Validation of an LC-MS/MS Method for the Quantification of Caffeine and Theobromine Using Non-Matched Matrix Calibration Curve


 ,
, 

Abstract
:1. Introduction
2. Results and Discussion
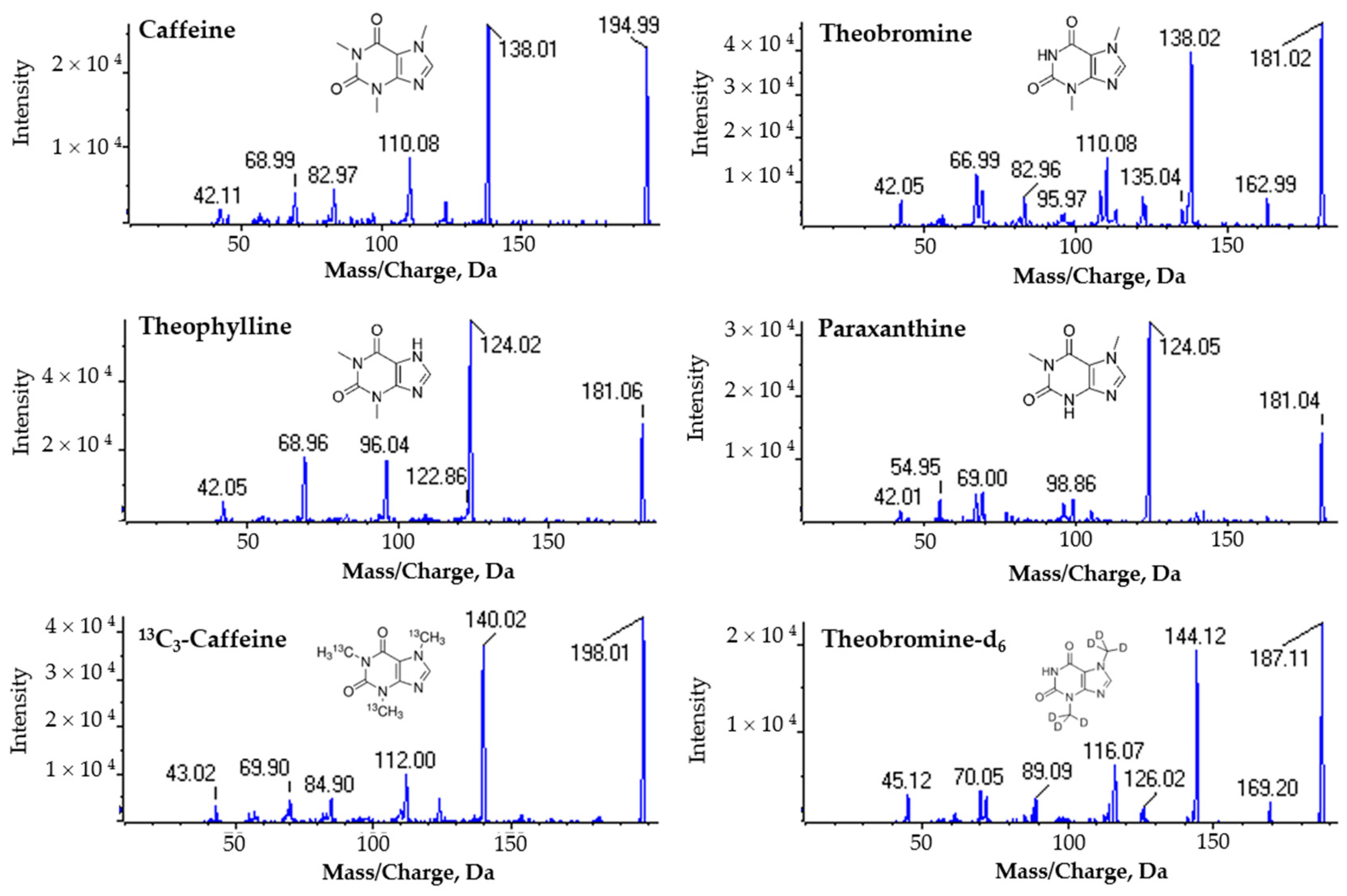
2.1. Optimization of LC-MS Conditions
2.2. Method Validation
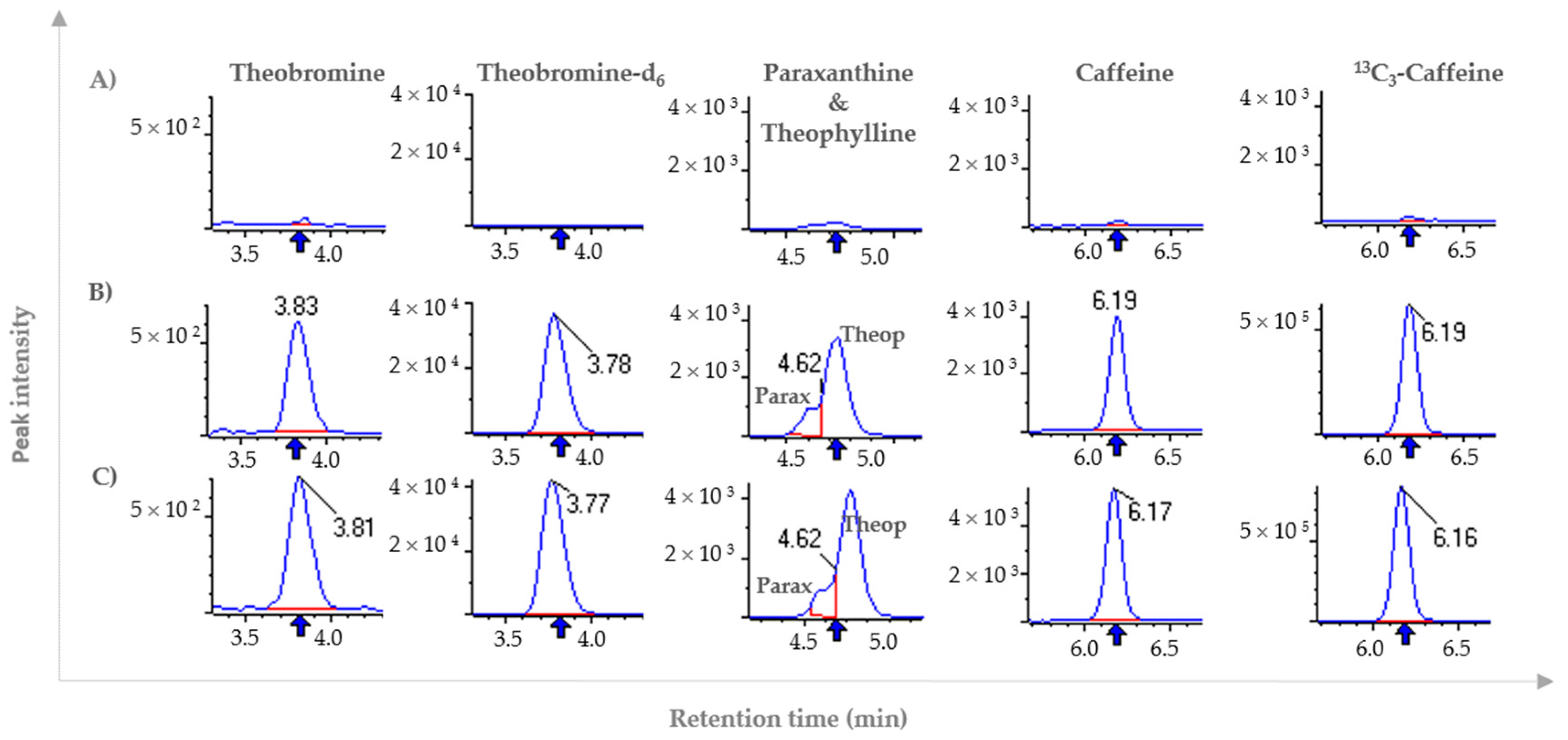
2.2.1. Selectivity
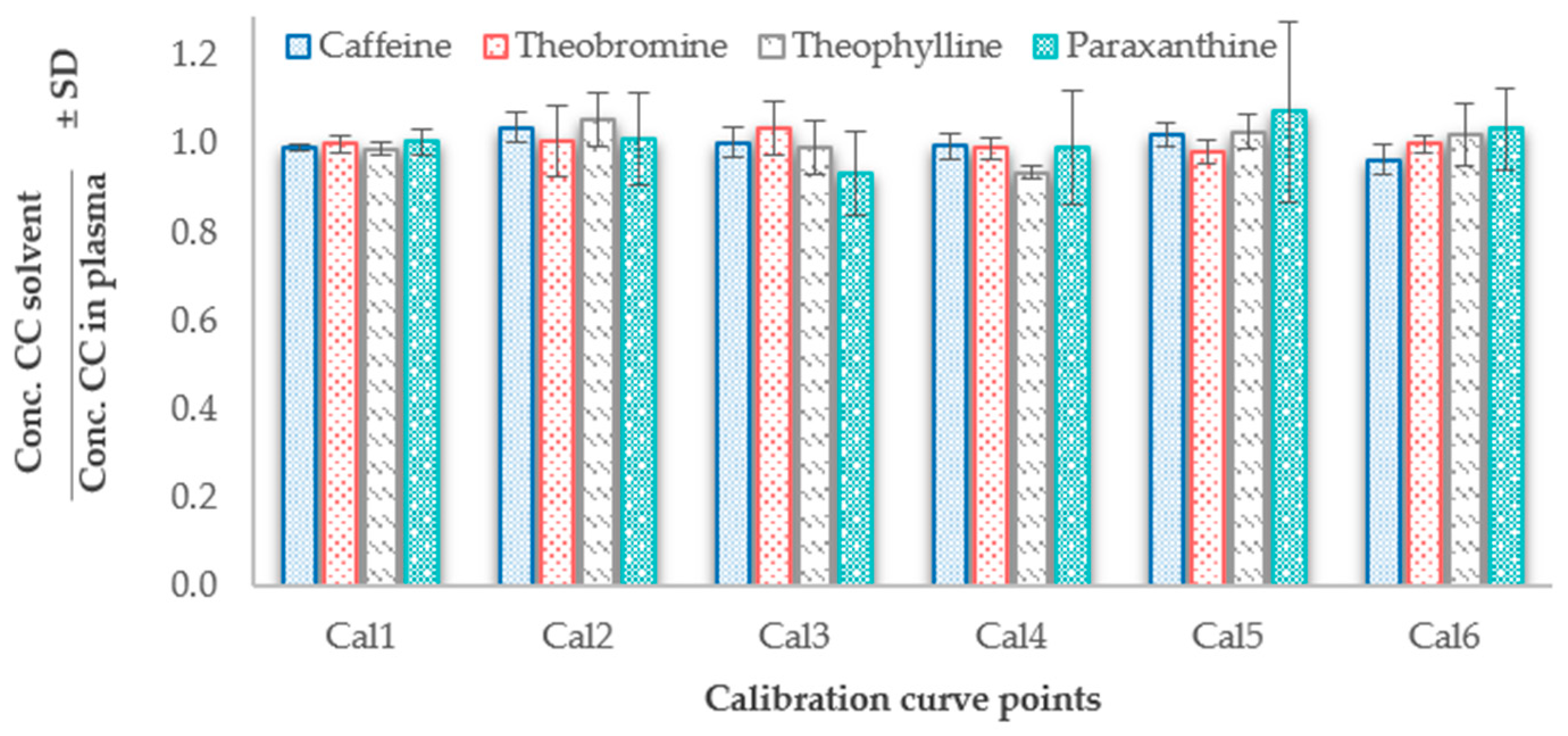
2.2.2. Linearity, Limits of Detection and Quantification
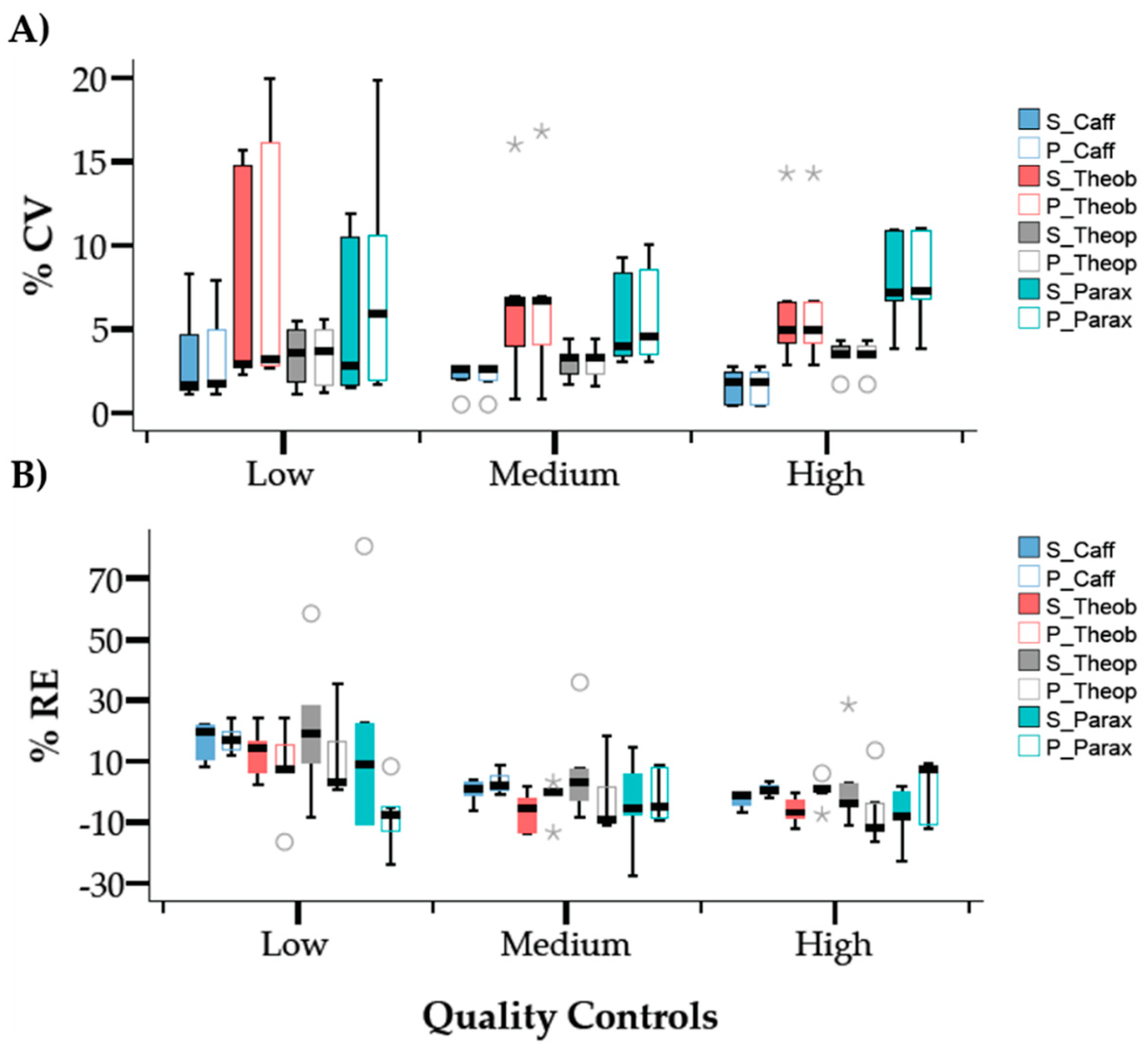
2.2.3. Precision and Accuracy
2.2.4. Extraction Efficiency and Matrix Effect
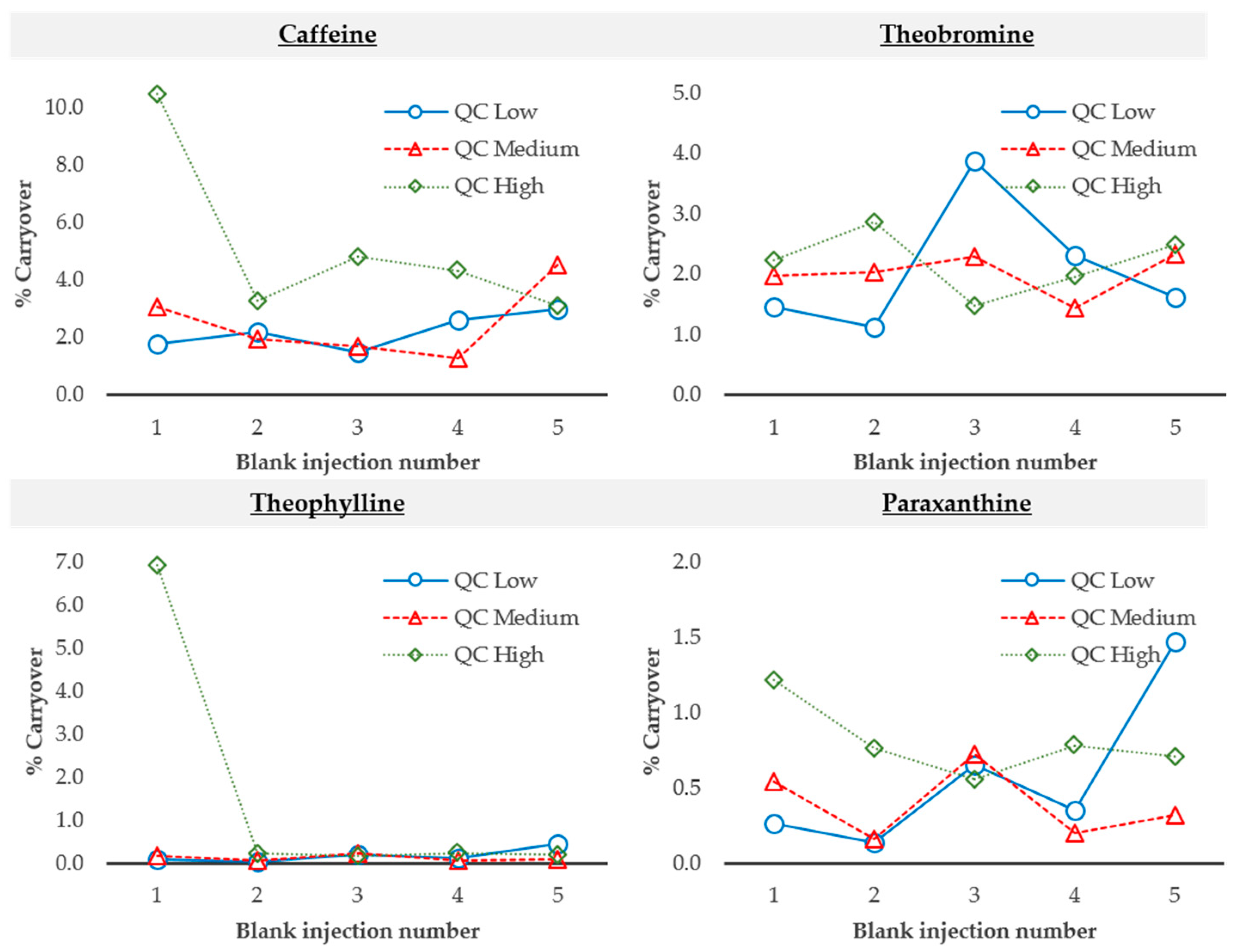
2.2.5. Carryover
2.3. Applicability of the Method in Human Plasma Samples
3. Materials and Methods
3.1. Chemicals
3.2. Compound Optimization for the MRM Acquisition Method
3.3. Liquid Chromatography-Mass Spectrometry Instrumentation
3.4. Method Validation
3.4.1. Selectivity
3.4.2. Linearity and Limits of Detection and Quantification
3.4.3. Intra and Inter-Day Precision and Accuracy
3.4.4. Extraction Efficiency and Matrix Effect
3.4.5. Carryover
3.5. Plasma Samples
3.6. Plasma Samples Preparation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McLellan, T.M.; Caldwell, J.A.; Lieberman, H.R. A review of caffeine’s effects on cognitive, physical and occupational performance. Neurosci. Biobehav. Rev. 2016, 71, 294–312. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A. Interindividual Differences in Caffeine Metabolism and Factors Driving Caffeine Consumption. Pharmacol. Rev. 2018, 70, 384–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, D.C.; Knight, C.A.; Hockenberry, J.; Teplansky, R.; Hartman, T.J. Beverage caffeine intakes in the U.S. Food Chem. Toxicol. 2014, 63, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrot, P.; Jordan, S.; Eastwood, J.; Rotstein, J.; Hugenholtz, A.; Feeley, M. Effects of caffeine on human health. Food Addit. Contam. 2003, 20, 1–30. [Google Scholar] [CrossRef] [PubMed]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies). Scientific Opinion on the safety of caffeine. Efsa J. 2015, 13, 4102. [Google Scholar] [CrossRef]
- Verster, J.C.; Koenig, J. Caffeine intake and its sources: A review of national representative studies. Crit. Rev. Food Sci. Nutr. 2018, 58, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A. Are we dependent upon coffee and caffeine? A review on human and animal data. Neurosci. Biobehav. Rev. 1999, 23, 563–576. [Google Scholar] [CrossRef]
- George, A.J. Central nervous system stimulants. Baillieres Best Pract. Res. Clin. Endocrinol. Metab. 2000, 14, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Cappelletti, S.; Piacentino, D.; Sani, G.; Aromatario, M. Caffeine: Cognitive and physical performance enhancer or psychoactive drug? Curr. Neuropharmacol. 2015, 13, 71–88. [Google Scholar] [CrossRef]
- Maia, L.; de Mendonca, A. Does caffeine intake protect from Alzheimer’s disease? Eur. J. Neurol. 2002, 9, 377–382. [Google Scholar] [CrossRef]
- Arendash, G.W.; Cao, C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. 1), S117–S126. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Zhang, S.M.; Hernan, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann. Neurol. 2001, 50, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Yang, J.; Wang, Y. Low, but not high, dose caffeine is a readily available probe for adenosine actions. Mol. Aspects Med. 2017, 55, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.W. Caffeine analogs: Biomedical impact. Cell Mol. Life Sci. 2007, 64, 2153–2169. [Google Scholar] [CrossRef] [PubMed]
- Costenla, A.R.; Cunha, R.A.; de Mendonca, A. Caffeine, adenosine receptors, and synaptic plasticity. J. Alzheimers Dis. 2010, 20 (Suppl. 1), S25–S34. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.W.; Shi, D.; Nikodijevic, O.; Jacobson, K.A. The role of adenosine receptors in the central action of caffeine. Pharmacopsychoecologia 1994, 7, 201–213. [Google Scholar] [PubMed]
- Ribeiro, J.A.; Sebastiao, A.M. Caffeine and adenosine. J. Alzheimers Dis. 2010, 20 (Suppl. 1), S3–S15. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Hu, Z.Y.; Parker, R.B.; Laizure, S.C. Measurement of caffeine and its three primary metabolites in human plasma by HPLC-ESI-MS/MS and clinical application. Biomed. Chromatogr. 2017, 31, e3900. [Google Scholar] [CrossRef]
- Tang-Liu, D.D.; Williams, R.L.; Riegelman, S. Disposition of caffeine and its metabolites in man. J. Pharmacol. Exp. Ther. 1983, 224, 180–185. [Google Scholar]
- Orru, M.; Guitart, X.; Karcz-Kubicha, M.; Solinas, M.; Justinova, Z.; Barodia, S.K.; Zanoveli, J.; Cortes, A.; Lluis, C.; Casado, V.; et al. Psychostimulant pharmacological profile of paraxanthine, the main metabolite of caffeine in humans. Neuropharmacology 2013, 67, 476–484. [Google Scholar] [CrossRef]
- Boulenger, J.P.; Patel, J.; Marangos, P.J. Effects of caffeine and theophylline on adenosine and benzodiazepine receptors in human brain. Neurosci. Lett. 1982, 30, 161–166. [Google Scholar] [CrossRef]
- Turnbull, D.; Rodricks, J.V.; Mariano, G.F.; Chowdhury, F. Caffeine and cardiovascular health. Regul. Toxicol. Pharmacol. 2017, 89, 165–185. [Google Scholar] [CrossRef]
- Del Brutto, O.H.; Mera, R.M.; Zambrano, M. Cardiovascular health and caffeine consumption. A population-based study in rural Ecuador. Int. J. Cardiol. 2014, 172, 284–285. [Google Scholar] [CrossRef]
- Chou, T.M.; Benowitz, N.L. Caffeine and coffee: Effects on health and cardiovascular disease. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1994, 109, 173–189. [Google Scholar] [CrossRef]
- de Mendonca, A.; Cunha, R.A. Therapeutic opportunities for caffeine in Alzheimer’s disease and other neurodegenerative disorders. J. Alzheimers Dis. 2010, 20 (Suppl. 1), S1–S2. [Google Scholar] [CrossRef]
- Costa, J.; Lunet, N.; Santos, C.; Santos, J.; Vaz-Carneiro, A. Caffeine exposure and the risk of Parkinson’s disease: A systematic review and meta-analysis of observational studies. J. Alzheimers Dis. 2010, 20 (Suppl. 1), S221–S238. [Google Scholar] [CrossRef]
- Khadrawy, Y.A.; Salem, A.M.; El-Shamy, K.A.; Ahmed, E.K.; Fadl, N.N.; Hosny, E.N. Neuroprotective and Therapeutic Effect of Caffeine on the Rat Model of Parkinson’s Disease Induced by Rotenone. J. Diet Suppl. 2017, 14, 553–572. [Google Scholar] [CrossRef]
- Prediger, R.D. Effects of caffeine in Parkinson’s disease: From neuroprotection to the management of motor and non-motor symptoms. J. Alzheimers Dis. 2010, 20 (Suppl. 1), S205–S220. [Google Scholar] [CrossRef]
- Dewar, L.; Heuberger, R. The effect of acute caffeine intake on insulin sensitivity and glycemic control in people with diabetes. Diabetes Metab. Syndr. 2017, 11 (Suppl. 2), S631–S635. [Google Scholar] [CrossRef]
- Urry, E.; Jetter, A.; Holst, S.C.; Berger, W.; Spinas, G.A.; Langhans, W.; Landolt, H.P. A case-control field study on the relationships among type 2 diabetes, sleepiness and habitual caffeine intake. J. Psychopharmacol. 2017, 31, 233–242. [Google Scholar] [CrossRef]
- Inoue, M.; Tsugane, S. Coffee Drinking and Reduced Risk of Liver Cancer: Update on Epidemiological Findings and Potential Mechanisms. Curr. Nutr. Rep. 2019, 8, 182–186. [Google Scholar] [CrossRef]
- Schmit, S.L.; Rennert, H.S.; Rennert, G.; Gruber, S.B. Coffee Consumption and the Risk of Colorectal Cancer. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.J.; Bae, S.H.; Park, J.B.; Kwon, M.J.; Jang, S.M.; Zheng, Y.F.; Lee, Y.S.; Lee, S.J.; Bae, S.K. Simultaneous quantification of caffeine and its three primary metabolites in rat plasma by liquid chromatography-tandem mass spectrometry. Food Chem. 2013, 141, 2735–2742. [Google Scholar] [CrossRef]
- Lopez-Sanchez, R.D.C.; Lara-Diaz, V.J.; Aranda-Gutierrez, A.; Martinez-Cardona, J.A.; Hernandez, J.A. HPLC Method for Quantification of Caffeine and Its Three Major Metabolites in Human Plasma Using Fetal Bovine Serum Matrix to Evaluate Prenatal Drug Exposure. J. Anal. Methods Chem. 2018, 2018, 2085059. [Google Scholar] [CrossRef]
- Gassner, A.L.; Schappler, J.; Feinberg, M.; Rudaz, S. Derivation of uncertainty functions from validation studies in biological fluids: Application to the analysis of caffeine and its major metabolites in human plasma samples. J. Chromatogr. A 2014, 1353, 121–130. [Google Scholar] [CrossRef]
- Van Eeckhaut, A.; Lanckmans, K.; Sarre, S.; Smolders, I.; Michotte, Y. Validation of bioanalytical LC-MS/MS assays: Evaluation of matrix effects. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2198–2207. [Google Scholar] [CrossRef]
- Krautbauer, S.; Buchler, C.; Liebisch, G. Relevance in the Use of Appropriate Internal Standards for Accurate Quantification Using LC-MS/MS: Tauro-Conjugated Bile Acids as an Example. Anal. Chem. 2016, 88, 10957–10961. [Google Scholar] [CrossRef]
- Wakamatsu, A.; Ochiai, S.; Suzuki, E.; Yokota, Y.; Ochiai, M.; Kotani, Y.; Sasahara, S.; Nakanaga, K.; Hashimoto, Y.; Ueno, S.; et al. Proposed selection strategy of surrogate matrix to quantify endogenous substances by Japan Bioanalysis Forum DG2015-15. Bioanalysis 2018, 10, 1349–1360. [Google Scholar] [CrossRef] [Green Version]
- Alvi, S.N.; Hammami, M.M. Validated HPLC method for determination of caffeine level in human plasma using synthetic plasma: Application to bioavailability studies. J. Chromatogr. Sci. 2011, 49, 292–296. [Google Scholar] [CrossRef]
- Gu, H.; Liu, G.; Wang, J.; Aubry, A.F.; Arnold, M.E. Selecting the correct weighting factors for linear and quadratic calibration curves with least-squares regression algorithm in bioanalytical LC-MS/MS assays and impacts of using incorrect weighting factors on curve stability, data quality, and assay performance. Anal. Chem. 2014, 86, 8959–8966. [Google Scholar]
- Almeida, A.M.; Castel-Branco, M.M.; Falcao, A.C. Linear regression for calibration lines revisited: Weighting schemes for bioanalytical methods. J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2002, 774, 215–222. [Google Scholar] [CrossRef]
- del Campo, M.; Mollenhauer, B.; Bertolotto, A.; Engelborghs, S.; Hampel, H.; Simonsen, A.H.; Kapaki, E.; Kruse, N.; Le Bastard, N.; Lehmann, S.; et al. Recommendations to standardize preanalytical confounding factors in Alzheimer’s and Parkinson’s disease cerebrospinal fluid biomarkers: An update. Biomark. Med. 2012, 6, 419–430. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Matrix | Slope (1) | Y-Intercept (1) | R2 (1) | LOD (3) (ng/mL) | LOQ (3) (ng/mL) |
|---|---|---|---|---|---|---|
| Caffeine | Plasma | 0.0015 ± 0.0002 | 0.0010 ± 0.0004 | 0.9985 ± 0.0013 | 1.21 | 3.68 |
| Solvent | 0.0016 ± 0.0002 | 0.0007 ± 0.0003 | 0.9983 ± 0.0005 | 0.92 | 2.79 | |
| p-value (2) | 0.631 | 0.293 | ||||
| Theobromine | Plasma | 0.0028 ± 0.0011 | 0.0030 ± 0.0031 | 0.9984 ± 0.0016 | 1.39 | 4.22 |
| Solvent | 0.0030 ± 0.0012 | 0.0006 ± 0.0008 | 0.9990 ± 0.0007 | 1.38 | 4.19 | |
| p-value (2) | 0.816 | 0.13 | ||||
| Theophylline | Plasma | 0.0011 ± 0.0001 | 0.0005 ± 0.0002 | 0.9943 ± 0.0008 | 1.95 | 5.91 |
| Solvent | 0.0010 ± 0.0001 | 0.0005 ± 0.0002 | 0.9957 ± 0.0028 | 1.85 | 5.61 | |
| p-value (2) | 0.260 | 0.791 | ||||
| Paraxanthine | Plasma | 0.00022 ± 0.00004 | 0.00041 ± 0.00038 | 0.9932 ± 0.0088 | 1.32 | 3.99 |
| Solvent | 0.00024 ± 0.00003 | 0.00001 ± 0.00006 | 0.9947 ± 0.0024 | 1.47 | 4.46 | |
| p-value (2) | 0.430 | 0.052 |
| Caffeine (Solvent) | Caffeine (Plasma) | |||
| Quality Control | Precision (CV%) | Accuracy (RE %) | Precision (CV%) | Accuracy (RE %) |
| Low | 3.3 | 16.4 | 3.4 | 17.4 |
| Medium | 2.1 | 0.1 | 2.1 | 3.2 |
| High | 1.6 | −2.6 | 1.6 | 0.7 |
| Theobromine (Solvent) | Theobromine (Plasma) | |||
| Quality Control | Precision (CV%) | Accuracy (RE %) | Precision (CV%) | Accuracy (RE %) |
| Low | 7.3 | 12.8 | 8.4 | 7.5 |
| Medium | 6.6 | −6.6 | 6.8 | −2.2 |
| High | 6.5 | −6.1 | 6.4 | 0.1 |
| Theophylline (Solvent) | Theophylline (Plasma) | |||
| Quality Control | Precision (CV%) | Accuracy (RE %) | Precision (CV%) | Accuracy (RE %) |
| Low | 3.5 | 21.3 | 3.3 | 11.4 |
| Medium | 3.0 | 7.1 | 3.0 | −2.0 |
| High | 3.4 | 2.3 | 3.4 | −6.3 |
| Paraxanthine (Solvent) | Paraxanthine (Plasma) | |||
| Quality Control | Precision (CV%) | Accuracy (RE %) | Precision (CV%) | Accuracy (RE %) |
| Low | 4.8 | 18.1 | 7.3 | −8.1 |
| Medium | 5.5 | −4.1 | 5.9 | −1.2 |
| High | 8.0 | −7.8 | 8.0 | 0.4 |
| Molecule | Parameter | Low | Medium | High | |||
| Mean | %CV | Mean | %CV | Mean | %CV | ||
| Caffeine | EE (%) | 72.9 | 9.0 | 79.1 | 1.7 | 78.9 | 5.5 |
| ME (%) | 112.3 | 14.8 | 99.1 | 7.1 | 101.4 | 1.3 | |
| Theobromine | EE (%) | 84.4 | 14.5 | 88.7 | 4.6 | 91.7 | 1.6 |
| ME (%) | 116.4 | 21.4 | 94.1 | 8.9 | 93.8 | 5.7 | |
| Theophylline | EE (%) | 84.9 | 18.3 | 86.3 | 1.7 | 90.3 | 2.1 |
| ME (%) | 103.4 | 16.0 | 98.0 | 4.8 | 94.2 | 8.6 | |
| Paraxanthine | EE (%) | 85.6 | 2.6 | 98.2 | 14.6 | 92.5 | 16.0 |
| ME (%) | 117.9 | 27.1 | 93.9 | 7.9 | 101.3 | 16.6 | |
| Mean | %CV | ||||||
| 13C3-Caffeine | EE (%) | 77.7 | 10.1 | ||||
| ME (%) | 107.4 | 10.3 | |||||
| Theobromine-d6 | EE (%) | 89.1 | 15.6 | ||||
| ME (%) | 104.1 | 14.6 | |||||
| Consumption (mg/day) | Absolute Quantification (ng/mL) | |||||
|---|---|---|---|---|---|---|
| Sample Code | Caffeine | Theobromine | Caffeine | Theobromine | Theophylline | Paraxanthine |
| 1 | 0.0 | 0.0 | <LOQ | <LOQ | ND | 7.6 |
| 2 | 0.6 | 5.0 | <LOQ | <LOQ | ND | <LOQ |
| 3 | 27.1 | 24.6 | 13.0 | 26.3 | <LOQ | 123.0 |
| 4 | 51.7 | 63.8 | 19.3 | 125.8 | 10.8 | 180.0 |
| 5 | 52.4 | 5.0 | 9.6 | 47.8 | <LOQ | 112.1 |
| 6 | 53.2 | 75.0 | <LOQ | 142.0 | ND | 11.2 |
| 7 | 57.9 | 0.0 | 190.0 | 378.8 | 53.2 | 1014.8 |
| 8 | 100.0 | 0.0 | 9.7 | 93.2 | < LOQ | 23.0 |
| Name | Q1 | Q3 | RT (min) | CE | CXP | DP |
|---|---|---|---|---|---|---|
| Theophylline & Paraxanthine | 181.1 | 124.1 | 4.7 | 27 | 10 | 90 |
| Theobromine | 181.1 | 138.1 | 3.8 | 25 | 10 | 66 |
| Theobromine-d6 | 187.3 | 144.2 | 3.8 | 25 | 10 | 91 |
| Caffeine | 195.2 | 138.0 | 6.2 | 27 | 8 | 71 |
| 13C3-Caffeine | 198.2 | 140.2 | 6.2 | 27 | 10 | 61 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendes, V.M.; Coelho, M.; Tomé, A.R.; Cunha, R.A.; Manadas, B. Validation of an LC-MS/MS Method for the Quantification of Caffeine and Theobromine Using Non-Matched Matrix Calibration Curve. Molecules 2019, 24, 2863. https://doi.org/10.3390/molecules24162863
Mendes VM, Coelho M, Tomé AR, Cunha RA, Manadas B. Validation of an LC-MS/MS Method for the Quantification of Caffeine and Theobromine Using Non-Matched Matrix Calibration Curve. Molecules. 2019; 24(16):2863. https://doi.org/10.3390/molecules24162863
Chicago/Turabian StyleMendes, Vera M., Margarida Coelho, Angelo R. Tomé, Rodrigo A. Cunha, and Bruno Manadas. 2019. "Validation of an LC-MS/MS Method for the Quantification of Caffeine and Theobromine Using Non-Matched Matrix Calibration Curve" Molecules 24, no. 16: 2863. https://doi.org/10.3390/molecules24162863
APA StyleMendes, V. M., Coelho, M., Tomé, A. R., Cunha, R. A., & Manadas, B. (2019). Validation of an LC-MS/MS Method for the Quantification of Caffeine and Theobromine Using Non-Matched Matrix Calibration Curve. Molecules, 24(16), 2863. https://doi.org/10.3390/molecules24162863