Google Scholar]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of Proteins in Membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Grubmuller, H.; Groll, V. Solvate 1.0.
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates ofN-acetylalanyl-N?-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef]
- Durrant, J.D.; Votapka, L.; Sørensen, J.; Amaro, R.E. POVME 2.0: An Enhanced Tool for Determining Pocket Shape and Volume Characteristics. J. Chem. Theory Comput. 2014, 10, 5047–5056. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Not available |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Best Resolution (Å) | Number of Structures | Class A Branch | |
|---|---|---|---|
| A2A adenosine receptor | 1.7 | 24 | α |
| Protease-activated receptor 1 | 2.2 | 1 | δ |
| Protease-activated receptor 2 | 2.8 | 2 | δ |
| β1 adrenergic receptor | 2.1 | 3 | α |
| D4 dopamine receptor | 2.1 | 1 | α |
| Complement component 5a receptor 1 | 2.2 | 1 | γ |
| δ opioid receptor | 1.8 | 2 | γ |
| A2A AR Inactive Conformation | A2A AR Intermediate Active Conformation | |||||
|---|---|---|---|---|---|---|
| SuMD time (ns) | Reached the allosteric site | RMSDmin (Å) | SuMD time (ns) | Reached the allosteric site | RMSDmin (Å) | |
| Replica 1 | 10.8 | No | 10.03 | 15.4 | Yes | 0.2 |
| Replica 2 | 23.6 | Yes | 0.17 | 12.0 | Yes | 0.1 |
| Replica 3 | 20.6 | Yes | 0.04 | 2.4 | No | 24.2 |
| Replica 4 | 20.8 | Yes | 0.18 | 15.6 | Yes | 0.1 |
| Replica 5 | 18.2 | Yes | 0.40 | 4.6 | No | 17.1 |
| SuMD Time (ns) | Reached the Orthosteric Site | Adenosine Binding Mode | X-ray Binding Mode after 100 ns of MD | RMSDmin (Å) | |
|---|---|---|---|---|---|
| Replica 1a | 7.2 | No | No (Meta-binding site on ECL2) | - | 14.3 |
| Replica 2a | 31.8 | Yes | No (Distorted binding mode) | Yes | 0.4 |
| Replica 3a | 40.8 | Yes | Yes | Yes | 0.4 |
| Replica 4a | 32.4 | Yes | No (Ribose Up) | Yes (Ribose syn conformation) | 2.5 |
| Replica 5a | 29.4 | Yes | No (Ribose Up) | Yes (Ribose syn conformation) | 2.7 |
| Replica 6a | 15.6 | No | No (Meta-binding site on ECL2) | - | 12.2 |
| Replica 7a | 28.2 | Yes | No (Ribose Up) | Yes (Ribose syn conformation) | 0.4 |
| Replica 8a | 32.4 | Yes | Yes (Ribose syn conformation) | Yes (Ribose syn conformation) | 2.7 |
| Replica 9a | 24.0 | Yes | No (Distorted binding mode) | Yes (Ribose syn conformation) | 2.3 |
| Replica 10a | 10.2 | No | No (Distorted binding mode) | - | 15.3 |
| SuMD Time (ns) | Reached the Orthosteric Site | Adenosine BINDING Mode | X-ray Binding Mode after 100 ns of MD | RMSDmin (Å) | |
|---|---|---|---|---|---|
| Replica 1i | 9.0 | No | No (Meta-binding site on ECL2) | - | 16.1 |
| Replica 2i | 16.8 | No | No (Meta-binding site on ECL2) | - | 13.6 |
| Replica 3i | 16.2 | Yes | No (Receptor Vestibule) | No (Receptor Vestibule) | 5.5 |
| Replica 4i | 31.2 | Yes | No (Receptor Vestibule) | No (Adenosine unbinding) | 6.1 |
| Replica 5i | 37.8 | Yes | No (Receptor Vestibule) | No (Receptor Vestibule) | 6.6 |
| Replica 6i | 24.6 | No | No (Meta-binding site on ECL2 | - | 15.4 |
| Replica 7i | 7.8 | No | No (Meta-binding site on ECL2) | - | 14.7 |
| Replica 8i | 7.8 | No | No (Meta-binding site on ECL2) | - | 13.8 |
| Replica 9i | 8.4 | Yes | No (Receptor Vestibule) | No (Adenosine unbinding) | 7.9 |
| Replica 10i | 45.6 | Yes | No (Receptor Vestibule) | Yes | 0.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bissaro, M.; Bolcato, G.; Deganutti, G.; Sturlese, M.; Moro, S. Revisiting the Allosteric Regulation of Sodium Cation on the Binding of Adenosine at the Human A2A Adenosine Receptor: Insights from Supervised Molecular Dynamics (SuMD) Simulations. Molecules 2019, 24, 2752. https://doi.org/10.3390/molecules24152752
Bissaro M, Bolcato G, Deganutti G, Sturlese M, Moro S. Revisiting the Allosteric Regulation of Sodium Cation on the Binding of Adenosine at the Human A2A Adenosine Receptor: Insights from Supervised Molecular Dynamics (SuMD) Simulations. Molecules. 2019; 24(15):2752. https://doi.org/10.3390/molecules24152752
Chicago/Turabian StyleBissaro, Maicol, Giovanni Bolcato, Giuseppe Deganutti, Mattia Sturlese, and Stefano Moro. 2019. "Revisiting the Allosteric Regulation of Sodium Cation on the Binding of Adenosine at the Human A2A Adenosine Receptor: Insights from Supervised Molecular Dynamics (SuMD) Simulations" Molecules 24, no. 15: 2752. https://doi.org/10.3390/molecules24152752
APA StyleBissaro, M., Bolcato, G., Deganutti, G., Sturlese, M., & Moro, S. (2019). Revisiting the Allosteric Regulation of Sodium Cation on the Binding of Adenosine at the Human A2A Adenosine Receptor: Insights from Supervised Molecular Dynamics (SuMD) Simulations. Molecules, 24(15), 2752. https://doi.org/10.3390/molecules24152752