1. Introduction
Prostate cancer (PCa), other than skin cancer, is the most common cancer and is the second leading cause of cancer deaths, behind lung cancer, among men in the United States [
1]. The American Cancer Society estimates about 174,650 new cases of prostate cancer and about 31,620 deaths from this disease in 2019. It is also estimated that about 1 in 9 men will be diagnosed with prostate cancer during their lifetime. Therefore, early detection of primary disease and its metastases is critical for clinical staging, prognosis, and therapy management.
Currently, prostate cancer is diagnosed in early stages by elevated serum prostate-specific antigen (PSA) levels, by clinical symptoms, and by imaging techniques such as magnetic resonance imaging (MRI), computed tomography (CT) and transrectal ultrasound, and finally confirmed by prostate biopsy. However, PSA levels can be elevated in benign conditions, and cancer lesions may be missed by imaging. Since the prostate cancer metastasizes to bones, single-photon emission computed tomography (SPECT) bone scintigraphy using
99mTc-labeled methylene diphosphate (
99mTc-MDP) has been used for the past several decades [
2]. However, this technique is limited because of its low specificity and often requires correlation of the results with other supplemental modalities.
Recently, positron emission tomography (PET) using imaging pharmaceuticals and in combination with other modalities has been found useful in the detection of biochemical relapse, recurrence, and extent of metastatic disease. Several imaging pharmaceuticals have been proposed for molecular imaging of prostate cancer, including
18F-NaF (
18F-sodium fluoride) for bone imaging due to prostate cancer metastasis, choline (
11C-Choline and
18F-Choline) as a marker of membrane cell proliferation, incorporation of
11C-Acetate into intracellular phosphatidylcholine membrane, and
18F-FACBC (
18F-fluciclovine; 1-amino-3-fluorocyclo-butane-1-carboxylic acid) to monitor amino acid transport [
3,
4,
5]. Numerous studies, however, have been reported suggesting limited sensitivity and specificity of these imaging pharmaceuticals in detecting prostate cancer in patients with low PSA levels [
6].
The need for receptor-targeted imaging pharmaceuticals has led to the discovery and development of numerous radiolabeled peptides and proteins that can target receptors that are known to overexpress on certain tumors [
7,
8,
9]. Two such receptors are identified in prostate cancer cells: prostate-specific membrane antigen (PSMA) and the gastrin-releasing peptide receptor (GRPR).
PSMA is a transmembrane protein that has significantly elevated expression in prostate cancer cells than in the benign prostatic tissues. Several PSMA-targeted PET imaging pharmaceuticals have been developed and evaluated in the past in preclinical and clinical environments [
10,
11,
12,
13]. This includes
68Ga- and
18F-AlF-labeled PSMA-11, PSMA-617, PSMA I&T, THP-PSMA,
18F-labeled DCFBC, DCFPyL, PSMA-1007,
99mTc-labeled MIP-1404,
123I- and
124I-labeled MIP-1095,
18F-AlF-labeled NOTA-DUPA-Pep, and other HBED analogs.
GRPR, a subtype of the bombesin receptor family, is an attractive target for imaging tumors with neuroendocrine origin including prostate, breast, and small cell lung cancers. For prostate cancer, especially, high-affinity GRPR expression has been identified in tissue biopsy samples and immortalized cell lines [
14,
15]. Bombesin (BBN) is a 14 amino acid amphibian peptide analog of the 27 amino acid mammalian GRP. BBN and GRP share a homologous seven amino acid amidated C-terminus, Trp-Ala-Val-Gly-His-Leu-Met-NH
2, which is necessary for binding as an agonist to the GRPR [
16]. Synthetic BBNs are modified versions of the above peptide sharing the common seven amino acid C-terminus. The N-terminus is free for conjugation with appropriate radiolabeled metal chelate for various applications.
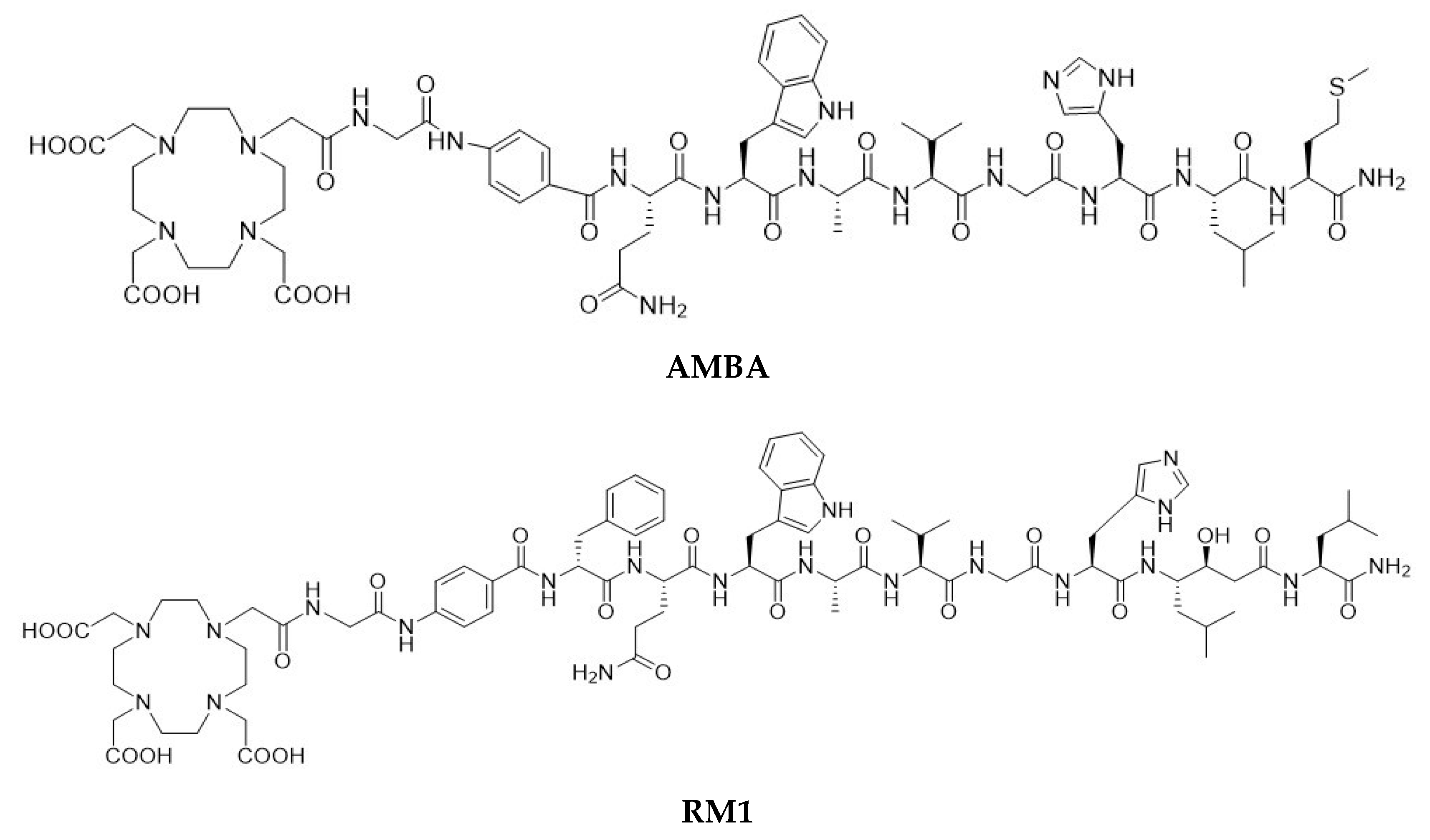
The first proof-of-concept GRPR targeting study was conducted using a
99mTc-labeled BBN (7–14) conjugate (RP527) for breast and prostate cancer imaging [
17]. Among various other BBN conjugates [
18,
19,
20,
21,
22], a BBN-like agonist peptide—AMBA (where AMBA = DO3A-CH
2CO-G[4-aminiobenzyl]-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH
2,
Figure 1)—labeled with
177Lu showed high affinity towards the gastrin-releasing peptide (GRP) receptors in a PC-3 cell line (measured IC
50 values were 4.75 ± 0.25 and 2.5 ± 0.5 nM for AMBA and Lu(AMBA), respectively [
23,
24]). Despite the general consensus related to the high tumor cell internalization rate of agonists for optimal imaging and in vivo therapy [
25], it has been recently shown that high-affinity somatostatin antagonists (both sst
2- and sst
3-selective analogs), despite their poor tumor cell internalization properties, can show clearer images and pharmacokinetic profiles [
26]. These observations led to a change in focus towards the discovery and development of GRPR antagonists for cancer imaging and therapy [
27].
Several classes of BBN antagonists have been designed by modifying the C-terminal residues of naturally amidated BBN agonists [
28,
29,
30,
31]. A series of potent radiolabeled antagonists,
111In- and
68Ga- labeled RM1 (where RM1 = DO3A-CH
2CO-G-[4-aminobenzyl]- D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH
2, where Sta is a statyl residue
Figure 1) and
111In-labeled RM2 peptides, showed higher tumor uptake and better pharmacokinetics than the corresponding agonists [
32,
33]. Additionally,
68Ga-,
111In-, and
18F-AlF-labeled NOTA-PEG
2-RM26 (where RM26 = D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH
2) conjugates demonstrated favorable properties for cell binding and internalization (IC
50 = 1.24 ± 0.29 nM for
111In- and 0.91 ± 0.19 nM for
68Ga-labeled NOTA- PEG
2-RM26), specific tumor uptake (8.1 ± 0.4%ID/g for
68Ga- and 5.7 ± 0.3%ID/g for
111In-labeled NOTA- PEG
2-RM26), and imaging of GRPR-expression in PC-3 tumor-bearing mice [
34,
35]. A
68Ga-labeled GRPR antagonist (NOTA-PEG
3-RM26) was also shown to be a more promising candidate than a
68Ga-labeled agonist (NOTA-Aca-BBN
7-14) for imaging of prostate cancer [
36].
The GRPR targeting imaging pharmaceutical, RM1, is an attractive theranostic choice as the DO3A-CH
2CO- moiety forms stable chelates with trivalent metal ions,
68Ga,
111In, and
177Lu. For characterization, development, and translation of a potential imaging pharmaceutical into the clinic, it must be evaluated in a series of tests, including in vitro cell binding assays, buffer and serum stability studies, the biodistribution of the radiolabeled material, and finally imaging studies in preclinical animal models [
37]. In vitro cell binding, biodistribution, and imaging studies in preclinical models were reported for the two potential imaging pharmaceuticals (i.e.,
177Lu- and
68Ga-labeled AMBA [
23,
24,
38] and
111In- and
68Ga-labeled RM1 [
32]). Additionally, the feasibility and safety of
68Ga-labeled RM1 were tested in humans [
39]. Interestingly, there are no significant changes in the biological and stability properties of the agonist or antagonist by changing the metallic radionuclide in the imaging pharmaceutical. The in vitro mouse and human sera stability of
177Lu-labeled AMBA were reported previously [
23]; however, there are no data available related to mouse, canine, and human sera stability of
177Lu-labeled RM1 and canine serum stability of
177Lu-labeled AMBA. Determination of canine serum stability of
177Lu-labeled RM1 is important from the perspective that we have recently developed a canine prostate tumor model for evaluation of the GRPR receptor-specific peptides [
40] for cancer imaging and peptide receptor radiotherapy (PRRT).
The objectives of the present work were to study (1) in vitro stability of 177Lu-labeled RM1 (Antagonist) and AMBA (agonist) in acetate buffer with low (0.037–0.056 GBq/mL) radioconcentrations stored at room temperature and at 2–8 °C, (2) in vitro stability of 177Lu-labeled RM1 with high (~0.74 GBq/mL) radioconcentrations under 2–8 °C storage conditions (3) the effect of stabilizers, ascorbic acid, and gentisic acid, on the in vitro stability of high radioconcentration, acetate buffered, 177Lu-labeled RM1 samples stored at 2–8 °C, (4) in vitro stability of 177Lu-labeled RM1 and AMBA in mouse, canine, and human sera and canine serum, respectively , and (5 to compare the in vitro stability of 177Lu-labeled RM1 and AMBA in acetate buffer and mouse, canine, and human sera.
For this purpose, 177Lu radionuclide (with 6.7 d t1/2 and 113 keV gamma peak) than 68Ga (with 68 min half-life) was used as a radiotracer of choice to monitor the degradation of 177Lu-labeled AMBA and RM1 in acetate buffer, mouse, canine, and human sera. Because of the longer half-life of 177Lu, a single-labeled AMBA and RM1 could be used for multiple experiments leading to less variation in the data due to batch-to-batch variability of the radiolabeled materials.
We report herein our results related to (1) in vitro stability of low- radioconcentration samples of 177Lu-labeled RM1 and AMBA upon storage at room temperature and 2–8 °C and high-radioconcentration samples of 177Lu-labeled RM1 upon storage at 2–8 °C, (2) effect of stabilizers, ascorbic acid, and gentisic acid on the stability of high-radioconcentration samples of 177Lu-labeled RM1, (3) the comparison of half-lives of degradation of 177Lu-labeled AMBA and RM1, in acetate buffer, and (4) comparison of degradation kinetics of 177Lu-labeled RM1 antagonist and AMBA agonist in mouse, canine, and human sera.
2. Results and Discussion
2.1. Characterization of RM1, AMBA, Lu-RM1, and Lu-AMBA
The HPLC analysis of RM1 and AMBA and their lutetium chelates demonstrated >94% purity (
Figures S1–S4). The MALDI-TOF mass spectral analysis (
Figures S5–S8) gave m/e peaks for m + H of the compound (calculated m/e for molecular mass in the parenthesis) as follows: 1675.71 (1674.66) RM1; 1502.73 (1501.71) AMBA; 1847.67 (1846.77) Lu-RM1; and 1674.56 (1673.68) Lu-AMBA. Based on the retention times under similar HPLC conditions (
Figures S3 and S4), it can be concluded that the Lu-RM1 was slightly more lipophilic than Lu-AMBA, which was in good agreement with the log D measurements. The log D values (average of n = 3 with standard deviation in the parenthesis) determined were −2.46 (0.07) and −2.57 (0.04) for Lu-RM1 and Lu-AMBA, respectively.
2.2. Characterization of 177Lu-Labeled RM1 and AMBA
177Lu-labeled AMBA and RM1 were analyzed for radioactivity concentration and radiochemical purity (RCP) by using a dose calibrator and a reversed-phase high-performance liquid chromatography (RP-HPLC) method, respectively. The retention times (min), the radio concentration (GBq/mL), the molar activity (GBq/mmol), and the radiochemical purity (%RCP) were:
177Lu-labeled AMBA 13.9–14.0 min, 0.034 GBq/mL, 617 GBq/mmol, >99% and
177Lu-labeled RM1 14.9–15.2 min, 0.034 GBq/mL, 699.3 GBq/mmol, >99%, respectively. The radiochemical yield of
177Lu labeling of RM1 or AMBA was >99% as confirmed by the RCP data.
Figures S9 and S10 show chromatograms of Lu/
177Lu-labeled RM1 and AMBA, respectively.
2.3. Stability of Low-Radioconcentration Samples of 177Lu-Labeled RM1 and AMBA in Acetate Buffer
The final pH of the 177Lu-labeled RM1 and AMBA, post radiolabeling, was usually ~5 to 5.5, and the acetate buffer concentration was 0.1 M. Consequently, the degradation kinetics of 177Lu-labeled RM1 and AMBA were evaluated in this environment after storage at room temperature (25 °C) and 2–8 °C for 5 d. Both samples were analyzed using an RP-HPLC method at various time points.
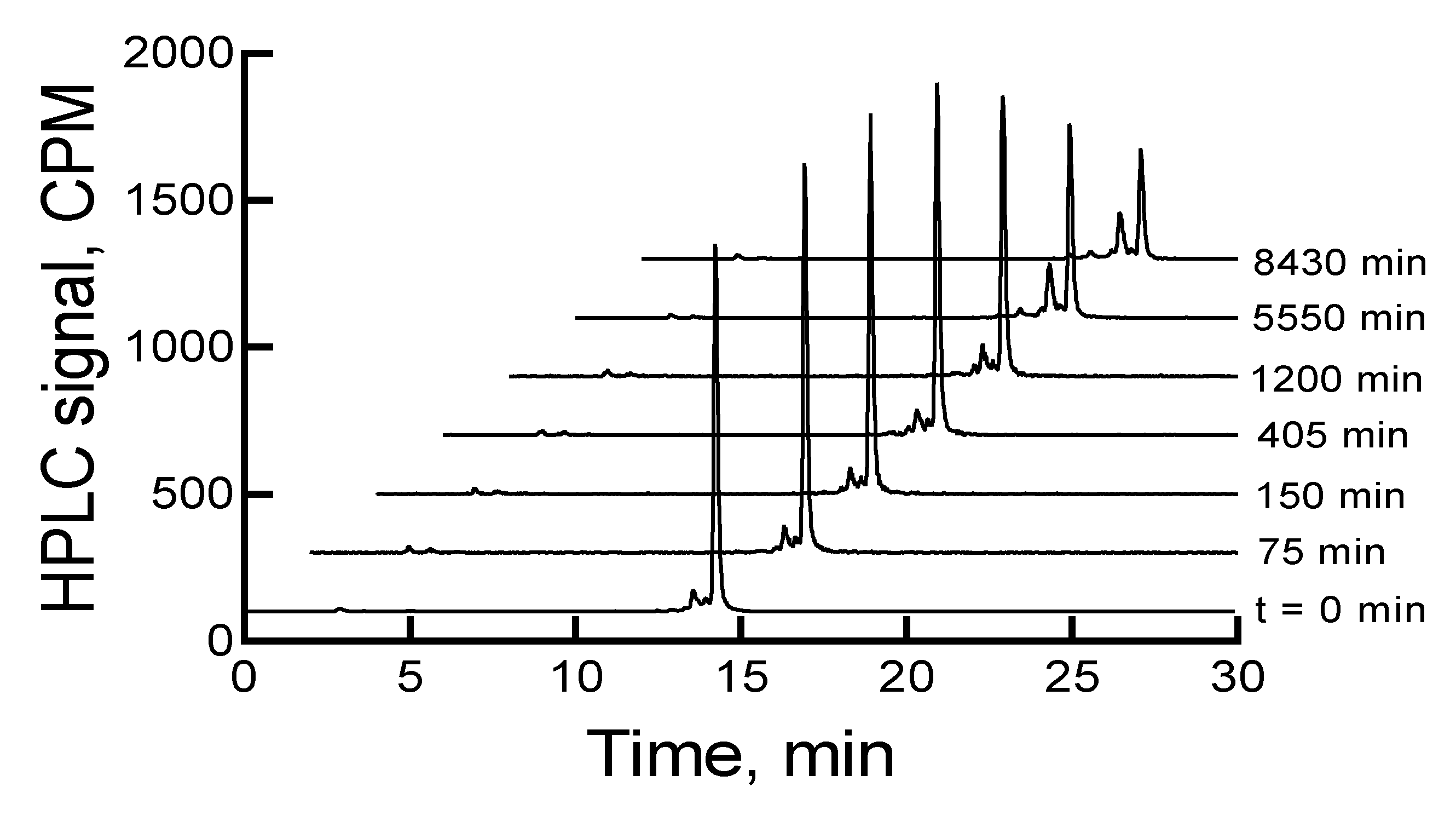
The HPLC analysis of
177Lu-labeled RM1, immediately after completion of the radiolabeling experiment, showed a major peak at ~15 min (~88.3%) in the HPLC chromatogram. There were three other minor peaks (percentages are given in the parenthesis) at 2.9 (1.0), 14.3 (~7), and 14.7 (3.6) min. When the samples were left at room temperature or 2–8 °C storage conditions, the main peak related to
177Lu-labeled RM1 decreased, and the 14.3 min peak increased. In almost 6 d, the main peaks decreased from 88.3% to 49% and 52%, and the 14.3 min peaks increased to 30% and 25% under room temperature and 2–8 °C storage conditions, respectively.
Figure 2 shows HPLC chromatograms of a
177Lu-labeled RM1 as a function of time for a sample stored at 2–8 °C. There were some other minor peaks (~1% to 4%) that eluted at lower retention times. Similar degradation patterns were observed for
177Lu-labeled AMBA; however, the AMBA agonist degraded at a relatively faster rate (i.e., 70% and 60% degradation upon storage at room temperature and 2–8 °C for 6 d, respectively). Additionally, the major degradant of
177Lu-labeled AMBA eluted at an earlier time than the major degradant of
177Lu-labeled RM1, suggesting that the AMBA degradant was probably more hydrophilic than the degradant of RM1.
The percent remaining of
177Lu-labeled RM1 was fitted into a first-order kinetics model (
Figure 3). The average (with standard deviation in the parenthesis for n = 3) t
1/2 (half-life) values for
177Lu-labeled RM1 and AMBA were calculated as 164.3 (3.6) and 195.6 (3.6) h (~7 and ~8 d) (
Table 1) and 87 (3.9) and 127.7 (2.1) h (~4 and ~5 d) at room temperature and 2–8 °C storage conditions, respectively. In summary, the
177Lu-labeled RM1 (antagonist) was found to degrade slower than the
177Lu-labeled AMBA (agonist) in both storage conditions.
2.4. Stability Evaluation and Stabilization of High-Radioconcentration 177Lu-Labeled RM1 Samples by Ascorbic Acid and Gentisic Acid
The 177Lu-labeled RM1 samples in acetate buffer, with 0.74 GBq/mL radio concentration, 10,175–12,765 GBq/mmol molar activity, and >99% RCP, were prepared for high-radioconcentration sample stability studies. The samples, when stored at 2–8 °C, degraded significantly faster than the samples with low radioconcentrations due to the radiolytic and oxidative degradation of the peptide moiety by a high level of radioactivity. For example, complete degradation of 177Lu-labeled RM1 was seen after ~3 d with several early eluting peaks in the HPLC, with a major peak being at 12.8 min with 46% peak area. From a time-dependent study, an average (with standard deviation in the parenthesis for n = 3) half-life of degradation of 177Lu-labeled RM1 was calculated as ~8.5 (0.2) h. Degradation of the high-radioconcentration sample in acetate buffer was at least 20 times faster than the degradation of the low-radioconcentration samples of 177Lu-labeled RM1.
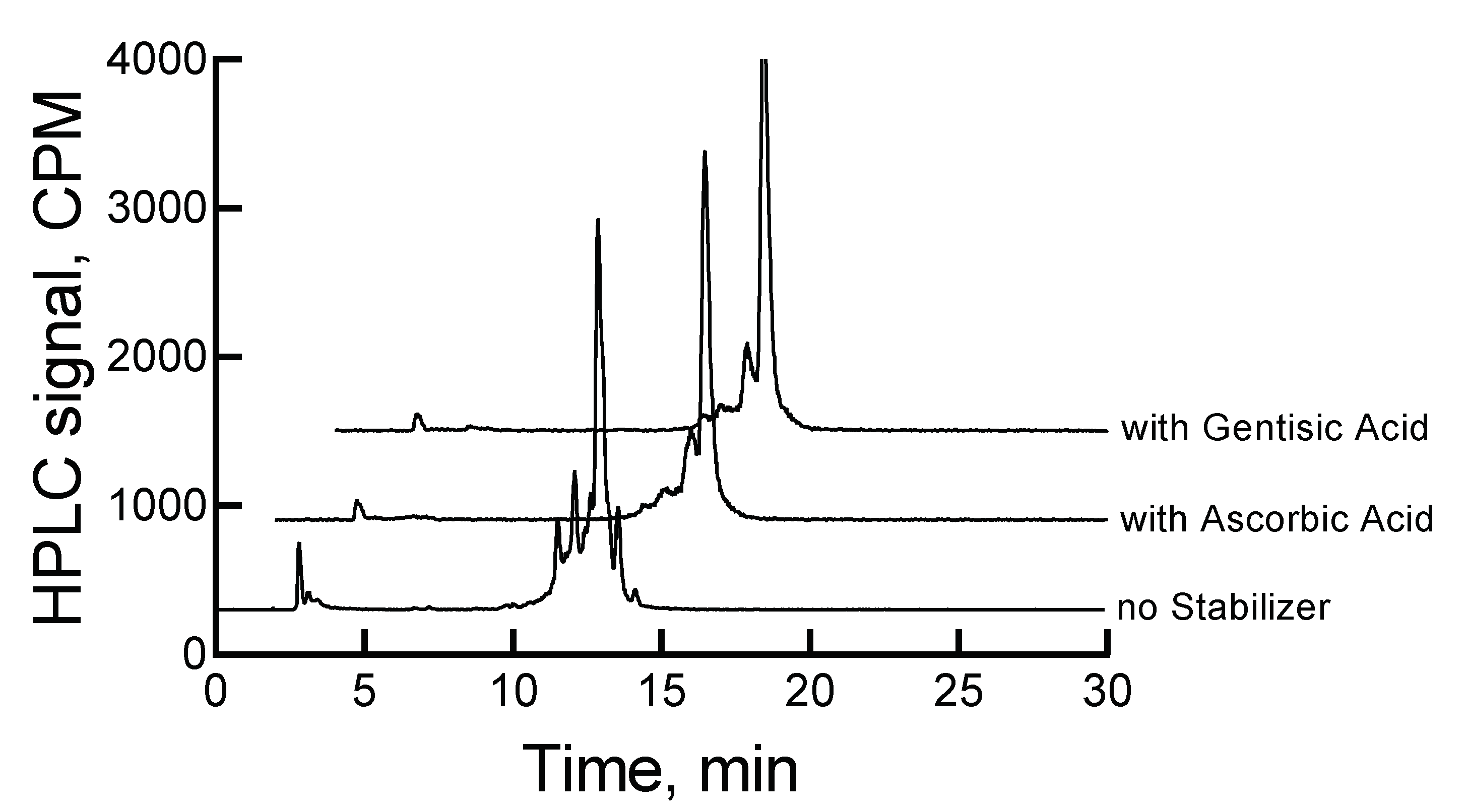
Traditionally, ascorbic acid and gentisic acid are known as radio stabilizers. Consequently, we investigated the effect of these two stabilizers on the stability of 177Lu-labeled RM1. An improvement in the stability of the high-radioconcentration sample of 177Lu-labeled RM1 was observed in the presence of 500 µg ascorbic acid when stored at 2–8 °C. For example, the percentages of 177Lu-labeled RM1 degraded were 9.7%, 21.5%, 34.8%, 87.1%, 89.5%, and 92% at 22, 46, 70, 118, 142, and 166 h, respectively. Several peaks (percentages given in parentheses) appeared at 12.3 (9%), 12.9 (19.9%), and 13.7 (43%) min after 166 h storage at 2–8 °C. Surprisingly, the peak eluting at 2.9 min corresponding to free 177Lu/Lu increased to 12.8% over the period of 7 d, even though the pH of the sample was constant throughout the study. A small amount (2.4%–3.5%) of the possible 177Lu(DO3A-amide) moiety was observed during the later days as well. An average (with standard deviation in the parenthesis for n = 3) half-life of degradation was calculated as 39.2 (0.6) h (~1.6 d) by fitting the percent remained versus time data into a first-order kinetics model. From the comparison of the control experiment, it can be concluded that the degradation of a high-radioconcentration sample of 177Lu-labeled RM1 was reduced at least by a factor of 5 in the presence of ascorbic acid.
A similar amount of gentisic acid (500 µg) was found to be more effective in preventing the radiolytic degradation of
177Lu-labeled RM1. For example, 3.9%, 13.3%, 18.3%, 32.2%, 36%, and 36.8% degradations at 22, 46, 70, 118, 142, and 166 h were observed. After 7 d storage of the mixture of
177Lu-labeled RM1 and gentisic acid, major degradation peaks seen were at 13.7 min (23.2%), 2.9 min (~6%) due to free
177Lu/Lu, and 3.2 min (~2%) due to
177Lu(DO3A-amide).
Figure 4 shows the HPLC chromatograms for
177Lu-labeled RM1 in the absence and presence of stabilizers at 46 h.
An average (with standard deviation in the parenthesis for n = 3) t
1/2 for degradation of
177Lu-labeled RM1 in the presence of 500 µg gentisic acid was calculated as 142.5 (1.4) h (~6 d), which was almost 17-fold improvement of the stability of
177Lu-labeled RM1 than without any stabilizer. In summary, gentisic acid appears to be more effective than ascorbic acid in retarding the degradation kinetics due to radiolysis and stabilizing RM1.
Table 1 provides a summary of the stability of low- and high-radioconcentration samples of
177Lu-labeled RM1 with and without stabilizers.
2.5. Stability of 177Lu-Labeled RM1 and AMBA in Mouse, Canine, and Human Sera
For mouse, canine, and human sera stability studies, 50 µL of 177Lu-labeled RM1 or AMBA was mixed with 450 µL of mouse or canine or human serum. The mixtures were incubated at 37 °C for 24 h, samples were processed at each time point, and analyzed by an RP-HPLC method.
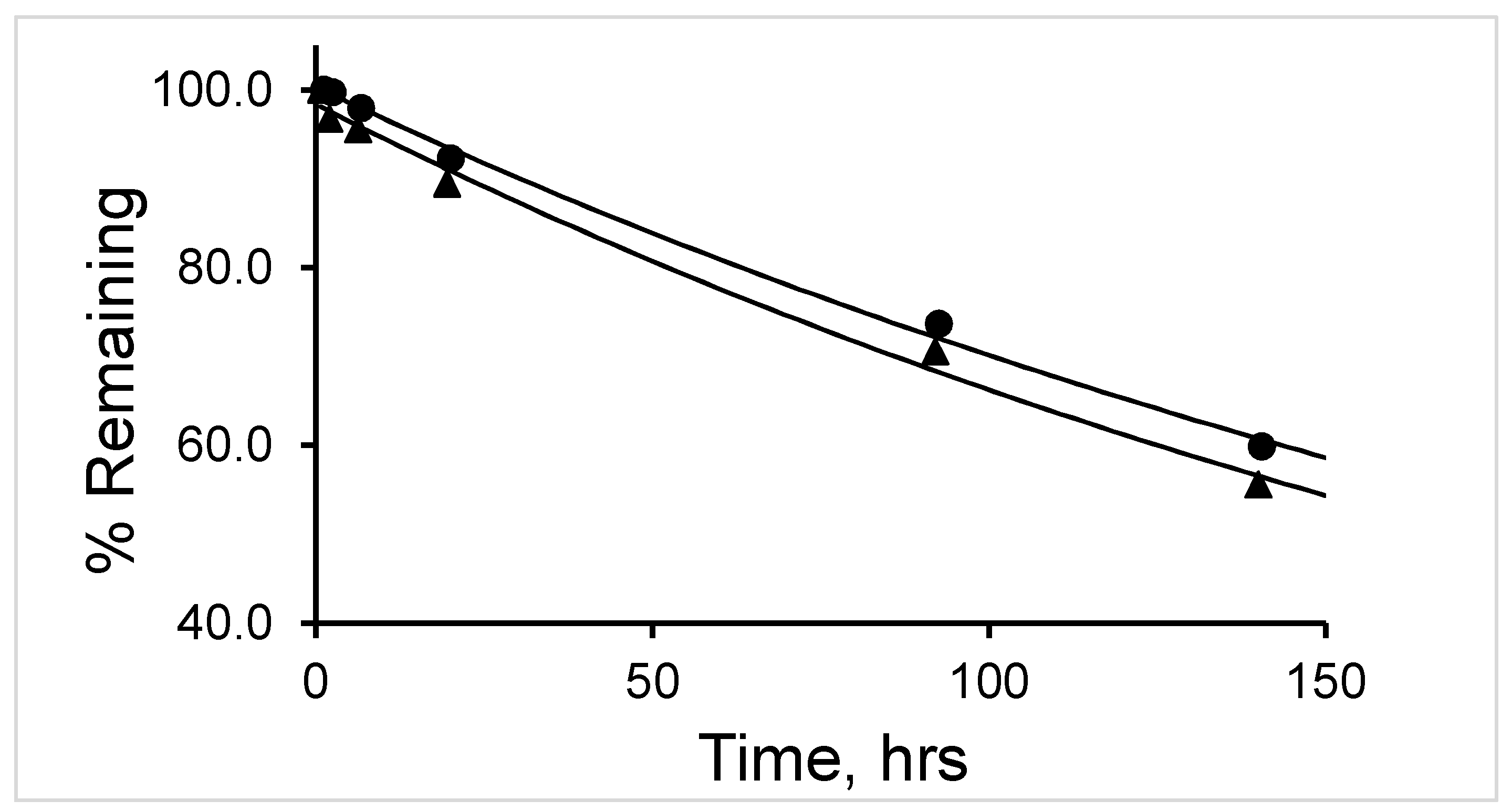
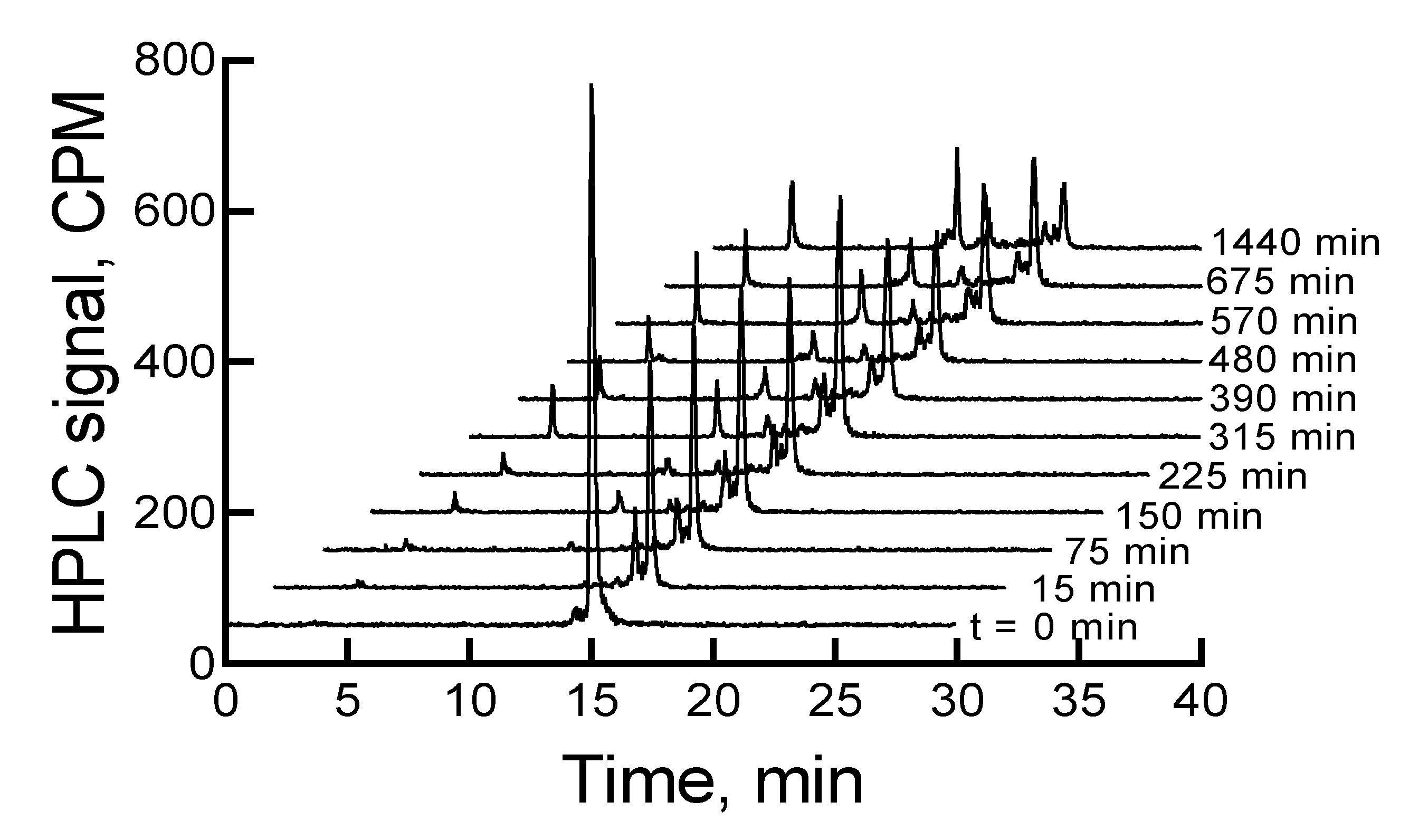
The
177Lu-labeled RM1 degraded in mouse serum (expressed as percent remaining) with time, that is, 91.9, 85.3, 78.7, 73.6, 71.1, 69.6, 61.7, and 59.1 after 75, 150, 225, 315, 390, 480, 570, and 675 min incubation at 37 °C (
Figure 5). On the contrary, only 10% to 12% of
177Lu-labeled RM1 was degraded within the same time period in pH 5 sodium acetate buffer environment when it was stored at room temperature or a 2–8 °C storage condition. In the mouse serum incubated
177Lu-labeled RM1 samples, new peaks appeared at lower retention times of 3.4, 10.1, 11.7, 12.3, and 12.9 min (or 0.22, 0.66, 0.77, 0.81, and 0.85 relative retention times (RRTs)). With the exception of the degradant with 0.77 and 0.85 RRTs, which remained unchanged, the percent of other degradant generally increased, suggesting the formation of less lipophilic metabolites of
177Lu-labeled RM1. After 1440 min of incubation in mouse serum, complete degradation of the
177Lu-labeled RM1 was observed with three major peaks eluting at 3.3, 10.0, and 14.4 min with areas of 17.1%, 29%, and 22%, respectively.
The early eluting peak at 3.4 min most likely is the
177Lu(DO3A-amide) moiety, since we have established that free
177Lu/
177Lu-EDTA elutes at 2.9 min under these HPLC conditions. Furthermore, this is also consistent with our previously published in vitro serum stability of
153Gd-labeled DO3A conjugated peptides [
37].
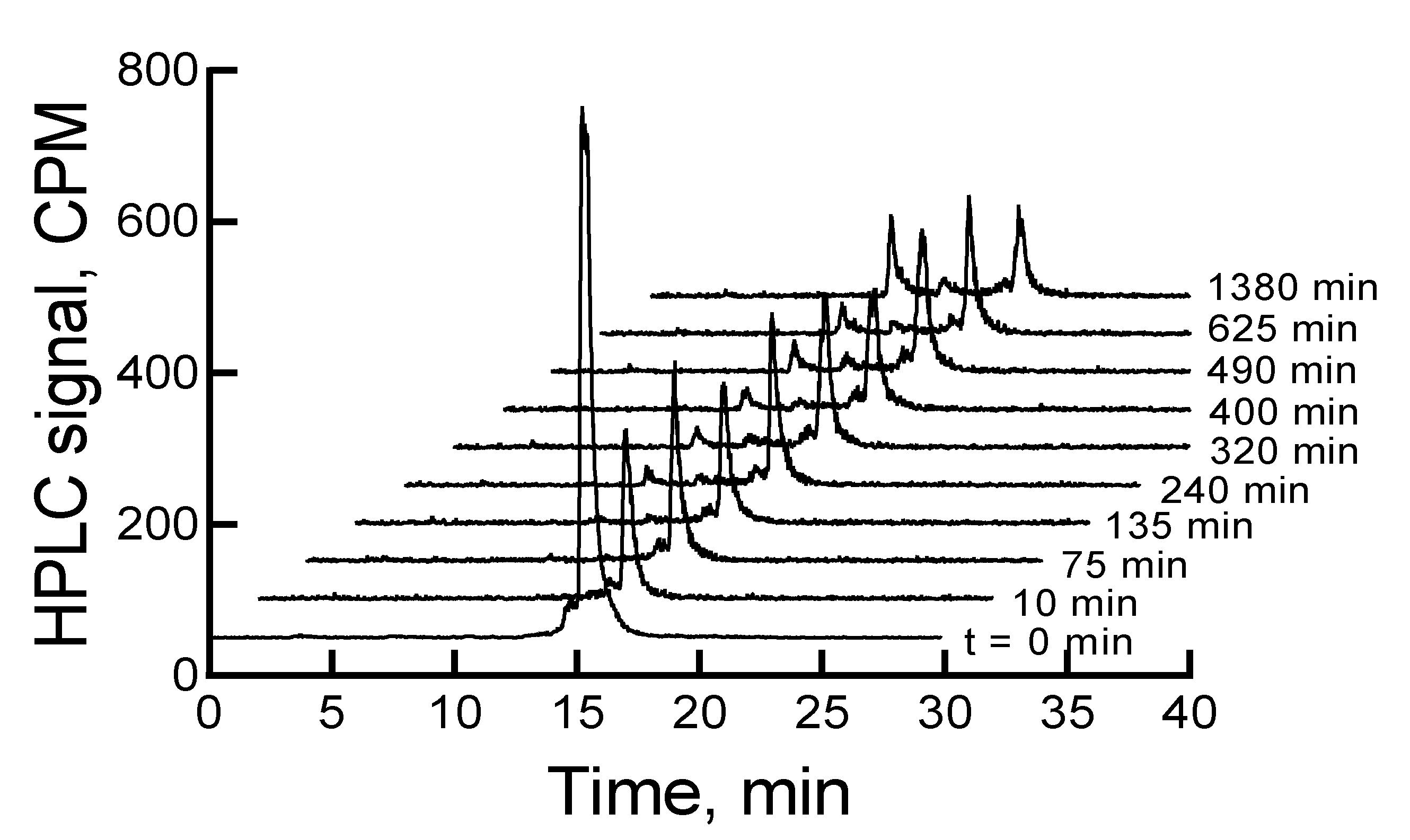
When the mixture of canine serum and
177Lu-labeled RM1 or AMBA was incubated for 75, 135, 240, 320, 400, 490, 625, and 1380 min, the
177Lu-labeled RM1 degraded with time. The percent remaining was calculated from the RP-HPLC analysis of
177Lu-labeled RM1 as 99.98, 94.6, 87.2, 85.3, 82.5, 77.8, 75.0, 53.5 at 75, 135, 240, 320, 400, 490, 625, and 1380 min, respectively (
Figure 6). The degradants in canine serum were similar to the degradants observed in mouse serum eluting at 10.1, 12.3, and 12.9 min. The
177Lu-labeled AMBA degraded in canine serum faster than
177Lu-labeled RM1. For example, the percent remaining of
177Lu-labeled AMBA were 89.5, 80.7, 75.0, 70.9, 66.5, 62.1, 55, and 18.4 at 75, 135, 240, 320, 400, 490, 625, and 1380 min, respectively. The peaks at lower retention times appeared at 3.2, 6.2, 9.4, 11, and 12.1 min and showed a gradual increase with longer incubation of the samples.
The 177Lu-labeled RM1 and human serum mixture samples were incubated for 24 h, and samples were analyzed using RP-HPLC. Contrary to the mouse and canine sera studies, slow degradation of 177Lu-labeled RM1 (<10%) was observed in human serum in 24 h. However, the metabolites eluting at 3.4, 10.1, and 12.2 min (although lower percentages) were observed to appear at later time points of incubation. It is interesting to note that the metabolites in the mouse, canine, and human sera samples had similar properties, suggesting that the degradation of 177Lu-labeled RM1 follows common degradation pathways/steps independent of the source of the serum. Most of the radioactivity was in the form of bound 177Lu either to the peptide fragment conjugated DO3A or DO3A-amide. No significant amount of free 177Lu was detected in the serum stability studies.
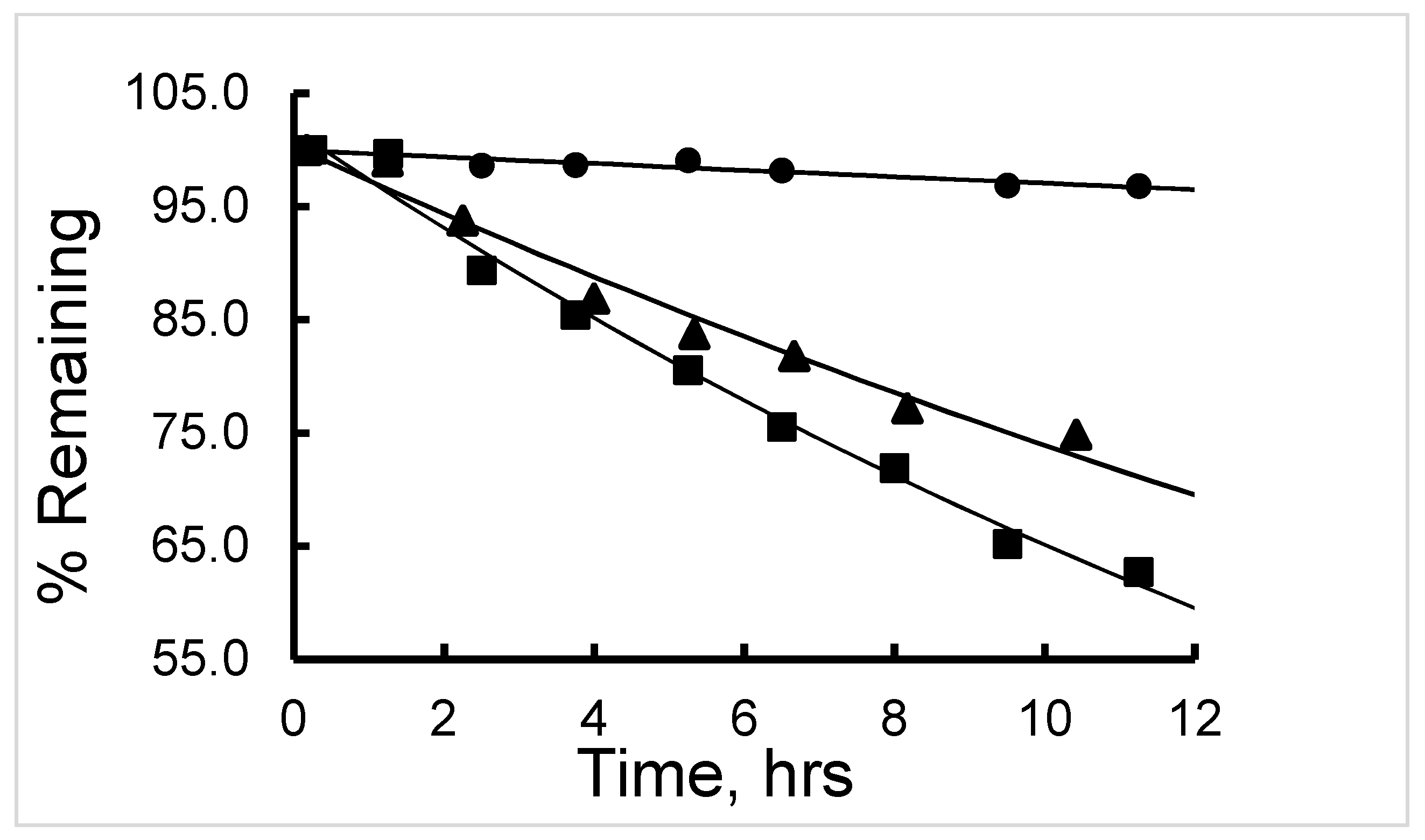
The half-life (t
1/2) of degradation of
177Lu-labeled RM1 (in mouse, canine, and human sera) and
177Lu-labeled AMBA (in canine serum) were calculated by fitting the percent remaining of
177Lu-labeled peptide versus incubation time data into a first-order kinetics model (
Figure 7). Average (with standard deviation in the parenthesis for n = 3) t
1/2 (in h) values for degradation of
177Lu-labeled RM1 were calculated as 16.4 (0.6), 18.9 (0.8), and 141 (5) h (
Table 2) in mouse, canine and human sera, respectively. An average (with standard deviation in the parenthesis for n = 3) t
1/2 (in h) for degradation of
177Lu-labeled AMBA in canine serum was determined as 10.1 (0.5) h in the present work, and half-lives of degradation in mouse and human sera were reported as 3.1 and 38.8 h, respectively [
23,
24]. In summary, (1) the half-life of degradation (stability) of
177Lu-labeled RM1 and AMBA followed the order: human > canine > ~mouse, and more interestingly, (2) in general, the antagonist,
177Lu-labeled RM1, was slower to degrade than the agonist,
177Lu-labeled AMBA, in mouse, canine, and human sera.
Determination of serum stability of a new chemical entity (NCE), such as a peptide or a small molecule containing amide bonds, is an important step in the development of a potential imaging pharmaceutical. Various esterases appear to have the most significant effect on drugs circulating in the serum [
41]. Carboxylesterases that will hydrolyze ester or amide groups in peptides or small molecules have not been detected in human, dog, or monkey plasma. However, rats, rabbits, and other small animals possess this class of esterases, which may be responsible for faster degradation of
177Lu-labeled RM1 and AMBA in mouse serum. Faster degradation of
177Lu-labeled AMBA than RM1 may be due to the presence of methionine amino acid in the former. It was found previously that methionine residue in AMBA is readily oxidized to its biologically inactive methionine sulfoxide (Met(O)), forming
177Lu-AMBA-Met(O) [
42].
3. Materials and Methods
3.1. General
Commercial mouse and human serum from MP Biomedicals (Solon, OH, USA) and canine serum from Immunoreagents, Inc. (Raleigh, NC, USA) were used in the in vitro serum stability studies. Buffers were prepared, and pH was controlled using sodium acetate (Sigma Aldrich, St. Louis, MO, USA), sodium hydroxide, and hydrochloric acid (Fisher Scientific, Fair Lawn, NJ, USA). Acetonitrile and Tris acetate containing ethylenediaminetetraacetic acid (EDTA) (all from Fisher) were used for mobile phase preparations. Ascorbic acid (Sigma Aldrich) and gentisic acid (Acros Organics, Molinons, France, distributed by Fisher Scientific) were used as stabilizers. Octanol (Sigma Aldrich, St. Louis, MO, USA) was used for log D determinations. Absolute ethanol for the precipitation of serum proteins in the degradation kinetics/stability experiments was purchased from Fisher Scientific. A sample of 177lutetium chloride (177LuCl3) in 0.05 M hydrochloric acid (molar activity > 555 GBq/mg Lu, produced by Missouri University Research Reactor, MURR, Columba, MO, USA) was supplied by Oak Ridge, TN, USA for radiolabeling of RM1 and AMBA. A block heater (Labnet International, Edison, NJ, USA) was used for controlling temperature of the reaction of 177LuCl3 with AMBA or RM1 and the maintenance of incubation temperature of 177Lu-labeled AMBA or RM1 and serum mixtures. All pH measurements were made with a combination glass electrode and a model S220 pH/Ion meter (Mettler Toledo, Columbus, OH, USA). Deionized MilliQ water (Millipore Sigma, Burlington, MA, USA) was used for all solution preparations.
3.2. Chemistry
Two GRPR targeting ligands, AMBA (agonist, a well-characterized gift from Bracco Diagnostics, Inc., Monroe Township, NJ, USA) and RM1 (antagonist, synthesized and purified by a modified procedure described elsewhere [
32]) as well as Lu-AMBA (a well-characterized gift from Bracco Diagnostics, Inc.,) and Lu-RM1 (obtained from in-situ synthesis) were analyzed by RP-HPLC and characterized by MALDI/TOF mass spectrometry. For the in situ synthesis of Lu-RM1, 1 mg (0.597 μmol) of an HPLC purified RM1 was dissolved in 1 mL 0.1 M sodium acetate buffer (pH of 4). The peptide solution was then mixed with a slight excess of lutetium acetate in 0.05 M HCl. The reaction mixture was heated at 95 °C in a heating block for 20–30 min. The pH of the reaction mixture was then slowly raised to ~ 5.5 (checked with a pH paper) using 0.1 M sodium acetate solution (pH of 7) with intermittent heating at 95 °C for 10–15 min. The final volume of the reaction mixture was made up to ~1 mL.
3.3. Radiochemistry
The 177Lu-labeled AMBA and RM1 were prepared by a standard procedure developed in our laboratories. In a typical experiment, a stock sample solution (1 mg/mL) of an HPLC purified AMBA or RM1 was prepared by dissolving an appropriate amount of the peptide chelating agent conjugate in 0.1 M sodium acetate buffer (pH of 4). A sample of a desired amount of radioactivity (177LuCl3) in 0.05 M HCl was then mixed with an excess (typically 10%–20%) of AMBA or RM1 (50 to 125 µg) in the acetate buffer medium. The reaction mixture was heated at 95 °C in the heating block for 20–30 min. The pH of the reaction mixture was then slowly raised to ~5.0 to 5.5 (checked with a pH paper) using 0.1 M sodium acetate solution with intermittent heating at 95 °C for 10–15 min.
3.4. Analytics
A dose calibrator (Capintec, Florham Park, NJ, USA) and a Wizard 2480 gamma counter (Perkin Elmer, Waltham, MA, USA) were used for radioactivity measurements in the samples. A model 1100 Agilent HPLC system (Agilent, Wilmington, DE, USA) was used for the analysis of 177Lu-labeled RM1 and AMBA and stability samples. The system consisted of a quaternary pump, degasser, column compartment capable of controlling the temperature at ambient or at 37 °C, auto-injector, a multiwavelength detector, and a Flow Ram radioisotope detector (Lab Logic, Sheffield, UK). The system was controlled by Laura software from Lab Logic. A Waters (Milford, MA, USA) Zorbax Bonus RP C18 column (4.6 × 250 mm, 5 µm) was used for the analysis of 177Lu-labeled AMBA and RM1 samples and for the separation of the 177Lu-labeled AMBA and RM1 from their in vitro metabolic degradants in mouse, human, and canine sera stability samples.
All mass spectral analyses were performed at the Campus Chemical Instrument Center of the Ohio State University (Columbus, OH, USA) using a Bruker UltrafleXtreme MALDI-TOF MS system. An RP-HPLC method, involving a gradient mobile phase system of 50 mM Tris acetate, 10 mM EDTA at pH 7.2 (solvent A), and acetonitrile (solvent B) with 1 mL/min flow rate, was used to monitor the extent of the reaction of 177Lu/Lu with AMBA and RM1; for the purity analysis of RM1, AMBA, Lu-RM1, Lu-AMBA, and of the final 177Lu-labeled samples; and for monitoring the degradation of 177Lu-labeled RM1 or AMBA samples in acetate buffer, mouse, canine, and human sera. The EDTA was incorporated to detect any unreacted 177Lu/Lu in the samples. The gradient program used was 0% B at the beginning, raising the percentage of B to 50% in 15 min, holding B at 50% for 4 min, followed by decreasing the percentage of B to 0% in 5 min and continuing 0% for an additional 9 min. The total run time was 30 min.
The log D values were determined by a traditional shake-flask method. In a typical experiment, 10 μL sample of
177Lu-labeled AMBA or RM1 with 0.037–0.052 GBq/mL radioconcentration and 814 GBq/mmol molar activity were added to a 4 mL mixture of 50:50 octanol:water. The mixture was shaken for one minute and mixed well. The sample was stored at room temperature for 5 min for separation of the two layers. Sample from each layer (25 μL) was added to a counting tube containing 250 μL water. The samples were mixed and counted. Triplicate measurements were made. The log D values were calculated using Equation (1):
3.5. Stability
All samples of 177Lu-labeled RM1 were prepared in 0.1 M acetate buffer, pH 5–5.5. The 177Lu-labeled RM1 samples, with a low radioconcentration, were stored at room temperature and at 2–8 °C storage conditions for 6 d for stability studies. The samples with a high radioconcentration, without and with a stabilizer (gentisic acid or ascorbic acid), were stored 2–8 °C for 2 and 7 d, respectively. For the effect of stabilizer (ascorbic acid or gentisic acid) on the stability of the high-radioconcentration samples of 177Lu-labeled RM1, the samples were mixed with 500 µg of ascorbic or gentisic acid post radiolabeling. The stability or degradation of the samples was monitored by a reduction in the percent HPLC peak area of 177Lu-labeled RM1 with time.
A literature procedure was followed for the serum stability studies of
177Lu-labeled RM1 and AMBA [
37,
43]. On the day of the experiment, a small portion of the frozen mouse, human or canine serum was thawed at room temperature. A stock sample of
177Lu-labeled RM1 or AMBA and the serum were equilibrated at 37 °C in the block heater initially. Several mixtures, containing 50 µL of
177Lu-labeled RM1 or AMBA (usually 1.48 to 2.22 MBq radioactivity) and 450 µL of the mouse or human or canine serum, were prepared in HPLC vials and incubated at 37 °C in the block heater. At a selected time point (typically 15, 75, 150, 225, 315, 390, 480, 570, 675, and 1440 min), a vial was taken out from the heating block, and degradation of the
177Lu-labeled AMBA or RM1 was stopped by adding cold (4 °C) absolute ethanol (500 µL). The sample was then centrifuged at 6000 rpm for 15 min, and 200 µL of the supernatant solution was isolated and analyzed by an RP- HPLC method given above. A control experiment was conducted where samples of
177Lu-labeled AMBA and RM1 were incubated in the water at 37 °C and analyzed by the RP-HPLC method.
Degradation of
177Lu-labeled RM1 or AMBA was monitored by a reduction in percent HPLC peak area with time. Half-lives of in vitro acetate buffer and serum degradation for each imaging pharmaceutical were calculated by fitting the percent peak remaining versus time to a first-order kinetics model (Equations (2) and (3)) [
44] using Excel or Graph Pad Prism version 5.03 software (Graph Pad Software, Inc., La Jolla, CA, USA).
where P
t is the percent peak area at time t, P
o is the percent peak area initially, t is the time elapsed, k is the first-order rate constant, and t
1/2 is the half-life of degradation reaction. All degradation kinetics experiments were conducted in triplicate, and average (n = 3) values (standard deviation given in the parenthesis) of half-life are reported.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}