
Sub-Atomic Resolution Crystal Structures Reveal Conserved Geometric Outliers at Functional Sites


Abstract
:1. Introduction
2. Results and Discussion
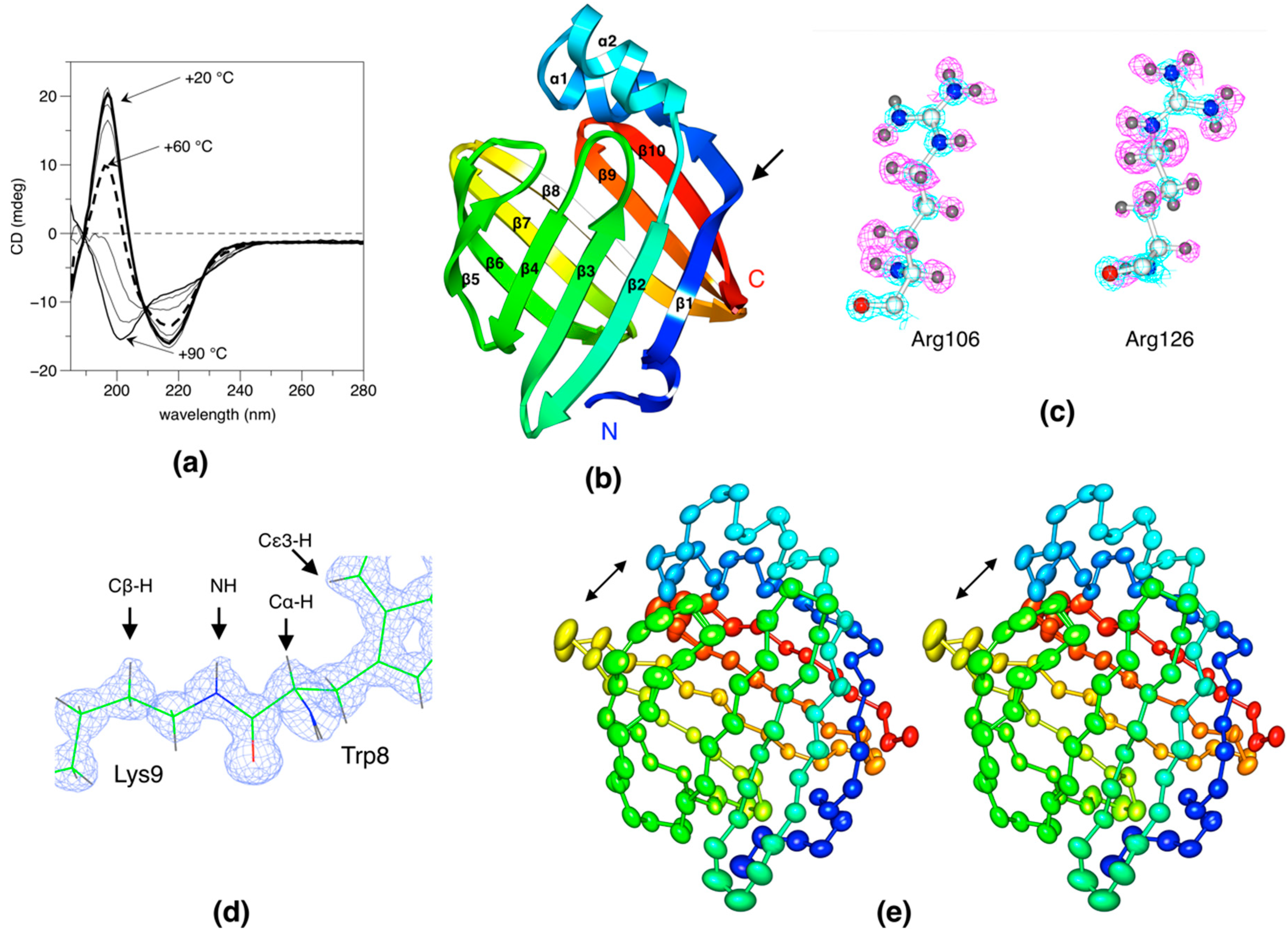
2.1. Overall Structure and Quality
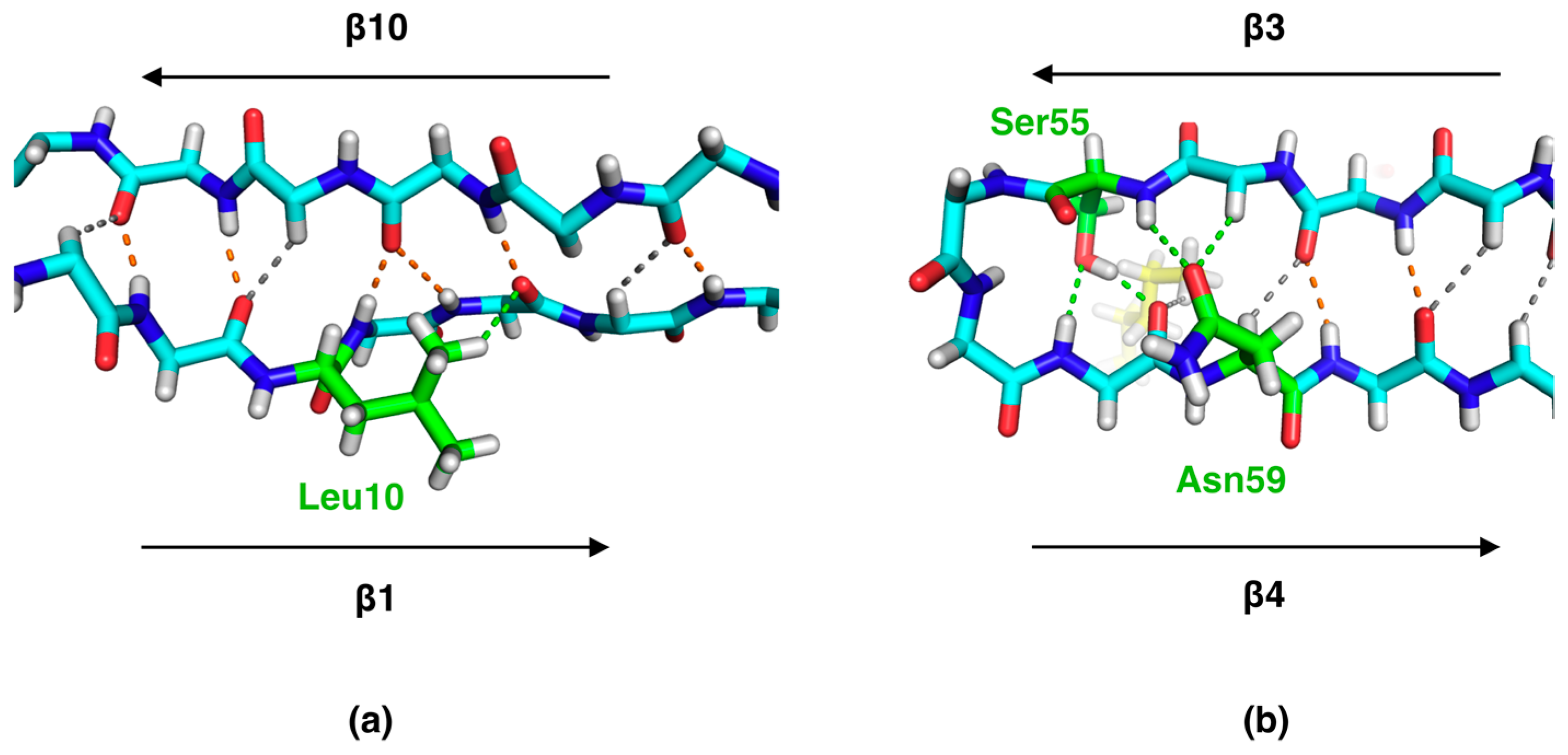
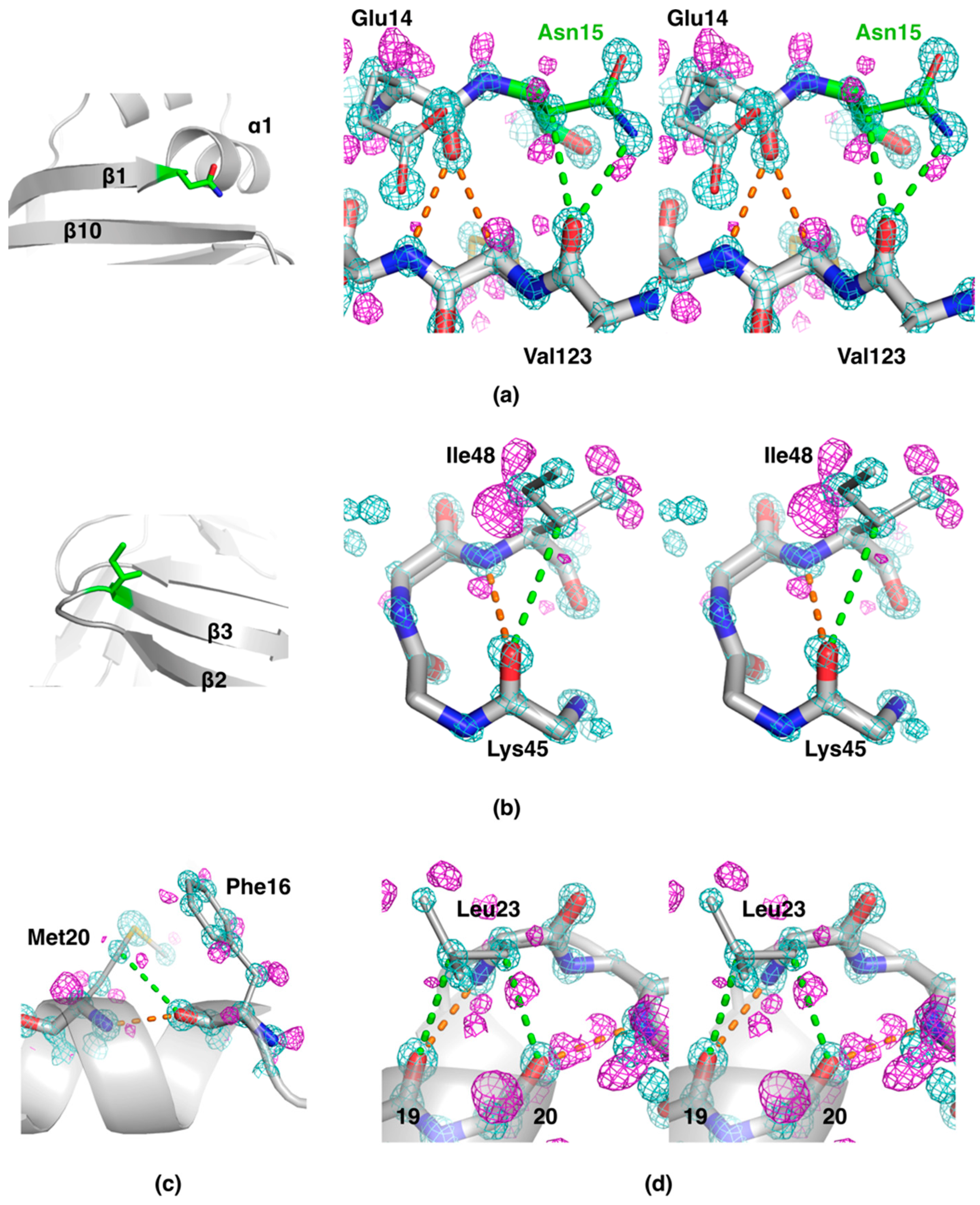
2.2. Side Chain-Backbone Interactions
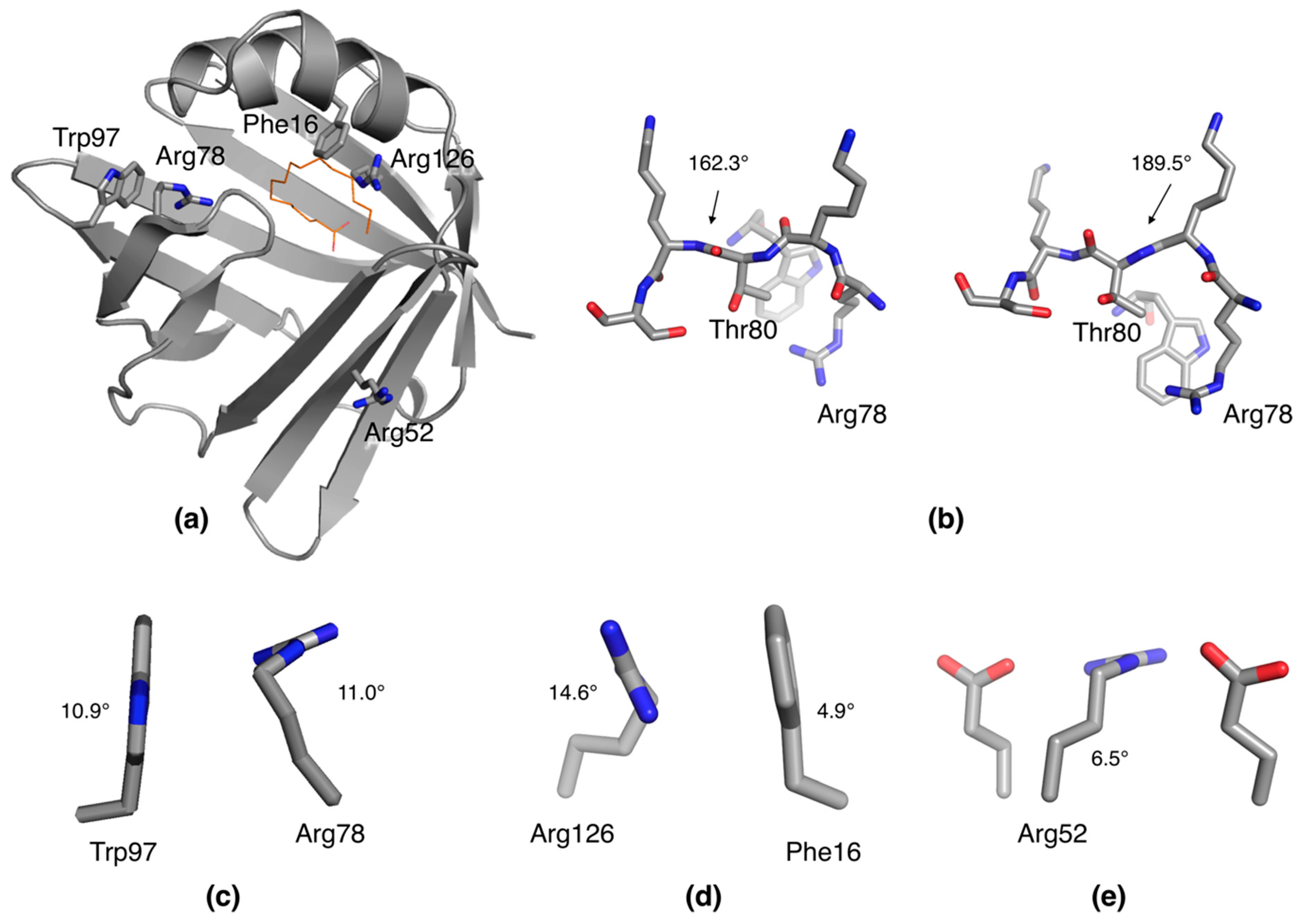
2.3. Unconventional Side Chain Conformations in P2
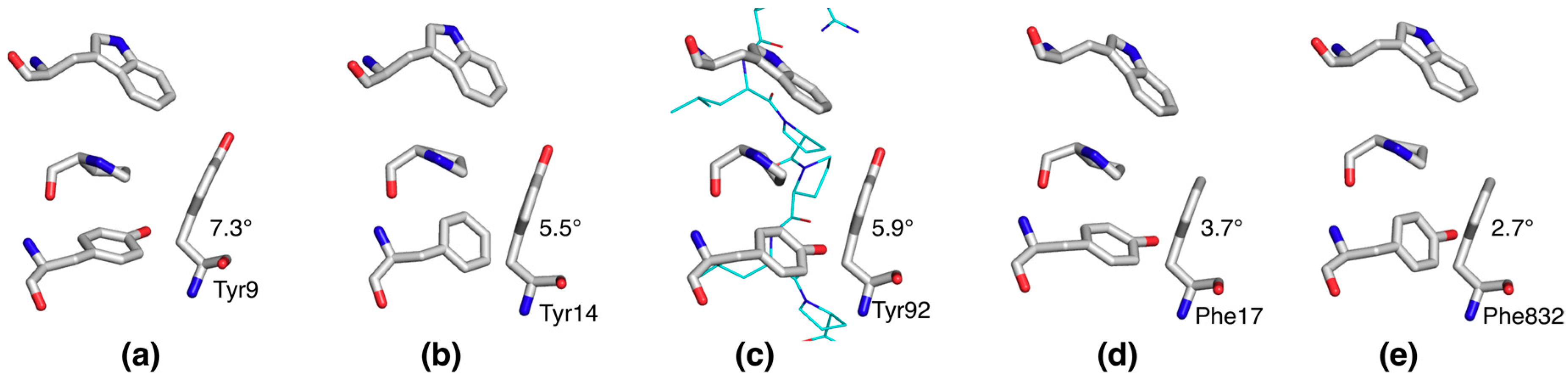
2.4. Distorted Side Chain Planarity in Other High-Resolution Structures
2.5. Deuteration as a Tool for Crystallization
3. Materials and Methods
3.1. Perdeuteration and Protein Purification
3.2. Crystallization
3.3. Data Collection and Processing
3.4. Structure Solution and Refinement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, X.; Hanson, B.L.; Langan, P.; Viola, R.E. The effect of deuteration on protein structure: A high-resolution comparison of hydrogenous and perdeuterated haloalkane dehalogenase. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, C.M.; Seeliger, D.; Sheldrick, G.M.; de Groot, B.L.; Wahl, M.C. The thermodynamic influence of trapped water molecules on a protein-ligand interaction. Angew. Chem. Int. Ed. Engl. 2009, 48, 5207–5210. [Google Scholar] [CrossRef] [PubMed]
- Howard, E.I.; Sanishvili, R.; Cachau, R.E.; Mitschler, A.; Chevrier, B.; Barth, P.; Lamour, V.; Van Zandt, M.; Sibley, E.; Bon, C.; et al. Ultrahigh resolution drug design I: Details of interactions in human aldose reductase-inhibitor complex at 0.66 A. Proteins 2004, 55, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.S.; Devedjiev, Y.; Derewenda, U.; Derewenda, Z.S. The PDZ2 domain of syntenin at ultra-high resolution: Bridging the gap between macromolecular and small molecule crystallography. J. Mol. Biol. 2004, 338, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Teeter, M.; Weckert, E.; Lamzin, V.S. Crystal structure of small protein crambin at 0.48 A resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Takeda, K.; Miki, K. Charge-density analysis of an iron-sulfur protein at an ultra-high resolution of 0.48 Å. Nature 2016, 534, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, M.G.; Mason, S.A.; Mossou, E.; Haertlein, M.; Forsyth, V.T. The cryofrozen atomic resolution X-ray crystal structure of perdeuterated Pyrococcus furiosus Rubredoxin (100K, 0.59A resolution). Protein Data Bank 2017. [Google Scholar] [CrossRef]
- Zenker, J.; Stettner, M.; Ruskamo, S.; Domenech-Estevez, E.; Baloui, H.; Medard, J.J.; Verheijen, M.H.; Brouwers, J.F.; Kursula, P.; Kieseier, B.C.; et al. A role of peripheral myelin protein 2 in lipid homeostasis of myelinating schwann cells. Glia 2014. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Harel, T.; Gambin, T.; Kousi, M.; Griffin, L.B.; Francescatto, L.; Ozes, B.; Karaca, E.; Jhangiani, S.N.; Bainbridge, M.N.; et al. Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep. 2015, 12, 1169–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.B.; Joo, J.; Hyun, Y.S.; Kwak, G.; Choi, Y.R.; Yeo, H.K.; Jwa, D.H.; Kim, E.J.; Mo, W.M.; Nam, S.H.; et al. A Mutation in PMP2 Causes Dominant Demyelinating Charcot-Marie-Tooth Neuropathy. PLoS Genet. 2016, 12, e1005829. [Google Scholar] [CrossRef] [PubMed]
- Motley, W.W.; Palaima, P.; Yum, S.W.; Gonzalez, M.A.; Tao, F.; Wanschitz, J.V.; Strickland, A.V.; Löscher, W.N.; De Vriendt, E.; Koppi, S.; et al. De novo PMP2 mutations in families with type 1 Charcot-Marie-Tooth disease. Brain 2016, 139, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Punetha, J.; Mackay-Loder, L.; Harel, T.; Coban-Akdemir, Z.; Jhangiani, S.N.; Gibbs, R.A.; Lee, I.; Terespolsky, D.; Lupski, J.R.; Posey, J.E. Identification of a pathogenic PMP2 variant in a multi-generational family with CMT type 1: Clinical gene panels versus genome-wide approaches to molecular diagnosis. Mol. Genet. Metab. 2018, 125, 302–304. [Google Scholar] [CrossRef]
- Ruskamo, S.; Nieminen, T.; Kristiansen, C.K.; Vatne, G.H.; Baumann, A.; Hallin, E.I.; Raasakka, A.; Joensuu, P.; Bergmann, U.; Vattulainen, I.; et al. Molecular mechanisms of Charcot-Marie-Tooth neuropathy linked to mutations in human myelin protein P2. Sci. Rep. 2017, 7, 6510. [Google Scholar] [CrossRef]
- Ruskamo, S.; Yadav, R.P.; Sharma, S.; Lehtimaki, M.; Laulumaa, S.; Aggarwal, S.; Simons, M.; Burck, J.; Ulrich, A.S.; Juffer, A.H.; et al. Atomic resolution view into the structure-function relationships of the human myelin peripheral membrane protein P2. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Majava, V.; Polverini, E.; Mazzini, A.; Nanekar, R.; Knoll, W.; Peters, J.; Natali, F.; Baumgärtel, P.; Kursula, I.; Kursula, P. Structural and functional characterization of human peripheral nervous system myelin protein P2. PLoS ONE 2010, 5, e10300. [Google Scholar] [CrossRef] [PubMed]
- Laulumaa, S.; Blakeley, M.P.; Raasakka, A.; Moulin, M.; Hartlein, M.; Kursula, P. Production, crystallization and neutron diffraction of fully deuterated human myelin peripheral membrane protein P2. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 1391–1395. [Google Scholar] [CrossRef]
- Kursula, P.; Laulumaa, S. Synchrotron diffraction images for the 0.86-Å crystal structure of hydrogenated human myelin protein P2. Zenodo 2019. [Google Scholar] [CrossRef]
- Kursula, P.; Laulumaa, S. Synchrotron diffraction images for the 0.72-Å crystal structure of perdeuterated human myelin protein P2. Zenodo 2019. [Google Scholar] [CrossRef]
- Manikandan, K.; Ramakumar, S. The occurrence of C--H.O hydrogen bonds in alpha-helices and helix termini in globular proteins. Proteins 2004, 56, 768–781. [Google Scholar] [CrossRef]
- Laulumaa, S.; Nieminen, T.; Raasakka, A.; Krokengen, O.C.; Safaryan, A.; Hallin, E.I.; Brysbaert, G.; Lensink, M.F.; Ruskamo, S.; Vattulainen, I.; et al. Structure and dynamics of a human myelin protein P2 portal region mutant indicate opening of the β barrel in fatty acid binding proteins. BMC Struct. Biol. 2018, 18, 8. [Google Scholar] [CrossRef]
- Flocco, M.M.; Mowbray, S.L. Planar stacking interactions of arginine and aromatic side-chains in proteins. J. Mol. Biol. 1994, 235, 709–717. [Google Scholar] [CrossRef]
- Wang, J.; Dauter, M.; Alkire, R.; Joachimiak, A.; Dauter, Z. Triclinic lysozyme at 0.65 A resolution. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 1254–1268. [Google Scholar] [CrossRef] [PubMed]
- Bacarizo, J.; Camara-Artigas, A. Atomic resolution structures of the c-Src SH3 domain in complex with two high-affinity peptides from classes I and II. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Gehmlich, K.; Pinotsis, N.; Hayess, K.; van der Ven, P.F.; Milting, H.; El Banayosy, A.; Körfer, R.; Wilmanns, M.; Ehler, E.; Fürst, D.O. Paxillin and ponsin interact in nascent costameres of muscle cells. J. Mol. Biol. 2007, 369, 665–682. [Google Scholar] [CrossRef] [PubMed]
- Kursula, P.; Kursula, I.; Lehmann, F.; Zou, P.; Song, Y.H.; Wilmanns, M. Crystal structure of the yeast Bzz1 first SH3 domain at 0.97-A resolution. Protein Data Bank 2006. [Google Scholar] [CrossRef]
- Hoelz, A.; Janz, J.M.; Lawrie, S.D.; Corwin, B.; Lee, A.; Sakmar, T.P. Crystal structure of the SH3 domain of betaPIX in complex with a high affinity peptide from PAK2. J. Mol. Biol. 2006, 358, 509–522. [Google Scholar] [CrossRef]
- Kursula, P.; Kursula, I.; Lehmann, F.; Song, Y.H.; Wilmanns, M. 0.97-A structure of the SH3 domain of bbc1. Protein Data Bank 2005. [Google Scholar] [CrossRef]
- Matsuoka, S.; Sugiyama, S.; Matsuoka, D.; Hirose, M.; Lethu, S.; Ano, H.; Hara, T.; Ichihara, O.; Kimura, S.R.; Murakami, S.; et al. Water-mediated recognition of simple alkyl chains by heart-type fatty-acid-binding protein. Angew. Chem. Int. Ed. Engl. 2015, 54, 1508–1511. [Google Scholar] [CrossRef]
- Kursula, I.; Wierenga, R.K. Crystal structure of triosephosphate isomerase complexed with 2-phosphoglycolate at 0.83-A resolution. J. Biol. Chem. 2003, 278, 9544–9551. [Google Scholar] [CrossRef] [PubMed]
- Gayathri, P.; Banerjee, M.; Vijayalakshmi, A.; Balaram, H.; Balaram, P.; Murthy, M.R. Biochemical and structural characterization of residue 96 mutants of Plasmodium falciparum triosephosphate isomerase: Active-site loop conformation, hydration and identification of a dimer-interface ligand-binding site. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Raasakka, A.; Myllykoski, M.; Laulumaa, S.; Lehtimäki, M.; Härtlein, M.; Moulin, M.; Kursula, I.; Kursula, P. Determinants of ligand binding and catalytic activity in the myelin enzyme 2’,3’-cyclic nucleotide 3’-phosphodiesterase. Sci. Rep. 2015, 5, 16520. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, A.; Pakendorf, T.; Reime, B.; Meyer, J.; Fischer, P.; Stübe, N.; Panneerselvam, S.; Lorbeer, O.; Stachnik, K.; Warmer, M. Status of the crystallography beamlines at PETRA III. Euro. Phys. J. Plus 2016, 131, 56. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Cryst. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Cianci, M.; Bourenkov, G.; Pompidor, G.; Karpics, I.; Kallio, J.; Bento, I.; Roessle, M.; Cipriani, F.; Fiedler, S.; Schneider, T.R. P13, the EMBL macromolecular crystallography beamline at the low-emittance PETRA III ring for high- and low-energy phasing with variable beam focusing. J. Synchrotron. Radiat. 2017, 24, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Karplus, P.A.; Diederichs, K. Linking crystallographic model and data quality. Science 2012, 336, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wing, R.A. Diamonds in the rough: A strong case for the inclusion of weak-intensity X-ray diffraction data. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1491–1497. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. D 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Merritt, E.A. Expanding the model: Anisotropic displacement parameters in protein structure refinement. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 1109–1117. [Google Scholar] [CrossRef]
- Schrödinger, L.L.C. The PyMOL Molecular Graphics System, version 1.2r3pre; Schrödinger: New York, NY, USA, 2009. [Google Scholar]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the P2 protein and its expression vector are available from the authors upon reasonable request. Raw diffraction images are available through zenodo.org, and the refined coordinates and structure factors from the Protein Data Bank. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | d-P2 | h-P2 |
|---|---|---|
| Space group | C2 | P41212 |
| Unit cell dimensions | a = 112.18 Å, b = 36.21 Å, c = 31.11 Å, α = γ = 90°, β = 97.03° | a = b = 57.93 Å, c = 101.32 Å, α = β = γ = 90° |
| Wavelength (Å) | 0.7443 | 0.8266 |
| Resolution range (Å) | 50–0.72 (0.74–0.72) | 30–0.86 (0.88–0.86) |
| <I/σ(I)> | 15.8 (0.9) | 14.7 (1.1) |
| Rsym (%) | 3.6 (121.4) | 6.3 (169.9) |
| Rmeas (%) | 3.9 (143.5) | 6.8 (183.0) |
| Completeness (%) | 91.0 (53.2) | 94.4 (87.1) |
| Redundancy | 4.7 (3.3) | 7.2 (7.0) |
| CC1/2 (%) | 99.9 (40.5) | 99.8 (43.3) |
| Wilson B factor (Å2) | 9.0 | 11.3 |
| Mosaicity (°) | 0.053 | 0.067 |
| Rcryst (%) | 10.4 | 9.9 |
| Rfree (%) | 11.1 | 11.8 |
| rmsd bond lengths (Å) | 0.020 | 0.020 |
| rmsd bond angles (°) | 1.8 | 1.9 |
| Average B factor (Å2); protein, ligand, solvent | 10.7, 14.1, 21.4 | 11.0, 11.6, 21.6 |
| Ramachandran favoured/allowed (%); Molprobity score (percentile) | 100/100; 1.30 (85th) | 99.2/100; 1.25 (86th) |
| Mean anisotropy;protein, ligand, solvent | 0.41 ± 0.13, 0.41 ± 0.14, 0.40 ± 0.15 | 0.49 ± 0.15, 0.46 ± 0.12, 0.41 ± 0.16 |
| PDB entry | 6S2M | 6S2S |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laulumaa, S.; Kursula, P. Sub-Atomic Resolution Crystal Structures Reveal Conserved Geometric Outliers at Functional Sites. Molecules 2019, 24, 3044. https://doi.org/10.3390/molecules24173044
Laulumaa S, Kursula P. Sub-Atomic Resolution Crystal Structures Reveal Conserved Geometric Outliers at Functional Sites. Molecules. 2019; 24(17):3044. https://doi.org/10.3390/molecules24173044
Chicago/Turabian StyleLaulumaa, Saara, and Petri Kursula. 2019. "Sub-Atomic Resolution Crystal Structures Reveal Conserved Geometric Outliers at Functional Sites" Molecules 24, no. 17: 3044. https://doi.org/10.3390/molecules24173044
APA StyleLaulumaa, S., & Kursula, P. (2019). Sub-Atomic Resolution Crystal Structures Reveal Conserved Geometric Outliers at Functional Sites. Molecules, 24(17), 3044. https://doi.org/10.3390/molecules24173044