
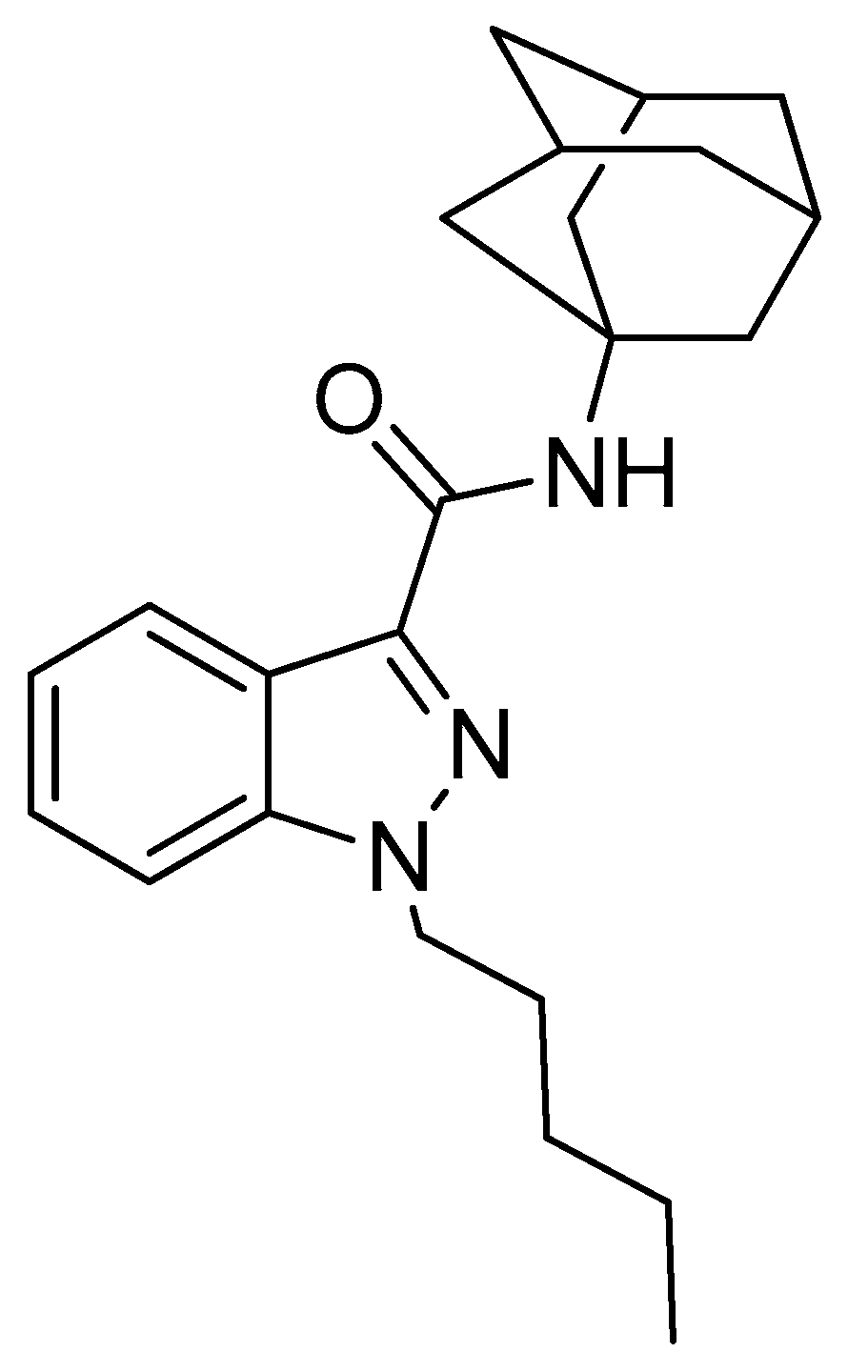
In Vitro Inhibitory Effects of APINACA on Human Major Cytochrome P450, UDP-Glucuronosyltransferase Enzymes, and Drug Transporters

 ,
,  , ,
, , 

Abstract
:1. Introduction
2. Results
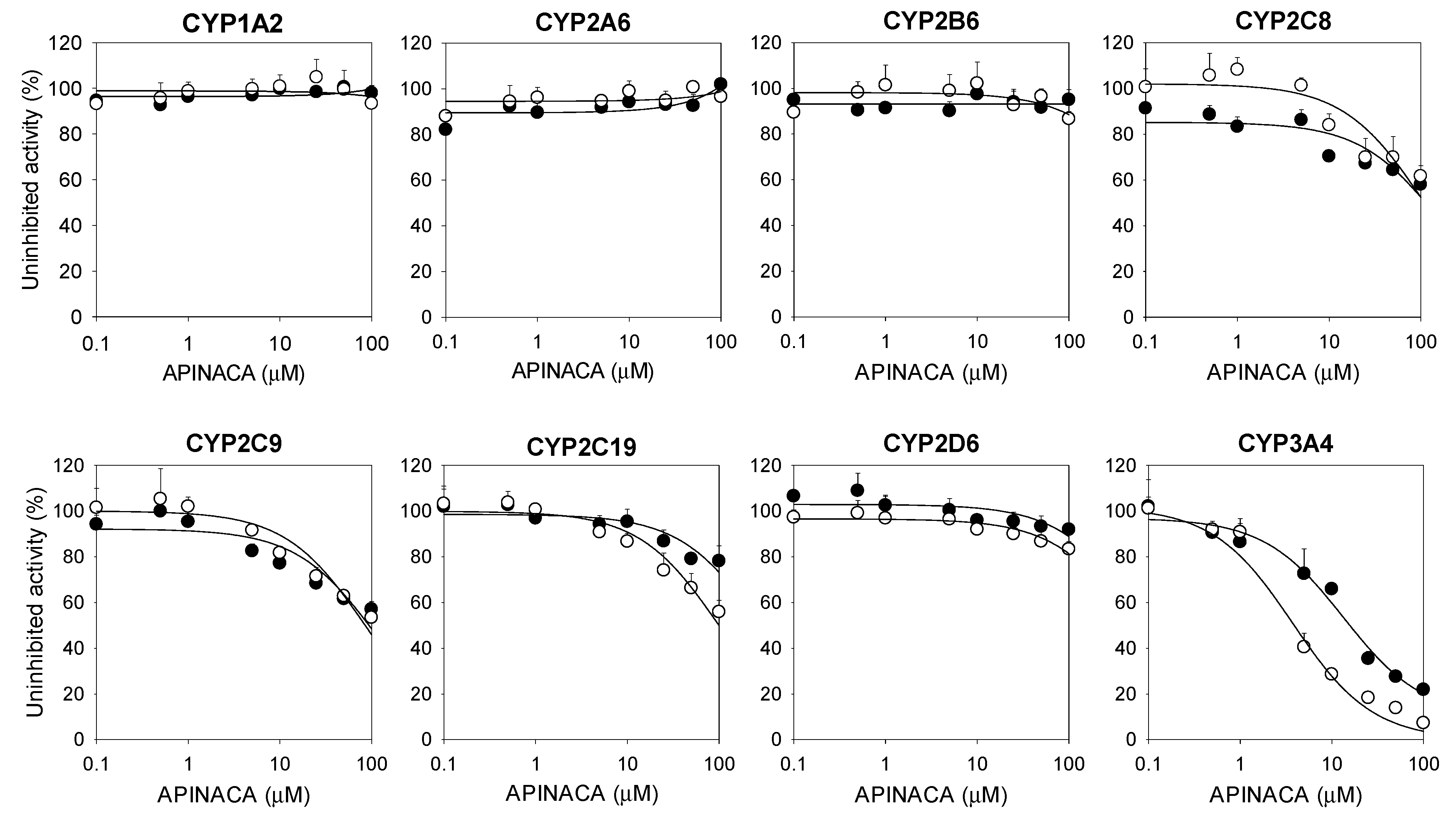
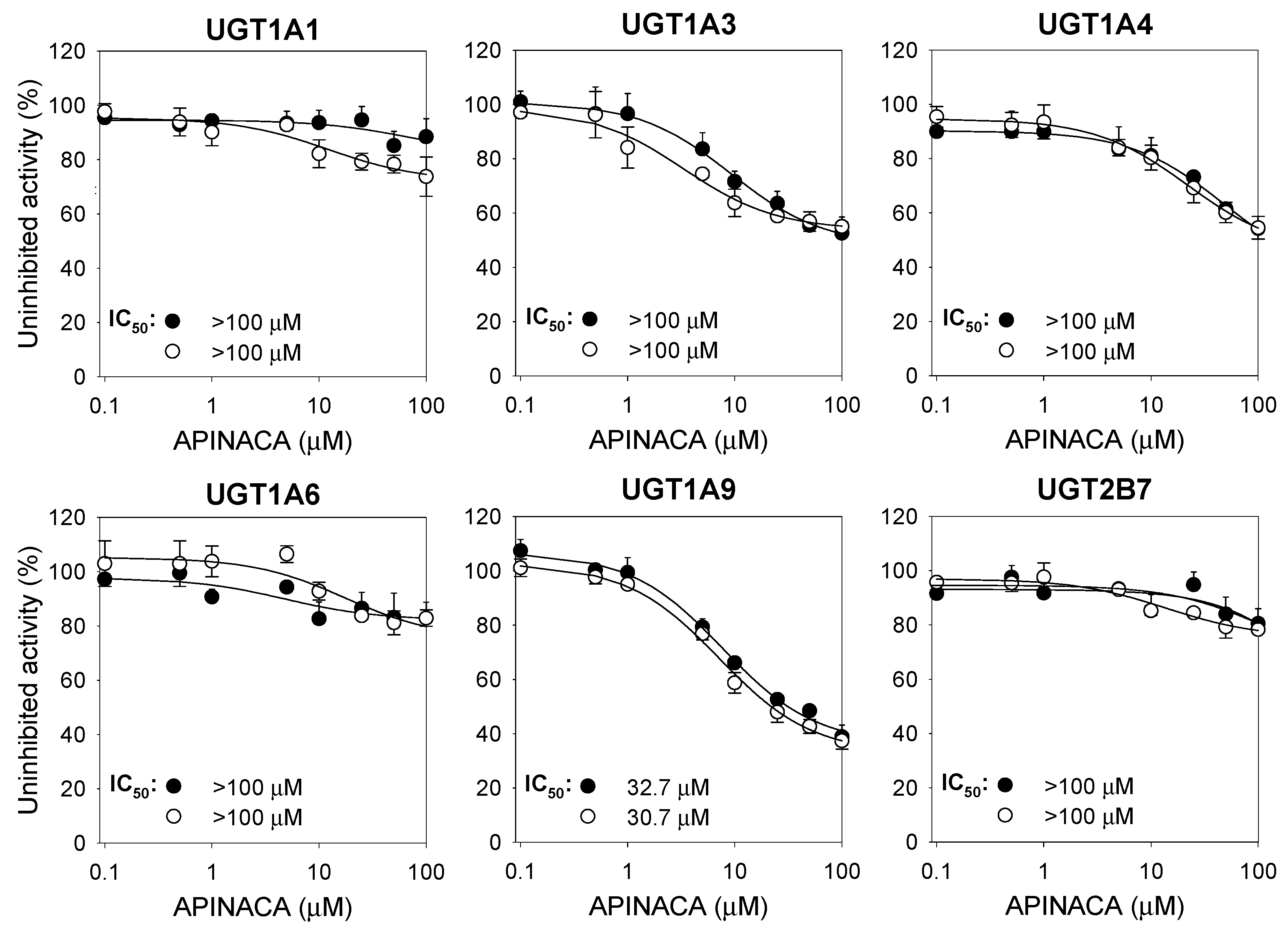
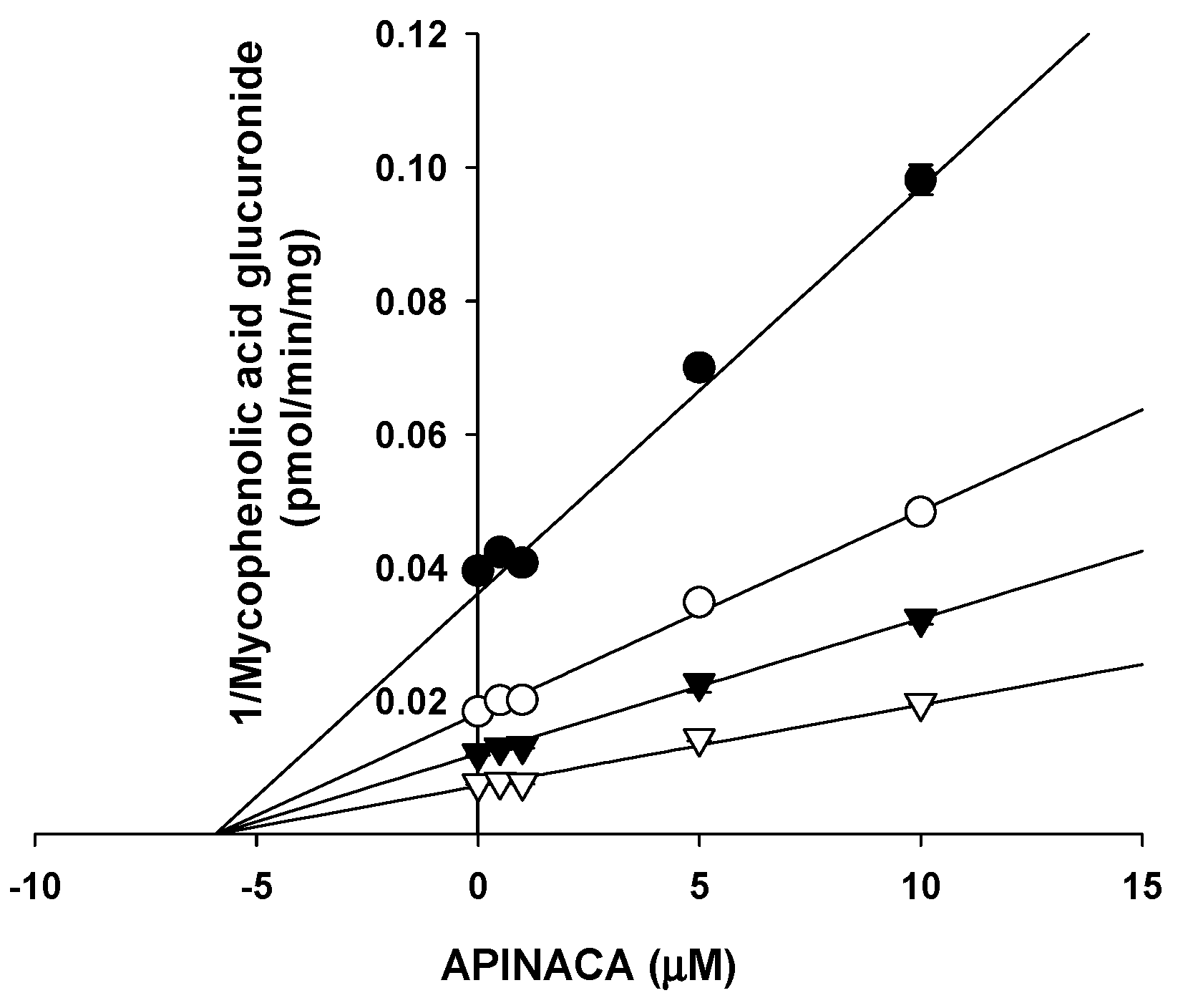
2.1. Inhibitory Effect of APINACA on CYP and UGT Enzymes in Human Liver Microsomes
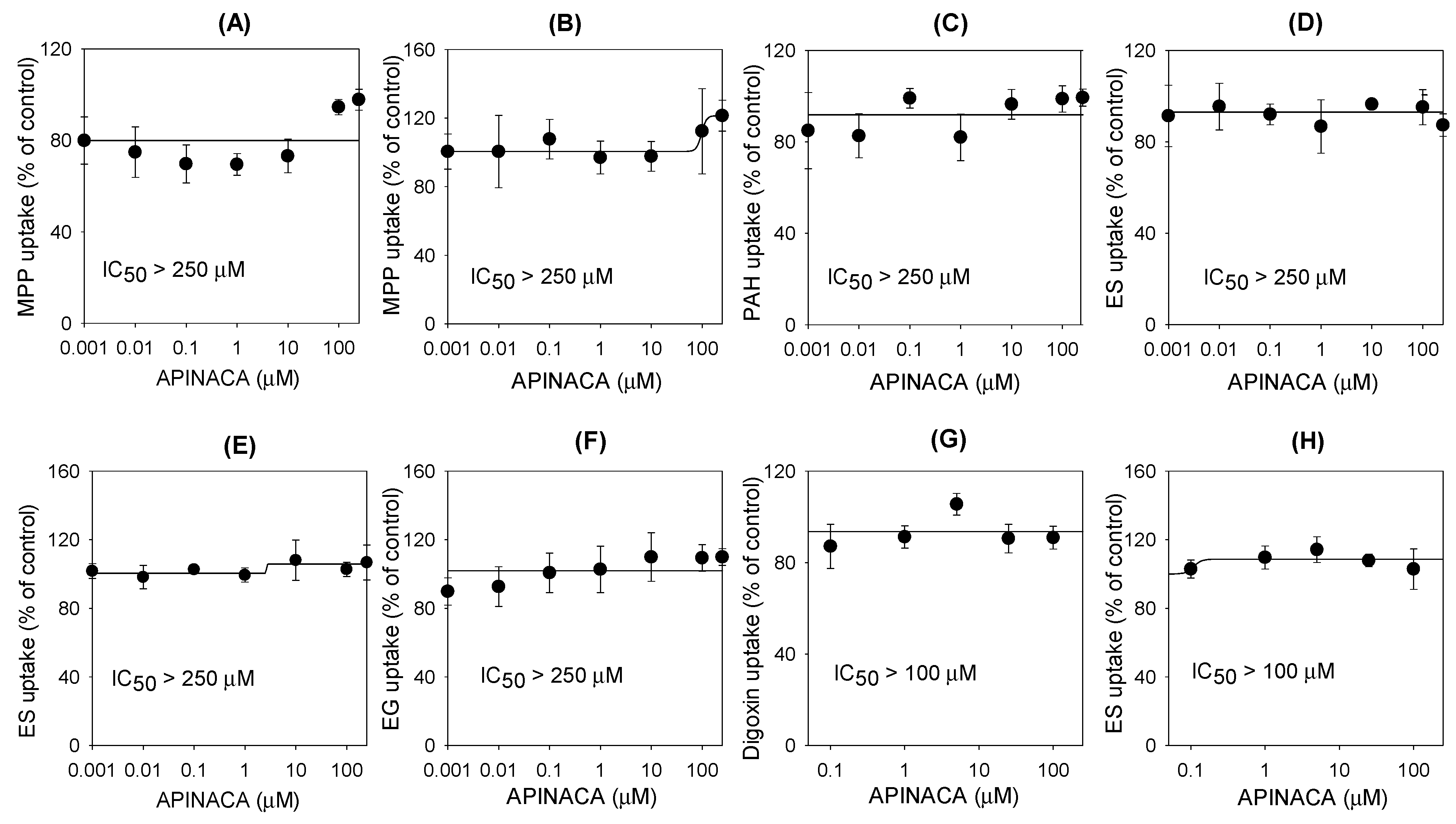
2.2. Inhibitory Effect of APINACA on Drug Transporters
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Inhibitory Effect of APINACA on Eight Major CYP Activities in Human Liver Microsomes
4.3. Inhibitory Effect of APINACA on Six Major UGT Activities
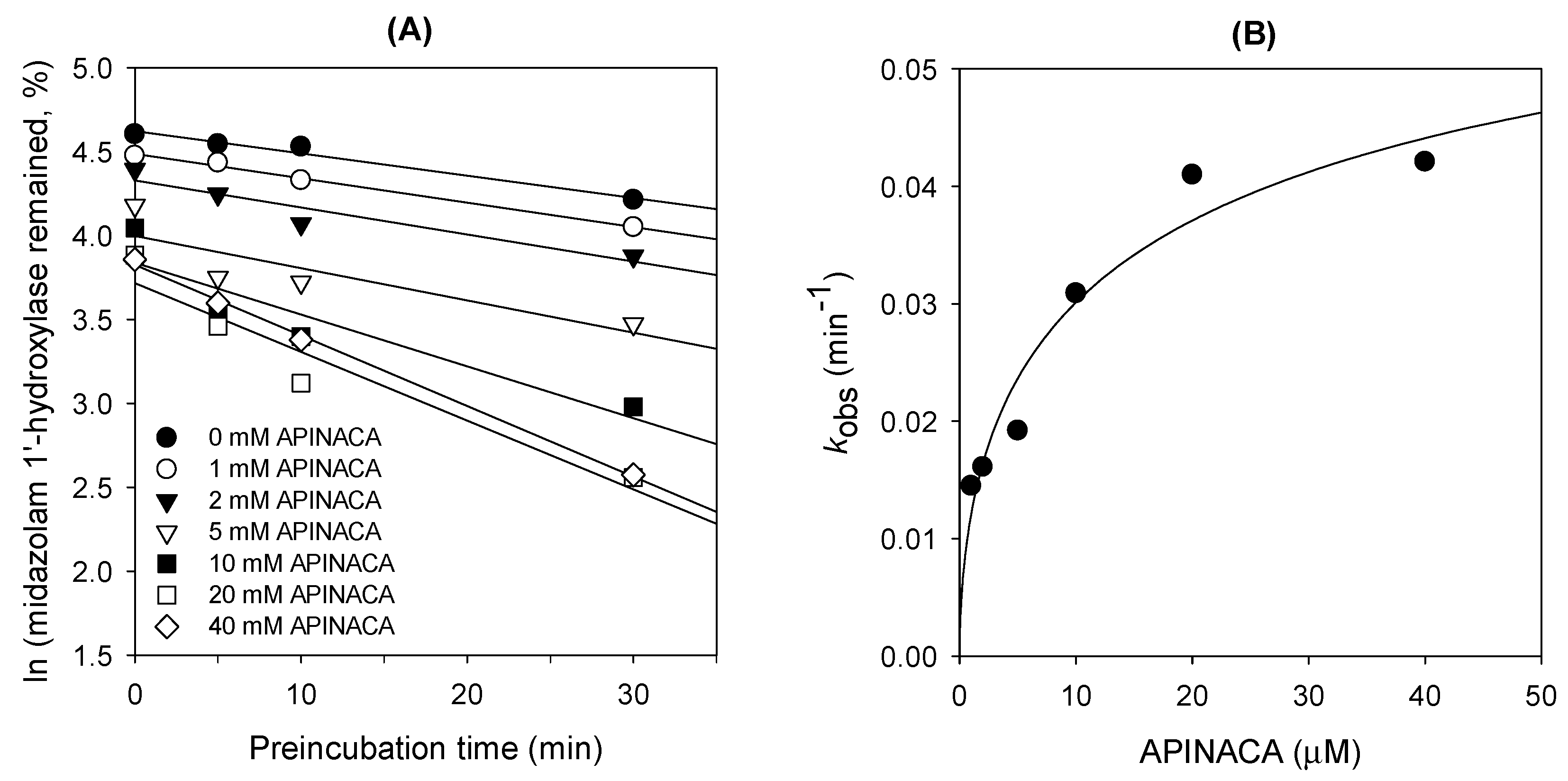
4.4. Time-Dependent Inhibition of CYP3A4 Activity by APINACA in Human Liver Microsomes
4.5. Enzyme Kinetic Analysis for the Inhibition of UGT1A9 by APINACA
4.6. Inhibitory Effect of APINACA on the Transport Activities of Efflux Transporters
4.7. Inhibitory Effect of APINACA on the Transport Activities of Solute Carrier Transporters
4.8. Data Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Le Boisselier, R.; Alexandre, J.; Lelong-Boulouard, V.; Debruyne, D. Focus on cannabinoids and synthetic cannabinoids. Clin. Pharmacol. Ther. 2017, 101, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Synthetic cannabinoids in Europe. Available online: http://emcdda.europa.eu/topics/pods/synthetic-cannabinoids (accessed on 6 June 2017).
- Gandhi, A.V.; Saxena, S.; Relles, D.; Sarosiek, K.; Kang, C.Y.; Chipitsyna, G.; Sendecki, J.A.; Yeo, C.J.; Arafat, H.A. Differential expression of cytochrome P450 omega-hydroxylase isoforms and their association with clinicopathological features in pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2013, 20, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Holm, N.B.; Nielsen, L.M.; Linnet, K. CYP3A4 Mediates Oxidative Metabolism of the Synthetic Cannabinoid AKB-48. AAPS J. 2015, 17, 1237–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, N.B.; Noble, C.; Linnet, K. JWH-018 omega-OH, a shared hydroxy metabolite of the two synthetic cannabinoids JWH-018 and AM-2201, undergoes oxidation by alcohol dehydrogenase and aldehyde dehydrogenase enzymes in vitro forming the carboxylic acid metabolite. Toxicol. Lett. 2016, 259, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.Y.; Kim, J.H.; Kim, D.K.; Lee, H.S. Synthetic cannabinoids are substrates and inhibitors of multiple drug-metabolizing enzymes. Arch. Pharm. Res. 2018, 41, 691–710. [Google Scholar] [CrossRef] [PubMed]
- Vikingsson, S.; Josefsson, M.; Green, H. Identification of AKB-48 and 5F-AKB-48 metabolites in authentic human urine samples using human liver microsomes and time of flight mass spectrometry. J. Anal. Toxicol. 2015, 39, 426–435. [Google Scholar] [CrossRef]
- Cerny, M.A. Prevalence of Non-Cytochrome P450-Mediated Metabolism in Food and Drug Administration-Approved Oral and Intravenous Drugs: 2006–2015. Drug Metab. Dispos. 2016, 44, 1246–1252. [Google Scholar] [CrossRef]
- Foti, R.S.; Dalvie, D.K. Cytochrome P450 and Non-Cytochrome P450 Oxidative Metabolism: Contributions to the Pharmacokinetics, Safety, and Efficacy of Xenobiotics. Drug Metab. Dispos. 2016, 44, 1229–1245. [Google Scholar] [CrossRef] [Green Version]
- Mao, Q.; Lai, Y.; Wang, J. Drug transporters in xenobiotic disposition and pharmacokinetic prediction. Drug Metab. Dispos. 2018, 46, 561–566. [Google Scholar] [CrossRef]
- Kong, T.Y.; Kim, J.H.; Kim, J.Y.; In, M.K.; Choi, K.H.; Kim, H.S.; Lee, H.S. Rapid analysis of drugs of abuse and their metabolites in human urine using dilute and shoot liquid chromatography-tandem mass spectrometry. Arch. Pharm. Res. 2017, 40, 180–196. [Google Scholar] [CrossRef]
- Salomone, A.; Palamar, J.J.; Gerace, E.; Di Corcia, D.; Vincenti, M. Hair Testing for Drugs of abuse and new psychoactive substances in a high-risk population. J. Anal. Toxicol. 2017, 41, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Kong, T.Y.; Cheong, J.C.; Kim, J.Y.; Lee, J.I.; Lee, H.S. Simultaneous determination of 75 abuse drugs including amphetamines, benzodiazepines, cocaine, opioids, piperazines, zolpidem and metabolites in human hair samples using liquid chromatography-tandem mass spectrometry. Biomed. Chromatogr. 2019, e4600. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kwon, S.S.; Kong, T.Y.; Cheong, J.C.; Kim, H.S.; In, M.K.; Lee, H.S. AM-2201 Inhibits Multiple Cytochrome P450 and Uridine 5′-Diphospho-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules 2017, 22, 443. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.Y.; Kim, J.H.; Kwon, S.S.; Cheong, J.C.; Kim, H.S.; In, M.K.; Lee, H.S. Inhibition of cytochrome P450 and uridine 5′-diphospho-glucuronosyltransferases by MAM-2201 in human liver microsomes. Arch. Pharm. Res. 2017, 40, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.Y.; Kwon, S.S.; Cheong, J.C.; Kim, H.S.; Kim, J.Y.; Lee, H.S. In Vitro Inhibitory Effects of Synthetic Cannabinoid EAM-2201 on Cytochrome P450 and UDP-Glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules 2018, 23, 920. [Google Scholar] [CrossRef] [PubMed]
- Ashino, T.; Hakukawa, K.; Itoh, Y.; Numazawa, S. Inhibitory effect of synthetic cannabinoids on CYP1A activity in mouse liver microsomes. J. Toxicol. Sci. 2014, 39, 815–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Yamaori, S.; Okamoto, Y.; Yamamoto, I.; Watanabe, K. Cannabidiol is a potent inhibitor of the catalytic activity of cytochrome P450 2C19. Drug Metab. Pharmacokinet. 2013, 28, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Yamaori, S.; Ebisawa, J.; Okushima, Y.; Yamamoto, I.; Watanabe, K. Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: Role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci. 2011, 88, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Yamaori, S.; Koeda, K.; Kushihara, M.; Hada, Y.; Yamamoto, I.; Watanabe, K. Comparison in the in vitro inhibitory effects of major phytocannabinoids and polycyclic aromatic hydrocarbons contained in marijuana smoke on cytochrome P450 2C9 activity. Drug Metab. Pharmacokinet. 2012, 27, 294–300. [Google Scholar] [CrossRef]
- Yamaori, S.; Kushihara, M.; Yamamoto, I.; Watanabe, K. Characterization of major phytocannabinoids, cannabidiol and cannabinol, as isoform-selective and potent inhibitors of human CYP1 enzymes. Biochem. Pharmacol. 2010, 79, 1691–1698. [Google Scholar] [CrossRef]
- Yamaori, S.; Maeda, C.; Yamamoto, I.; Watanabe, K. Differential inhibition of human cytochrome P450 2A6 and 2B6 by major phytocannabinoids. Forensic Toxicol. 2011, 29, 117–124. [Google Scholar] [CrossRef]
- Yamaori, S.; Okamoto, Y.; Yamamoto, I.; Watanabe, K. Cannabidiol, a major phytocannabinoid, as a potent atypical inhibitor for CYP2D6. Drug Metab. Dispos. 2011, 39, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Zendulka, O.; Dovrtelova, G.; Noskova, K.; Turjap, M.; Sulcova, A.; Hanus, L.; Jurica, J. Cannabinoids and Cytochrome P450 Interactions. Curr. Drug Metab. 2016, 17, 206–226. [Google Scholar] [CrossRef] [PubMed]
- Tournier, N.; Chevillard, L.; Megarbane, B.; Pirnay, S.; Scherrmann, J.M.; Decleves, X. Interaction of drugs of abuse and maintenance treatments with human P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2). Int. J. Neuropsychopharmacol. 2010, 13, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Holland, M.L.; Panetta, J.A.; Hoskins, J.M.; Bebawy, M.; Roufogalis, B.D.; Allen, J.D.; Arnold, J.C. The effects of cannabinoids on P-glycoprotein transport and expression in multidrug resistant cells. Biochem. Pharmacol. 2006, 71, 1146–1154. [Google Scholar] [CrossRef]
- Wagmann, L.; Maurer, H.H.; Meyer, M.R. Inhibition and stimulation of the human breast cancer resistance protein as in vitro predictor of drug-drug interactions of drugs of abuse. Arch. Toxicol. 2018, 92, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.R.; Orschiedt, T.; Maurer, H.H. Michaelis-Menten kinetic analysis of drugs of abuse to estimate their affinity to human P-glycoprotein. Toxicol. Lett. 2013, 217, 137–142. [Google Scholar] [CrossRef]
- Meyer, M.R.; Wagmann, L.; Schneider-Daum, N.; Loretz, B.; de Souza Carvalho, C.; Lehr, C.M.; Maurer, H.H. P-glycoprotein interactions of novel psychoactive substances - stimulation of ATP consumption and transport across Caco-2 monolayers. Biochem. Pharmacol. 2015, 94, 220–226. [Google Scholar] [CrossRef]
- Ossato, A.; Uccelli, L.; Bilel, S.; Canazza, I.; Di Domenico, G.; Pasquali, M.; Pupillo, G.; De Luca, M.A.; Boschi, A.; Vincenzi, F.; et al. Psychostimulant effect of the synthetic cannabinoid JWH-018 and AKB48: Behavioral, neurochemical, and dopamine transporter scan imaging studies in mice. Front. Psychiatry 2017, 8, 130. [Google Scholar] [CrossRef]
- Jeong, H.U.; Kwon, M.; Lee, Y.; Yoo, J.S.; Shin, D.H.; Song, I.S.; Lee, H.S. Organic anion transporter 3- and organic anion transporting polypeptides 1B1- and 1B3-mediated transport of catalposide. Drug Des. Devel. Ther. 2015, 9, 643–653. [Google Scholar]
- Seong, S.J.; Kang, W.Y.; Heo, J.K.; Jo, J.; Choi, W.G.; Liu, K.H.; Lee, S.; Choi, M.K.; Han, Y.H.; Lee, H.S.; et al. A comprehensive in vivo and in vitro assessment of the drug interaction potential of red ginseng. Clin. Ther. 2018, 40, 1322–1337. [Google Scholar] [CrossRef]
- Song, I.S.; Kong, T.Y.; Jeong, H.U.; Kim, E.N.; Kwon, S.S.; Kang, H.E.; Choi, S.Z.; Son, M.; Lee, H.S. Evaluation of the transporter-mediated herb-drug interaction potential of DA-9801, a standardized dioscorea extract for diabetic neuropathy, in human in vitro and rat in vivo. BMC Complement. Altern. Med. 2014, 14, 251. [Google Scholar] [CrossRef]
- Albaugh, D.R.; Fullenwider, C.L.; Fisher, M.B.; Hutzler, J.M. Time-dependent inhibition and estimation of CYP3A clinical pharmacokinetic drug-drug interactions using plated human cell systems. Drug Metab. Dispos. 2012, 40, 1336–1344. [Google Scholar] [CrossRef]
- Zhou, S.F. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef]
- Al Saabi, A.; Allorge, D.; Sauvage, F.L.; Tournel, G.; Gaulier, J.M.; Marquet, P.; Picard, N. Involvement of UDP-glucuronosyltransferases UGT1A9 and UGT2B7 in ethanol glucuronidation, and interactions with common drugs of abuse. Drug Metab. Dispos. 2013, 41, 568–574. [Google Scholar] [CrossRef]
- Miners, J.O.; Chau, N.; Rowland, A.; Burns, K.; McKinnon, R.A.; Mackenzie, P.I.; Tucker, G.T.; Knights, K.M.; Kichenadasse, G. Inhibition of human UDP-glucuronosyltransferase enzymes by lapatinib, pazopanib, regorafenib and sorafenib: Implications for hyperbilirubinemia. Biochem. Pharmacol. 2017, 129, 85–95. [Google Scholar] [CrossRef]
- Pattanawongsa, A.; Chau, N.; Rowland, A.; Miners, J.O. Inhibition of human UDP-glucuronosyltransferase enzymes by canagliflozin and dapagliflozin: Implications for drug-drug interactions. Drug Metab. Dispos. 2015, 43, 1468–1476. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, Y.; Jeong, H. Drug-Drug Interaction Potentials of Tyrosine Kinase Inhibitors via Inhibition of UDP-Glucuronosyltransferases. Sci. Rep. 2015, 5, 17778. [Google Scholar] [CrossRef]
- Lv, X.; Zhang, J.B.; Hou, J.; Dou, T.Y.; Ge, G.B.; Hu, W.Z.; Yang, L. Chemical probes for human UDP-glucuronosyltransferases: A comprehensive review. Biotechnol. J. 2019, 14, e1800002. [Google Scholar] [CrossRef]
- Meech, R.; Hu, D.G.; McKinnon, R.A.; Mubarokah, S.N.; Haines, A.Z.; Nair, P.C.; Rowland, A.; Mackenzie, P.I. The UDP-Glycosyltransferase (UGT) superfamily: New members, new functions, and novel paradigms. Physiol. Rev. 2019, 99, 1153–1222. [Google Scholar] [CrossRef]
- Karinen, R.; Tuv, S.S.; Oiestad, E.L.; Vindenes, V. Concentrations of APINACA, 5F-APINACA, UR-144 and its degradant product in blood samples from six impaired drivers compared to previous reported concentrations of other synthetic cannabinoids. Forensic Sci. Int. 2015, 246, 98–103. [Google Scholar] [CrossRef]
- Jeong, H.U.; Kong, T.Y.; Kwon, S.S.; Hong, S.W.; Yeon, S.H.; Choi, J.H.; Lee, J.Y.; Cho, Y.Y.; Lee, H.S. Effect of honokiol on cytochrome P450 and UDP-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules 2013, 18, 10681–10693. [Google Scholar] [CrossRef]
- Kwon, S.S.; Kim, J.H.; Jeong, H.U.; Cho, Y.Y.; Oh, S.R.; Lee, H.S. Inhibitory Effects of Aschantin on Cytochrome P450 and Uridine 5′-diphospho-glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules 2016, 21, 554. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CYPs | Enzyme Activities | IC50 (μM) | |
|---|---|---|---|
| No Preincubation | With Preincubation | ||
| 1A2 | Phenacetin O-deethylase | >100 | >100 |
| 2A6 | Coumarin 7-hydroxylase | >100 | >100 |
| 2B6 | Bupropion hydroxylase | >100 | >100 |
| 2C8 | Amodiaquine N-deethylase | >100 | >100 |
| 2C9 | Diclofenac 4′-hydroxylase | >100 | 85.0 |
| 2C19 | [S]-Mephenytoin 4′-hydroxylase | >100 | >100 |
| 2D6 | Bufuralol 1′-hydroxylase | >100 | >100 |
| 3A4 | Midazolam 1′-hydroxylase | 16.9 | 4.2 |
| Cells | Transporters | Probe Substrate | Transport Rate (pmol/min) (mean ± SD) | Fold Increase |
|---|---|---|---|---|
| HEK293 | Mock | 0.1 μM Methyl-4-phenylpyridinium | 0.60 ± 0.09 | 17.1 |
| OCT1 | 10.24 ± 0.98 | |||
| Mock | 0.1 μM Methyl-4-phenylpyridinium | 0.66 ± 0.11 | 25.5 | |
| OCT2 | 16.78 ± 0.43 | |||
| Mock | 0.1 μM para-aminohippuric acid | 1.22 ± 0.22 | 17.6 | |
| OAT1 | 21.49 ± 0.45 | |||
| Mock | 0.1 μM Estrone-3-sulfate | 0.98 ± 0.20 | 15.3 | |
| OAT3 | 14.96 ± 3.09 | |||
| Mock | 0.1 μM Estrone-3-sulfate | 0.76 ± 0.04 | 16.5 | |
| OATP1B1 | 12.58 ± 1.57 | |||
| Mock | 0.1 μM Estradiol-17β-d-glucuronide | 0.17 ± 0.02 | 13.4 | |
| OATP1B3 | 2.28 ± 0.08 | |||
| LLC-PK1 | Mock | 0.1 μM Digoxin | 0.14 ± 0.02 | 7.3 |
| MDR1 (P-gp) | 1.06 ± 0.07 | |||
| Mock | 0.1 μM Estrone-3-sulfate | 0.44 ± 0.07 | 4.7 | |
| BCRP | 2.09 ± 0.12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Choi, W.-G.; Kwon, M.; Lee, S.; Cho, Y.-Y.; Lee, J.Y.; Kang, H.C.; Song, I.-S.; Lee, H.S. In Vitro Inhibitory Effects of APINACA on Human Major Cytochrome P450, UDP-Glucuronosyltransferase Enzymes, and Drug Transporters. Molecules 2019, 24, 3000. https://doi.org/10.3390/molecules24163000
Kim S, Choi W-G, Kwon M, Lee S, Cho Y-Y, Lee JY, Kang HC, Song I-S, Lee HS. In Vitro Inhibitory Effects of APINACA on Human Major Cytochrome P450, UDP-Glucuronosyltransferase Enzymes, and Drug Transporters. Molecules. 2019; 24(16):3000. https://doi.org/10.3390/molecules24163000
Chicago/Turabian StyleKim, Sunjoo, Won-Gu Choi, Mihwa Kwon, Sowon Lee, Yong-Yeon Cho, Joo Young Lee, Han Chang Kang, Im-Sook Song, and Hye Suk Lee. 2019. "In Vitro Inhibitory Effects of APINACA on Human Major Cytochrome P450, UDP-Glucuronosyltransferase Enzymes, and Drug Transporters" Molecules 24, no. 16: 3000. https://doi.org/10.3390/molecules24163000
APA StyleKim, S., Choi, W. -G., Kwon, M., Lee, S., Cho, Y. -Y., Lee, J. Y., Kang, H. C., Song, I. -S., & Lee, H. S. (2019). In Vitro Inhibitory Effects of APINACA on Human Major Cytochrome P450, UDP-Glucuronosyltransferase Enzymes, and Drug Transporters. Molecules, 24(16), 3000. https://doi.org/10.3390/molecules24163000