
Specific Ion Effects of Dodecyl Sulfate Surfactants with Alkali Ions at the Air–Water Interface


Abstract
:1. Introduction
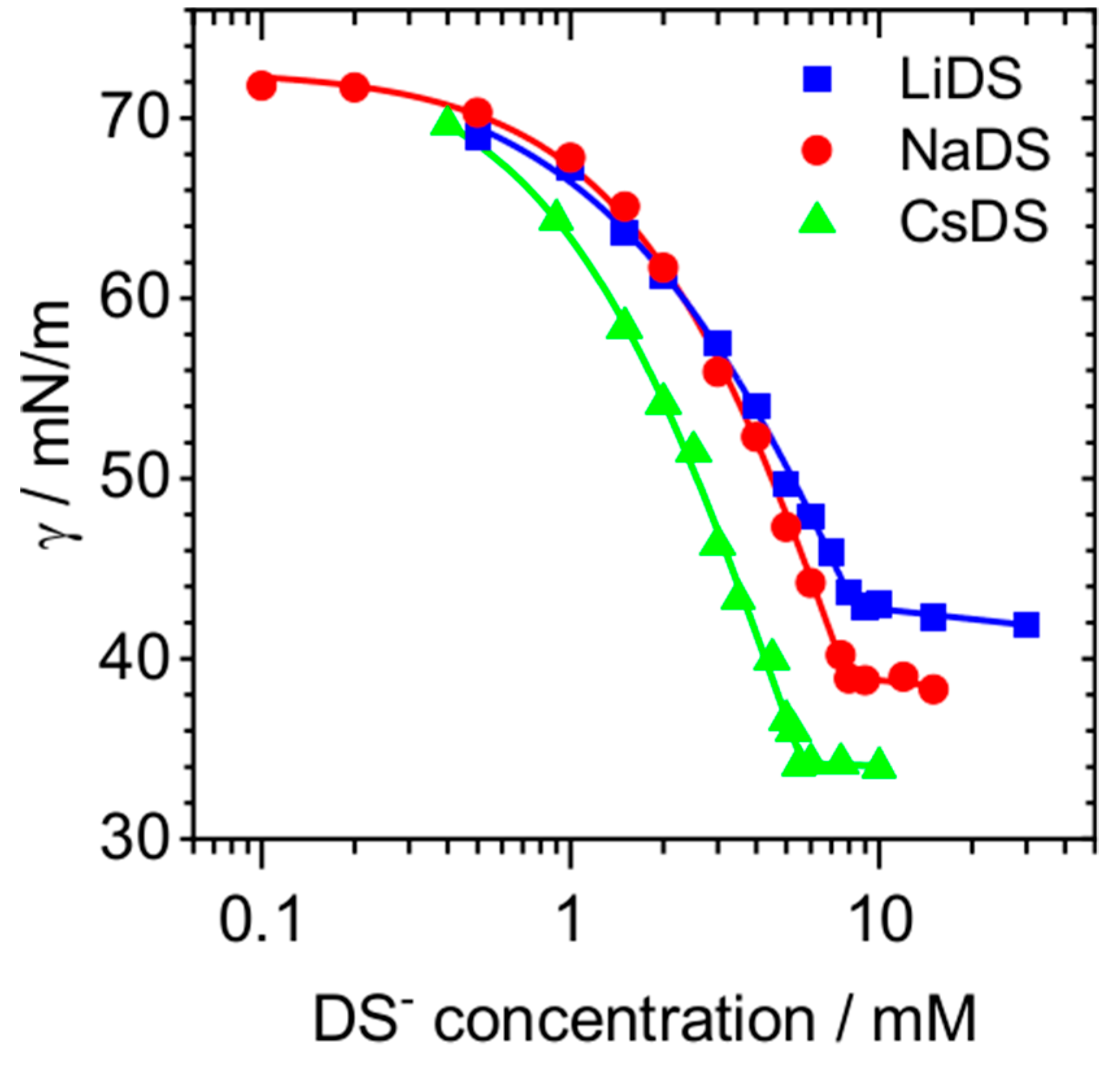
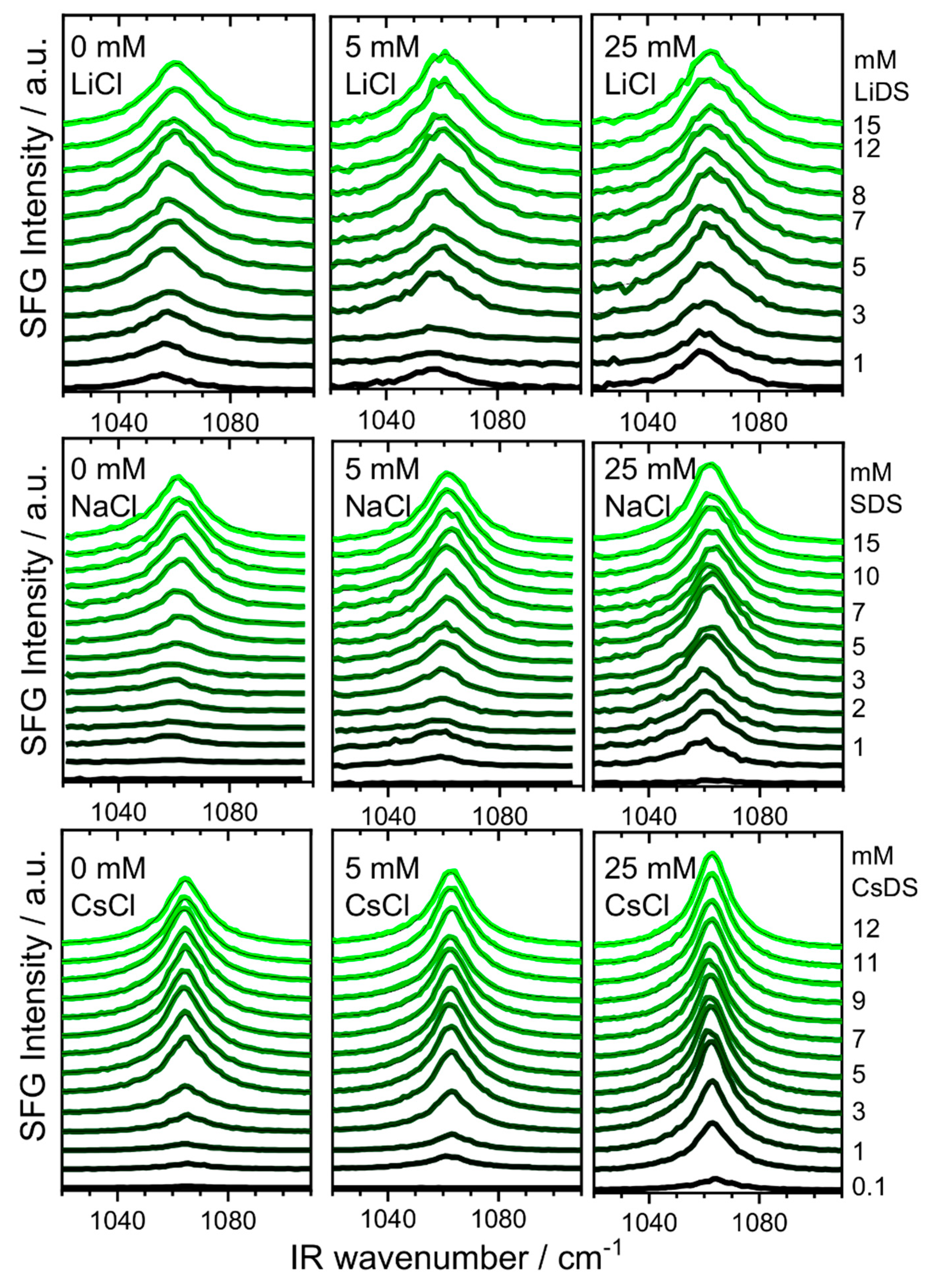
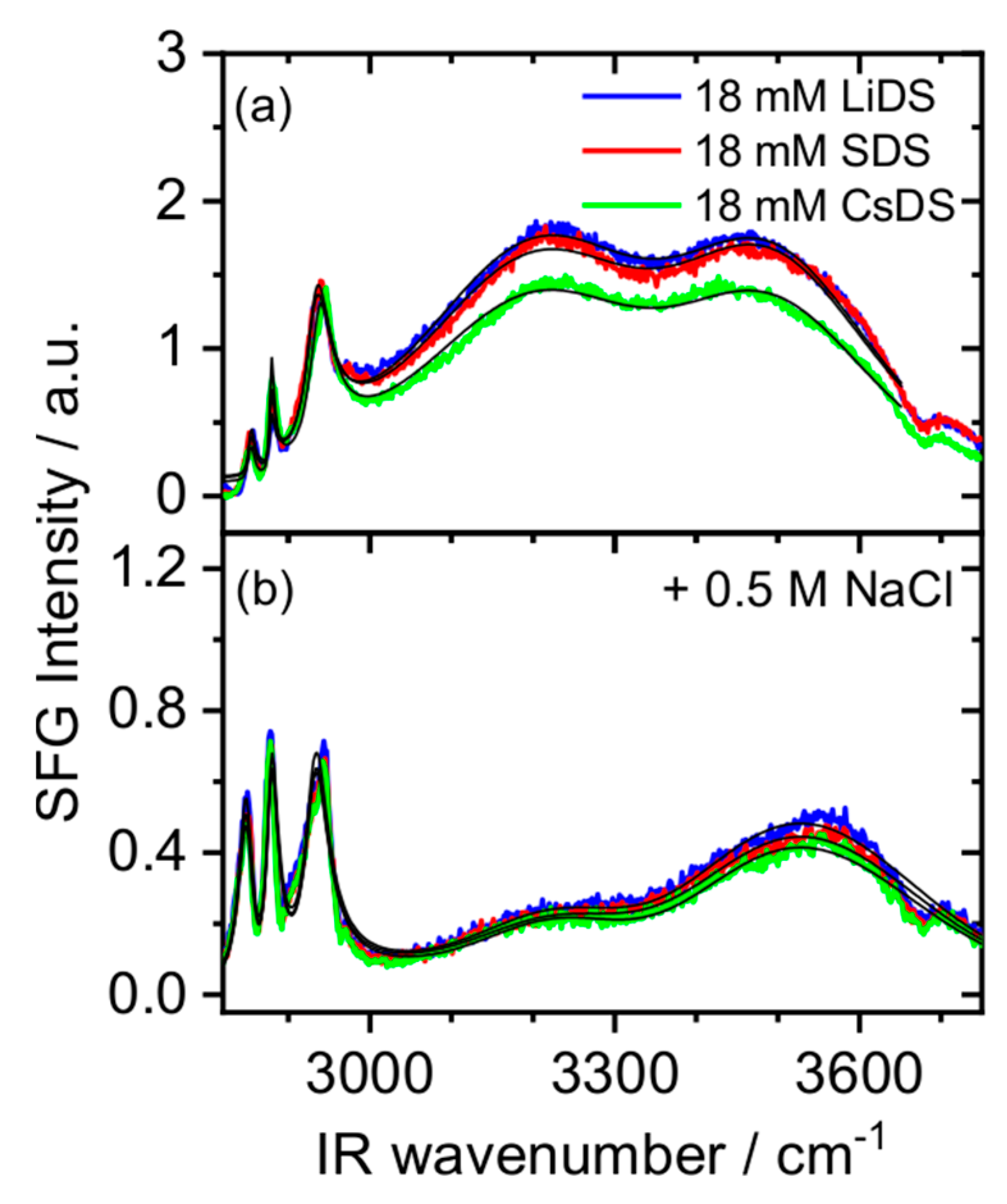
2. Results and Discussion
3. Materials and Methods
3.1. Sample Preparation
3.2. Sum Frequency Generation (SFG)
3.3. Surface Tensiometry
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hofmeister, F. On the understanding of the effects of salts. Arch. Exp. Pathol. Pharmakol. 1888, 24, 247–260. [Google Scholar] [CrossRef]
- Okur, H.I.; Hladílková, J.; Rembert, K.B.; Cho, Y.; Heyda, J.; Dzubiella, J.; Cremer, P.S.; Jungwirth, P. Beyond the Hofmeister Series: Ion-Specific Effects on Proteins and Their Biological Functions. J. Phys. Chem. B 2017, 121, 1997–2014. [Google Scholar] [CrossRef] [PubMed]
- Para, G.; Jarek, E.; Warszynski, P. The surface tension of aqueous solutions of cetyltrimethylammonium cationic surfactants in presence of bromide and chloride counterions. Coll. Surf. A Physicochem. Eng. Asp. 2005, 261, 65–73. [Google Scholar] [CrossRef]
- Para, G.; Jarek, E.; Warszynski, P. The Hofmeister series effect in adsorption of cationic surfactants—Theoretical description and experimental results. Adv. Coll. Int. Sci. 2006, 122, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Warszyński, P.; Lunkenheimer, K.; Czichocki, G. Effect of Counterions on the Adsorption of Ionic Surfactants at Fluid−Fluid Interfaces. Langmuir 2002, 18, 2506–2514. [Google Scholar] [CrossRef]
- Sthoer, A.; Hladílková, J.; Lund, M.; Tyrode, E. Molecular insight into carboxylic acid-alkali metal cations interactions: Reversed affinities and ion-pair formation revealed by non-linear optics and simulations. Phys. Chem. Chem. Phys. 2019, 21, 11329–11344. [Google Scholar] [CrossRef] [PubMed]
- Tyrode, E.; Corkery, R. Charging of Carboxylic Acid Monolayers with Monovalent Ions at Low Ionic Strengths: Molecular Insight Revealed by Vibrational Sum Frequency Spectroscopy. J. Phys. Chem. C 2018, 122, 28775–28786. [Google Scholar] [CrossRef] [Green Version]
- Mukerjee, P.; Mysels, K.; Kapauan, P. Counterion Specificity in the Formation of Ionic Micelles -Size, Hydration, and Hydrophobic Bonding Effects. J. Phys. Chem. 1967, 71, 4166–4175. [Google Scholar] [CrossRef]
- Schelero, N.; von Klitzing, R. Ion specific effects in foam films. Curr. Opin. Colloid Interface Sci. 2015, 20, 124–129. [Google Scholar] [CrossRef]
- Zhao, H. Protein Stabilization and Enzyme Activation in Ionic Liquids: Specific Ion Effects. J. Chem. Technol. Biot. 2016, 91, 25–50. [Google Scholar] [CrossRef]
- Itoh, T. Ionic Liquids as Tool to Improve Enzymatic Organic Synthesis. Chem. Rev. 2017, 117, 10567–10607. [Google Scholar] [CrossRef] [PubMed]
- Khuman, P.; Singh, W.B.K.; Devi, S.D.; Naorem, H. Viscosity-Temperature Behavior of Hydroxypropyl Cellulose Solution in Presence of an Electrolyte or a Surfactant: A Convenient Method to Determine the Cloud Point of Polymer Solutions. J. Polym. Sci. Pol. Chem. 2014, 51, 924–930. [Google Scholar] [CrossRef]
- Weißenborn, E.; Braunschweig, B. Hydroxypropyl cellulose as a green polymer for thermo-responsive aqueous foams. Soft Matter 2019, 15, 2876–2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Wickramasinghe, R.; Qian, X. Effects of Salt on the Lower Critical Solution Temperature of Poly (N-Isopropylacrylamide). J. Phys. Chem. B 2010, 114, 16594–16604. [Google Scholar] [CrossRef] [PubMed]
- Bruce, E.E.; Bui, P.T.; Rogers, B.A.; Cremer, P.S.; van der Vegt, N.F.A. Nonadditive Ion Effects Drive Both Collapse and Swelling of Thermoresponsive Polymers in Water. J. Am. Chem. Soc. 2019, 141, 6609–6616. [Google Scholar] [CrossRef]
- Zdrali, E.; Baer, M.D.; Okur, H.I.; Mundy, C.J.; Roke, S. The Diverse Nature of Ion Speciation at the Nanoscale Hydrophobic/Water Interface. J. Phys. Chem. B 2019, 123, 2414–2423. [Google Scholar] [CrossRef] [PubMed]
- Schelero, N.; Hedicke, G.; Linse, P.; Klitzing, R.V. Effects of counterions and co-ions on foam films stabilized by anionic dodecyl sulfate. J. Phys. Chem. B 2010, 114, 15523–15529. [Google Scholar] [CrossRef]
- Schulze-Zachau, F.; Bachmann, S.; Braunschweig, B. Effects of Ca2+ Ion Condensation on the Molecular Structure of Polystyrene Sulfonate at Air-Water Interfaces. Langmuir 2018, 34, 11714–11722. [Google Scholar] [CrossRef]
- Vlachy, N.; Jagoda-Cwiklik, B.; Vácha, R.; Touraud, D.; Jungwirth, P.; Kunz, W. Hofmeister series and specific interactions of charged headgroups with aqueous ions. Adv. Coll. Int. Sci. 2009, 146, 42–47. [Google Scholar] [CrossRef]
- Lukanov, B.; Firoozabadi, A. Specific Ion Effects on the Self-Assembly of Ionic Surfactants: A Molecular Thermodynamic Theory of Micellization with Dispersion Forces. Langmuir 2014, 30, 6373–6383. [Google Scholar] [CrossRef]
- Beierlein, F.R.; Clark, T.; Braunschweig, B.; Engelhardt, K.; Glas, L.; Peukert, W. Carboxylate Ion Pairing with Alkali-Metal Ions for β-Lactoglobulin and Its Role on Aggregation and Interfacial Adsorption. J. Phys. Chem. B 2015, 119, 5505–5517. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.D. Ions from the Hofmeister series and osmolytes: Effects on proteins in solution and in the crystallization process. Methods 2004, 34, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.D.; Neilson, G.W.; Enderby, J.E. Ions in water: Characterizing the forces that control chemical processes and biological structure. Biophys. Chem. 2007, 128, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Duignan, T.T.; Parsons, D.F.; Ninham, B.W. Collins’s rule, Hofmeister effects and ionic dispersion interactions. Chem. Phys. Lett. 2014, 608, 55–59. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Nguyen, A.V. New Evidence of Head-to-Tail Complex Formation of SDS-DOH Mixtures Adsorbed at the Air-Water Interface as Revealed by Vibrational Sum Frequency Generation Spectroscopy and Isotope Labelling. Langmuir 2019, 35, 4825–4833. [Google Scholar] [CrossRef] [PubMed]
- Yeon, C.; Kim, G.; Lim, J.W.; Yun, S.J. Highly conductive PEDOT: PSS treated by sodium dodecyl sulfate for stretchable fabric heaters. RSC Adv. 2017, 7, 5888–5897. [Google Scholar] [CrossRef]
- Qiao, B.; Liang, Y.; Wang, T.-J.; Jiang, Y. Surface modification to produce hydrophobic nano-silica particles using sodium dodecyl sulfate as a modifier. Appl. Surf. Sci. 2016, 364, 103–109. [Google Scholar] [CrossRef]
- Fauser, H.; Uhlig, M.; Miller, R.; von Klitzing, R. Surface Adsorption of Oppositely Charged SDS: C(12)TAB Mixtures and the Relation to Foam Film Formation and Stability. J. Phys. Chem. B 2015, 119, 12877–12886. [Google Scholar] [CrossRef] [PubMed]
- Windsor, R.; Neivandt, D.J.; Davies, P.B. Adsorption of Sodium Dodecyl Sulfate in the Presence of Poly(ethylenimine) and Sodium Chloride Studied Using Sum Frequency Vibrational Spectroscopy. Langmuir 2001, 17, 7306–7312. [Google Scholar] [CrossRef]
- Bain, C.D.; Davies, P.B.; Ward, R.N. In-Situ Sum-Frequency Spectroscopy of Sodium Dodecyl Sulfate and Dodecanol Coadsorbed at a Hydrophobic Surface. Langmuir 1994, 10, 2060–2063. [Google Scholar] [CrossRef]
- Gragson, D.E.; McCarty, B.M.; Richmond, G.L. Ordering of Interfacial Water Molecules at the Charged Air/Water Interface Observed by Vibrational Sum Frequency Generation. J. Am. Chem. Soc. 1997, 119, 6144–6152. [Google Scholar] [CrossRef]
- Messmer, M.C.; Conboy, J.C.; Richmond, G.L. Observation of Molecular Ordering at the Liquid-Liquid Interface by Resonant Sum Frequency Generation. J. Am. Chem. Soc. 1995, 117, 8039–8040. [Google Scholar] [CrossRef]
- Hommel, E.L.; Ma, G.; Allen, H.C. Broadband Vibrational Sum Frequency Generation Spectroscopy of a Liquid Surface. Anal. Sci. 2001, 17, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.M.; Tyrode, E. Study of the adsorption of sodium dodecyl sulfate (SDS) at the air/water interface: Targeting the sulfate headgroup using vibrational sum frequency spectroscopy. Phys. Chem. Chem. Phys. 2005, 7, 2635–2640. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar, H.B.; de Beer, A.G.F.; Strader, M.L.; Roke, S. The interfacial tension of nanoscopic oil droplets in water is hardly affected by SDS surfactant. J. Am. Chem. Soc. 2010, 132, 2122–2123. [Google Scholar] [CrossRef] [PubMed]
- Casson, B.D.; Bain, C.D. Phase transitions in mixed monolayers of sodium dodecyl sulfate and dodecanol at the air/water interface. J. Phys. Chem. B 1998, 102, 7434–7441. [Google Scholar] [CrossRef]
- Conboy, J.C.; Messmer, M.C.; Richmond, G.L. Investigation of surfactant conformation and order at the liquid-liquid interface by total internal reflection sum-frequency vibrational spectroscopy. J. Phys. Chem. 1996, 100, 7617–7622. [Google Scholar] [CrossRef]
- Zdrali, E.; Etienne, G.; Smolentsev, N.; Amstad, E.; Roke, S. The interfacial structure of nano-and micron-sized oil and water droplets stabilized with SDS and Span80. J. Chem. Phys. 2019, 150, 204704. [Google Scholar] [CrossRef]
- Sauerbeck, C. Untersuchung geladener kolloidaler Grenzflächen Mittels Nichtlinearer Lichtstreuung. Ph.D. Thesis, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany, 2018. [Google Scholar]
- Lu, J.R.; Marrocco, A.; Su, T.J.; Thomas, R.K.; Penfold, J. Adsorption of Dodecyl Sulfate Surfactants with Monovalent Metal Counterions at the Air-Water Interface Studied by Neutron Reflection and Surface Tension. J. Coll. Int. Sci. 1993, 158, 303–316. [Google Scholar] [CrossRef]
- Danov, K.D.; Kralchevsky, P.A.; Ananthapadmanabhan, K.P. Micelle-monomer equilibria in solutions of ionic surfactants and in ionic-nonionic mixtures: A generalized phase separation model. Adv. Coll. Int. Sci. 2014, 206, 17–45. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; Boxer, S.G. Vibrational Stark Effect Spectroscopy. J. Am. Chem. Soc. 1995, 117, 1449–1450. [Google Scholar] [CrossRef]
- Fried, S.D.; Boxer, S.G. Measuring electric fields and noncovalent interactions using the vibrational stark effect. Acc. Chem. Res. 2015, 48, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Hush, N.S.; Reimers, J.R. Vibrational Stark Spectroscopy. 1. Basic Theory and Application to the CO Stretch. J. Phys. Chem. 1995, 99, 15798–15805. [Google Scholar] [CrossRef]
- Zwaschka, G.; Wolf, M.; Campen, R.K.; Tong, Y. A Microscopic Model of the Electrochemical Vibrational Stark Effect: Understanding VSF Spectroscopy of (bi)Sulfate on Pt(111). Surf. Sci. 2018, 678, 78–85. [Google Scholar] [CrossRef]
- Braunschweig, B.; Mukherjee, P.; Dlott, D.D.; Wieckowski, A. Real-time investigations of Pt(111) surface transformations in sulfuric acid solutions. J. Am. Chem. Soc. 2010, 132, 14036–14038. [Google Scholar] [CrossRef]
- Andrews, S.S.; Boxer, S.G. Vibrational Stark Effects of Nitriles I. Methods and Experimental Results. J. Phys. Chem. A 2000, 104, 11853–11863. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces, 3rd ed.; Academic Press: Burlington, MA, USA, 2011. [Google Scholar]
- Kunz, W. Specific ion effects in colloidal and biological systems. Curr. Opin. Colloid Interface Sci. 2010, 15, 34–39. [Google Scholar] [CrossRef]
- Gonella, G.; Lütgebaucks, C.; de Beer, A.G.F.; Roke, S. Second Harmonic and Sum-Frequency Generation from Aqueous Interfaces Is Modulated by Interference. J. Phys. Chem. C Nanomater. Interfaces 2016, 120, 9165–9173. [Google Scholar] [CrossRef]
- Ohno, P.E.; Wang, H.-F.; Geiger, F.M. Second-order spectral lineshapes from charged interfaces. Nat. Commun. 2017, 8, 1032. [Google Scholar] [CrossRef]
- García Rey, N.; Weißenborn, E.; Schulze-Zachau, F.; Gochev, G.; Braunschweig, B. Quantifying Double-Layer Potentials at Liquid-Gas Interfaces from Vibrational Sum-Frequency Generation. J. Phys. Chem. C 2019, 123, 1279–1286. [Google Scholar] [CrossRef]
- Exerowa, D.; Zacharieva, M.; Cohen, R.; Platikanov, D. Dependence of the equilibrium thickness and double layer potential of foam films on the surfactant concentration. Colloid Polym. Sci. 1979, 257, 1089–1098. [Google Scholar] [CrossRef]
- Nakahara, H.; Shibata, O.; Moroi, Y. Examination of surface adsorption of sodium chloride and sodium dodecyl sulfate by surface potential measurement at the air/solution interface. Langmuir 2005, 21, 9020–9022. [Google Scholar] [CrossRef] [PubMed]
Sample Available: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CMC\mM | |||
|---|---|---|---|
| LiDS | −23.4 | 8.4 | 3.32 |
| LiDS + 25 mM LiCl | −28.2 | 5.3 | |
| SDS | −23.6 | 8.1 | 3.77 |
| SDS + 25 mM NaCl | −28.2 | 3.7 | |
| CsDS | −26.4 | 5.1 | 4.37 |
| CsDS + 25 mM CsCl | −30.7 | 2.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weißenborn, E.; Braunschweig, B. Specific Ion Effects of Dodecyl Sulfate Surfactants with Alkali Ions at the Air–Water Interface. Molecules 2019, 24, 2911. https://doi.org/10.3390/molecules24162911
Weißenborn E, Braunschweig B. Specific Ion Effects of Dodecyl Sulfate Surfactants with Alkali Ions at the Air–Water Interface. Molecules. 2019; 24(16):2911. https://doi.org/10.3390/molecules24162911
Chicago/Turabian StyleWeißenborn, Eric, and Björn Braunschweig. 2019. "Specific Ion Effects of Dodecyl Sulfate Surfactants with Alkali Ions at the Air–Water Interface" Molecules 24, no. 16: 2911. https://doi.org/10.3390/molecules24162911
APA StyleWeißenborn, E., & Braunschweig, B. (2019). Specific Ion Effects of Dodecyl Sulfate Surfactants with Alkali Ions at the Air–Water Interface. Molecules, 24(16), 2911. https://doi.org/10.3390/molecules24162911