
Enlargement of a Modular System—Synthesis and Characterization of an s-Triazine-Based Carboxylic Acid Ester Bearing a Galactopyranosyl Moiety and an Enormous Boron Load

 , ,
, , 
Abstract
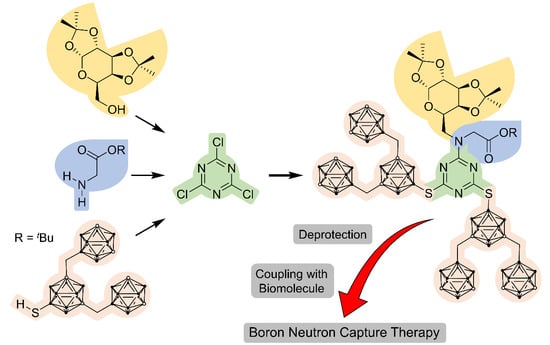
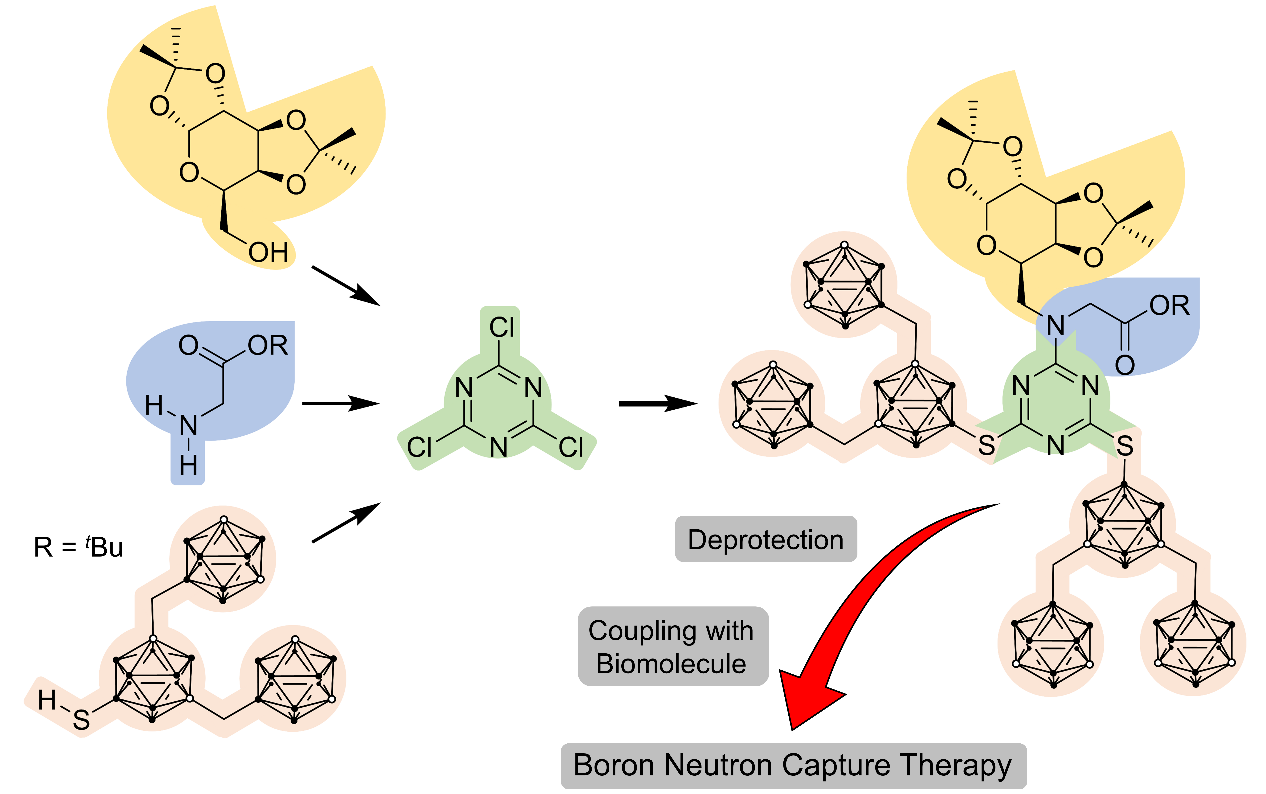
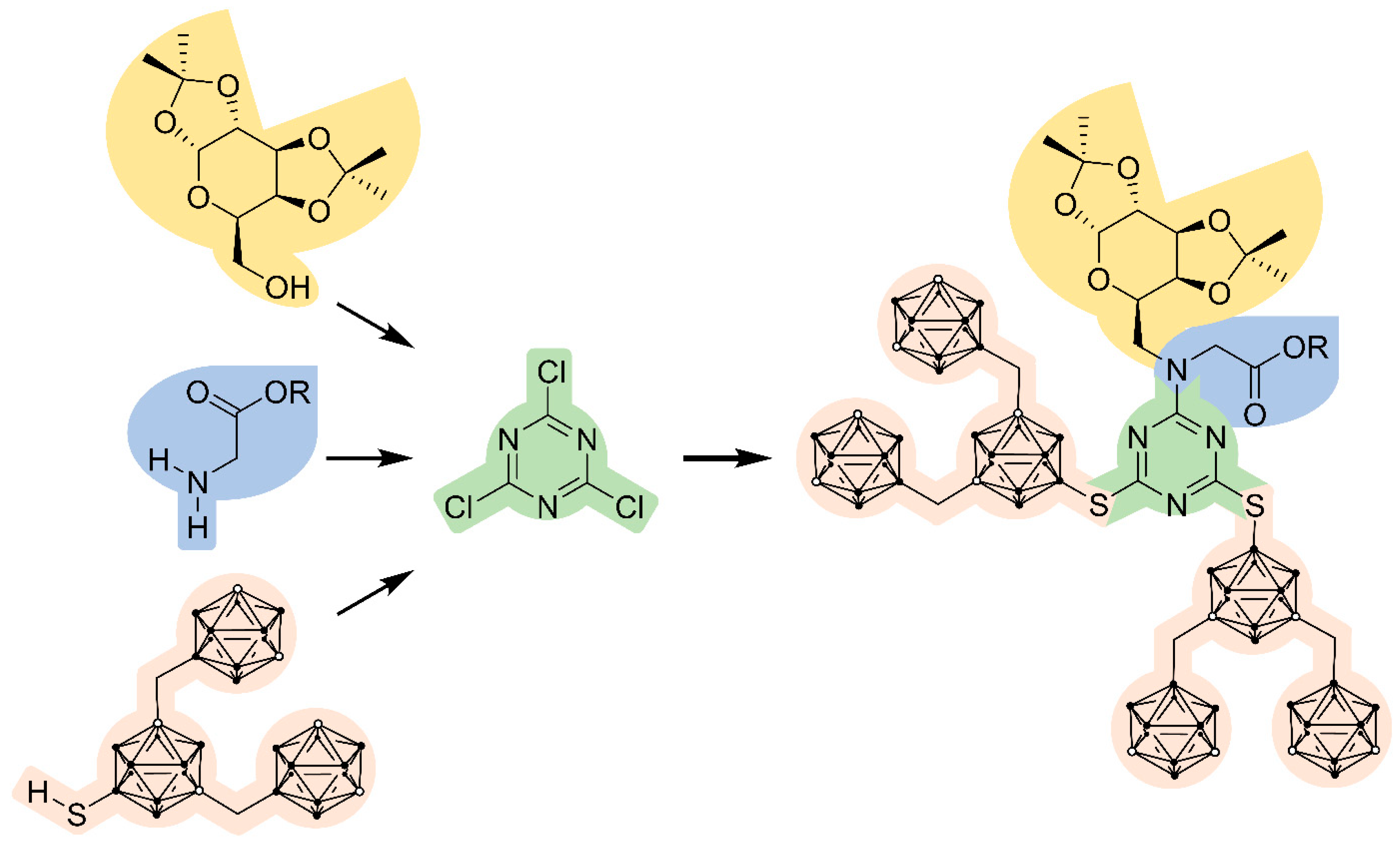
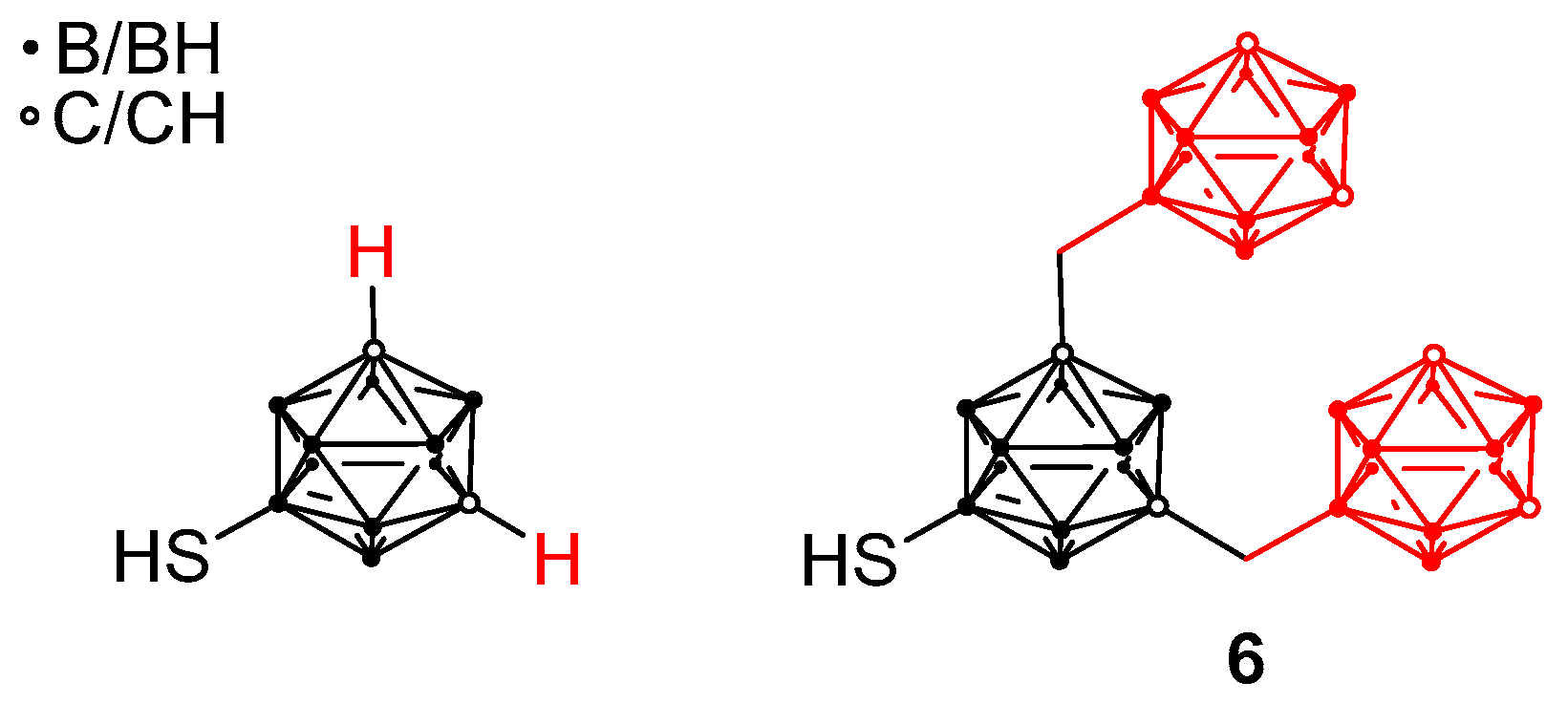
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
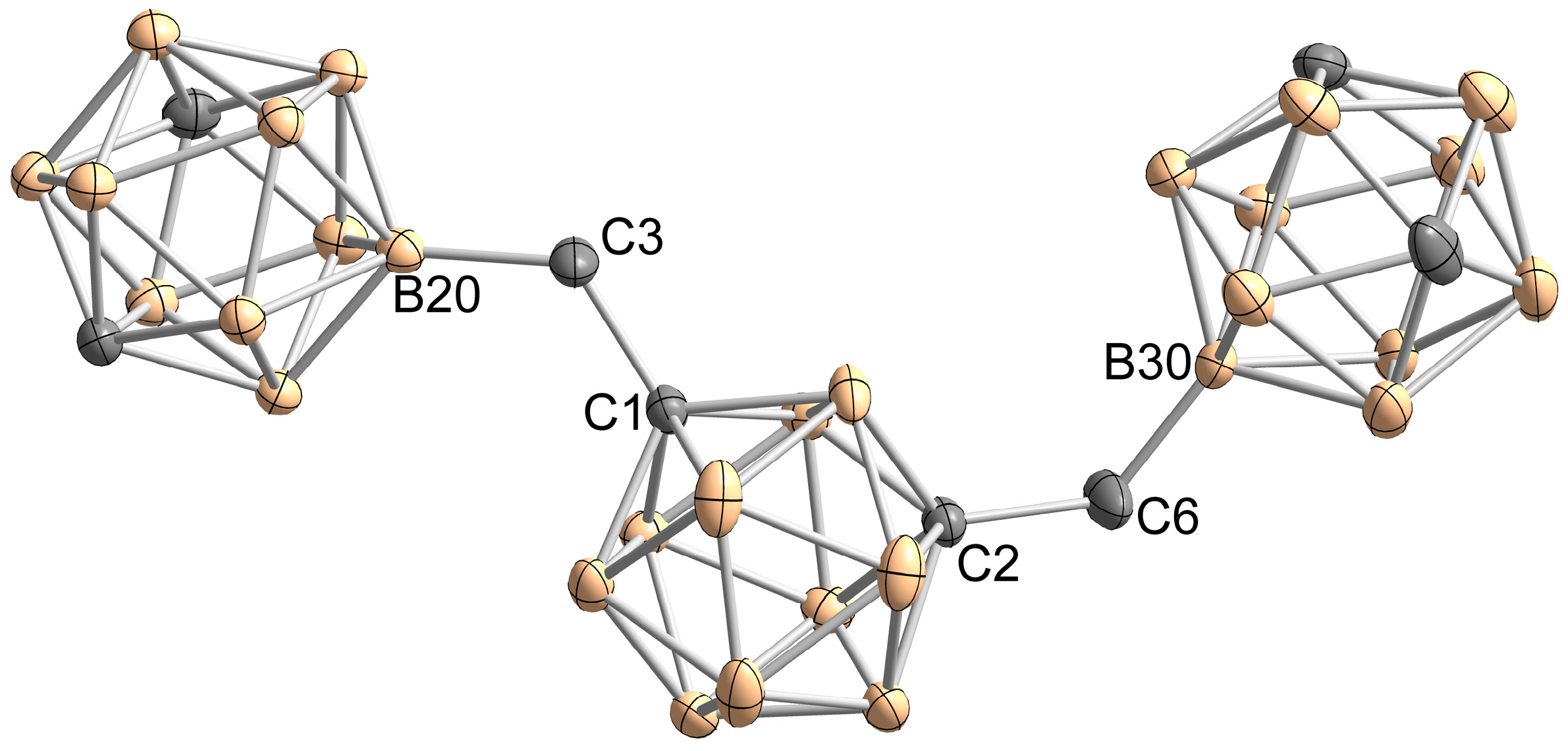
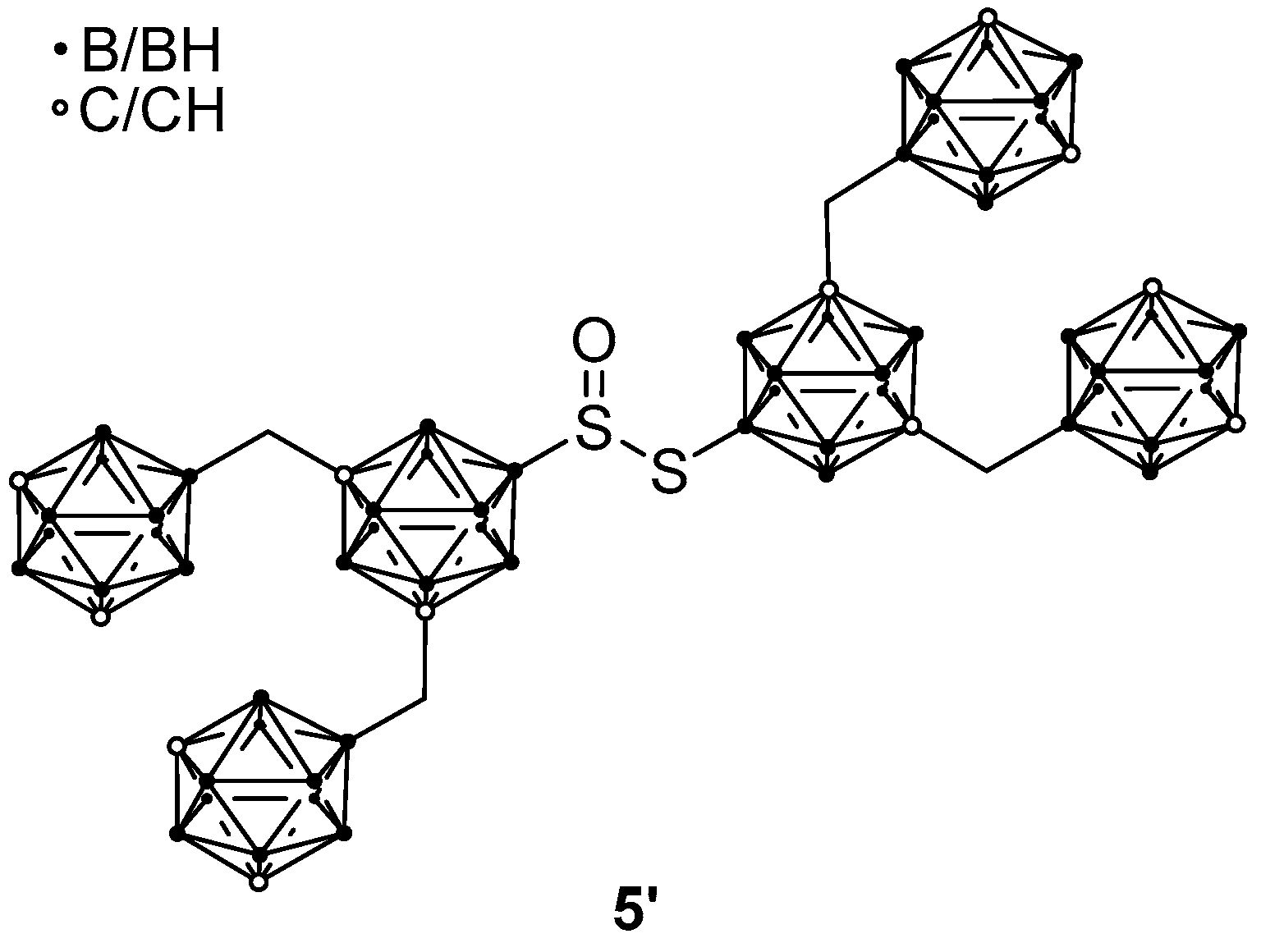
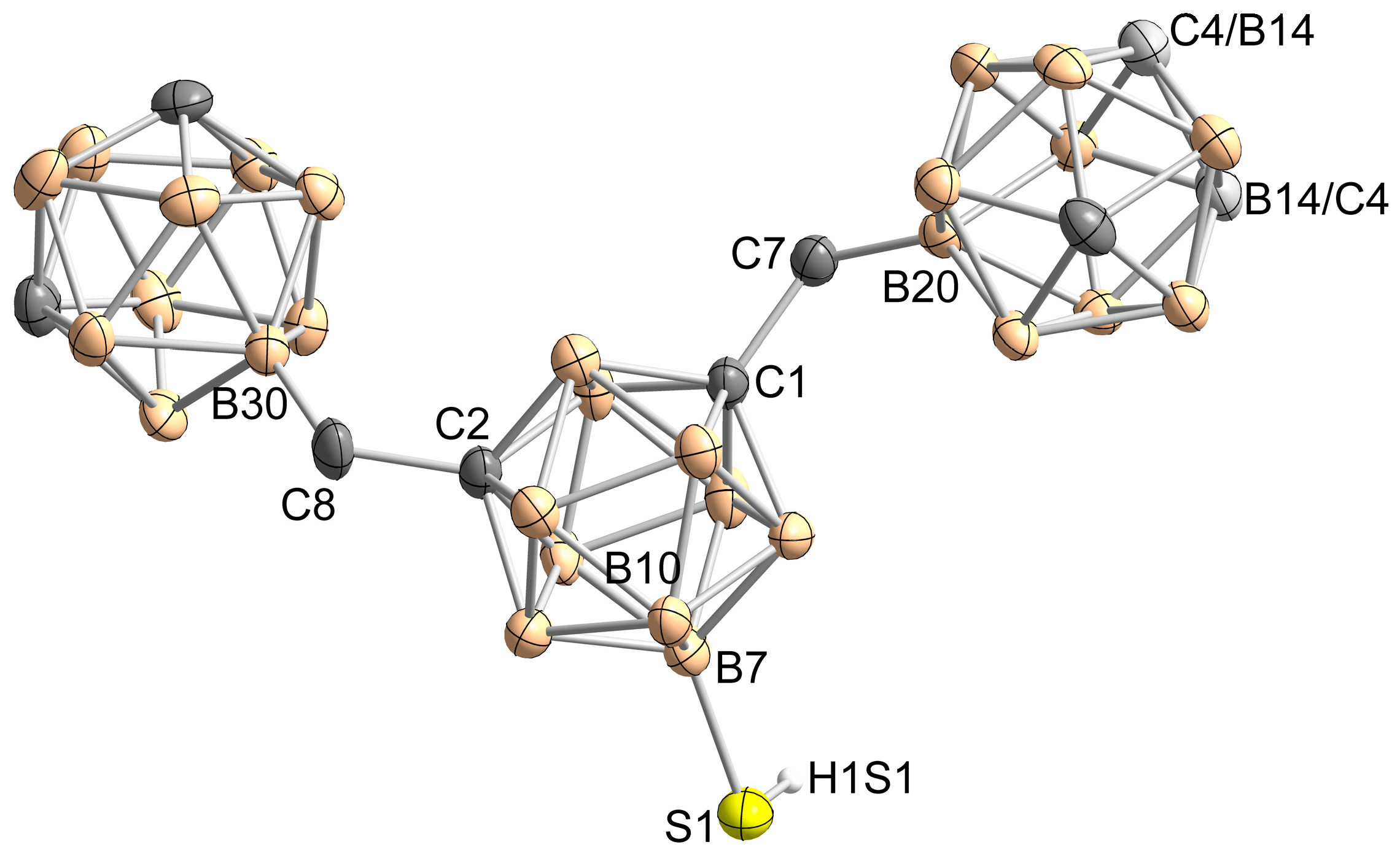
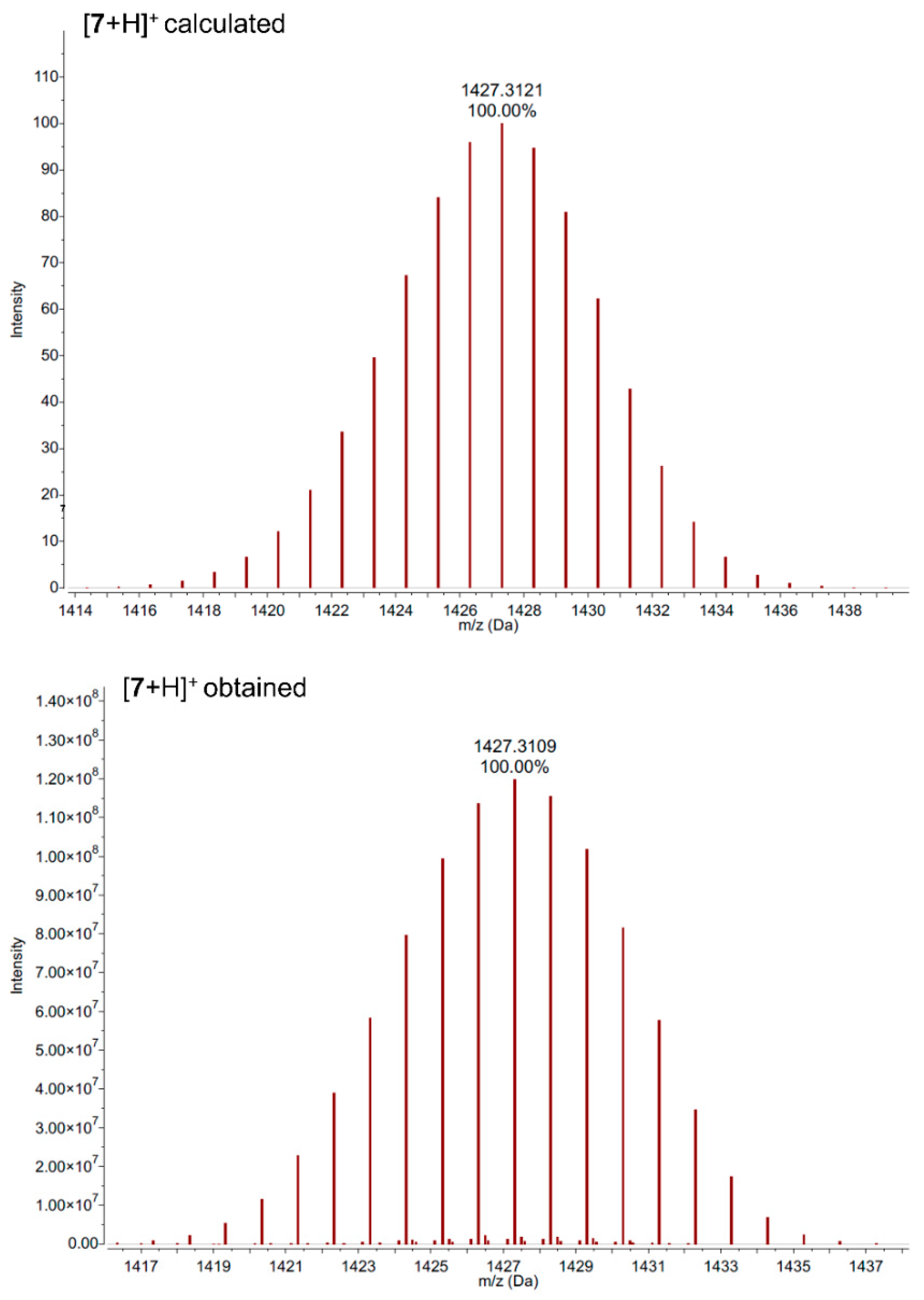
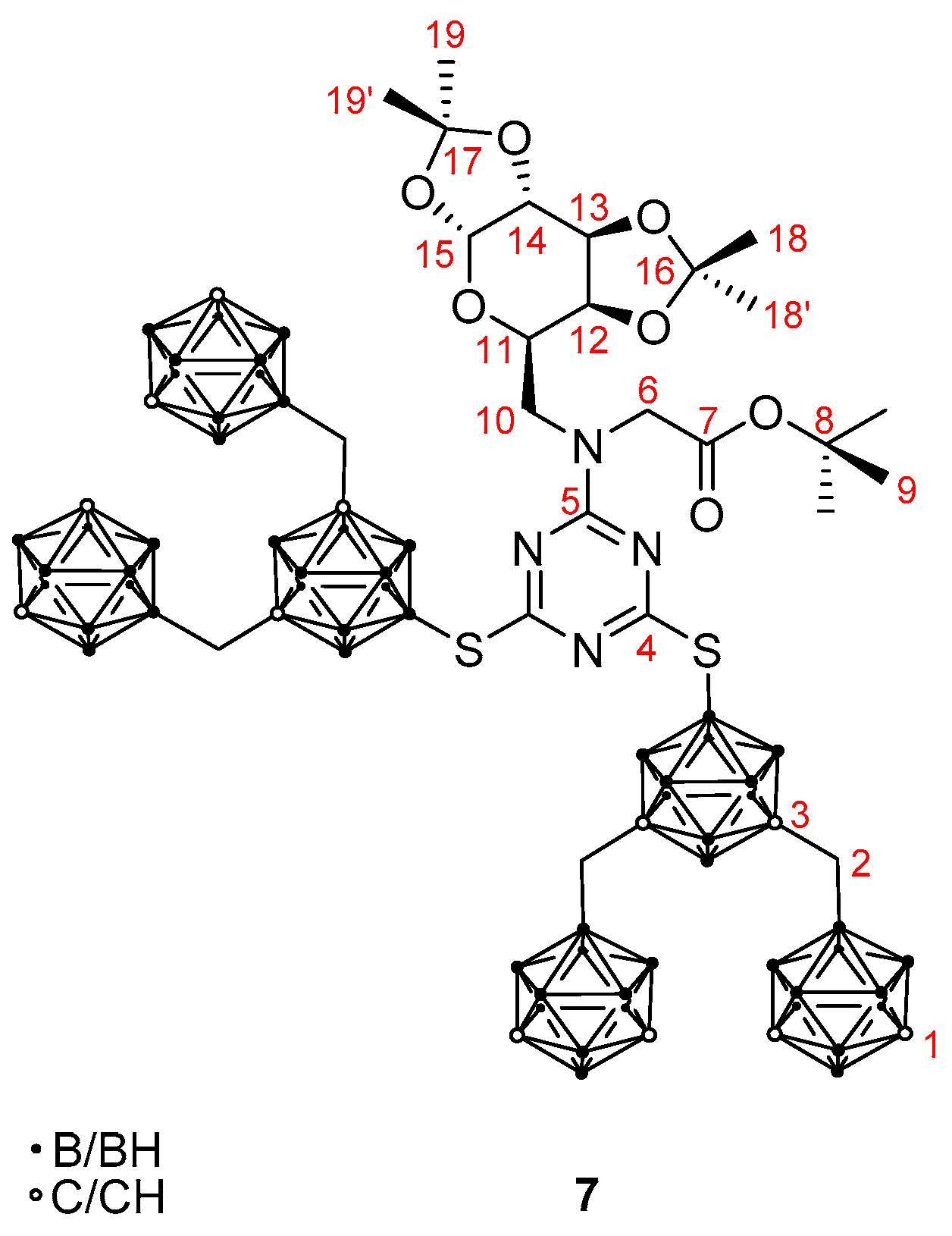
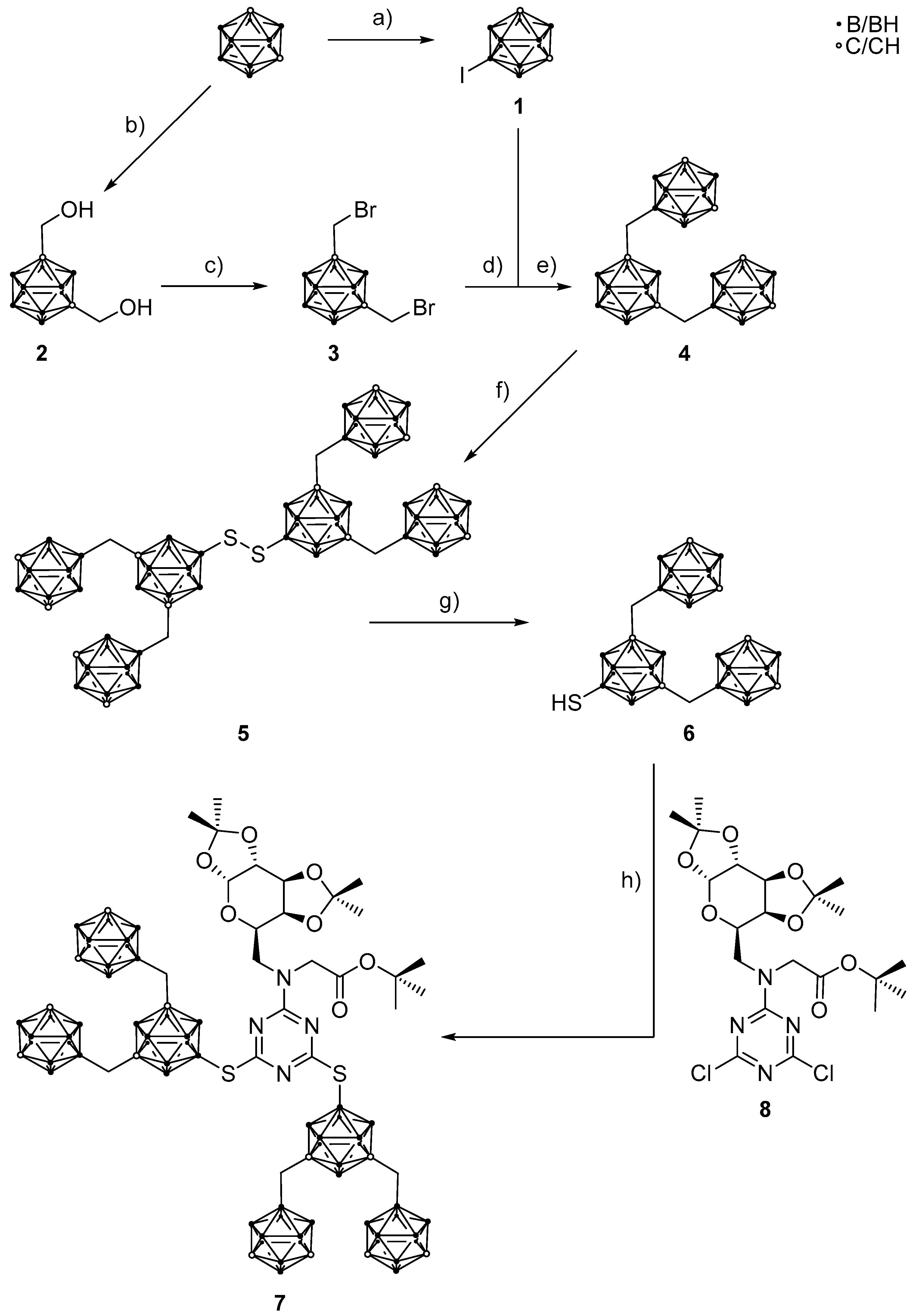
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Locher, G.L. Biological Effects and Therapeutic Possibilities of Neutrons. Am. J. Roentgenol. Radium Therapy 1936, 36, 1–18. [Google Scholar]
- Sweet, W.H. The Uses of Nuclear Disintegration in the Diagnosis and Treatment of Brain Tumor. N. Engl. J. Med. 1951, 245, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Hawthorne, M.F. The Role of Chemistry in the Development of Boron Neutron Capture Therapy of Cancer. Angew. Chem. Int. Ed. 1993, 32, 950–984. [Google Scholar] [CrossRef]
- Barth, R.F.; Mi, P.; Yang, W. Boron Delivery Agents for Neutron Capture Therapy of Cancer. Cancer Commun. 2018, 38, 35. [Google Scholar] [CrossRef] [PubMed]
- Sivaev, I.B.; Bregadze, V.V. Polyhedral Boranes for Medical Applications: Current Status and Perspectives. Eur. J. Inorg. Chem. 2009, 2009, 1433–1450. [Google Scholar] [CrossRef]
- Kabalka, G.W. Recent Developments in Boron Neutron Capture Therapy. Expert Opin. Ther. Pat. 1998, 8, 545–551. [Google Scholar] [CrossRef]
- Hatanaka, H. A revised Boron-Neutron Capture Therapy for Malignant Brain Tumors. J. Neurol. 1975, 209, 81–94. [Google Scholar] [CrossRef]
- Barth, R.F.; Soloway, A.H.; Fairchild, R.G. Boron Neutron Capture Therapy of Cancer. Cancer Res. 1990, 50, 1061–1070. [Google Scholar] [CrossRef]
- Soloway, A.H.; Tjarks, W.; Barnum, B.A.; Rong, F.-G.; Barth, R.F.; Codogni, I.M.; Wilson, J.G. The Chemistry of Neutron Capture Therapy. Chem. Rev. 1998, 98, 1515–1562. [Google Scholar] [CrossRef]
- Calabrese, G.; Daou, A.; Barbu, E.; Tsibouklis, J. Towards Carborane-functionalised Structures for the Treatment of Brain Cancer. Drug Discov. Today 2018, 23, 63–75. [Google Scholar] [CrossRef]
- Sears, V.F. Neutron Scattering Lengths and Cross Sections. Neutron News 1992, 3, 26–37. [Google Scholar] [CrossRef]
- Petry, W.; Neuhaus, J. Neutronen nach Maß. Physik J. 2007, 6, 31–37. [Google Scholar]
- Pan, X.Q.; Wang, H.; Shukla, S.; Sekido, M.; Adams, D.M.; Tjarks, W.; Barth, R.F.; Lee, R.J. Boron-Containing Folate Receptor-Targeted Liposomes as Potential Delivery Agents for Neutron Capture Therapy. Bioconjugate Chem. 2002, 13, 435–442. [Google Scholar] [CrossRef]
- Shukla, S.; Wu, G.; Chatterjee, M.; Yang, W.; Sekido, M.; Diop, L.A.; Müller, R.; Sudimack, J.J.; Lee, R.J.; Barth, R.F.; et al. Synthesis and Biological Evaluation of Folate Receptor-Targeted Boronated PAMAM Dendrimers as Potential Agents for Neutron Capture Therapy. Bioconjugate Chem. 2003, 14, 158–167. [Google Scholar] [CrossRef]
- Dubey, R.; Kushal, S.; Mollard, A.; Vojtovich, L.; Oh, P.; Levin, M.D.; Schnitzer, J.E.; Zharov, I.; Olenyuk, B.Z. Tumor Targeting, Trifunctional Dendritic Wedge. Bioconjugate Chem. 2015, 26, 78–89. [Google Scholar] [CrossRef]
- Feng, B.; Tomizawa, K.; Michiue, H.; Miyatake, S.-I.; Han, X.-J.; Fujimura, A.; Seno, M.; Kirihata, M.; Matsui, H. Delivery of Sodium Borocaptate to Glioma Cells using Immunoliposome Conjugated with Anti-EGFR Antibodies by ZZ-His. Biomaterials 2009, 30, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Michiue, H.; Sakurai, Y.; Kondo, N.; Kitamatsu, M.; Bin, F.; Nakajima, K.; Hirota, Y.; Kawabata, S.; Nishiki, T.-I.; Ohmori, I.; et al. The Acceleration of Boron Neutron Capture Therapy using Multi-linked Mercaptoundecahydrododecaborate (BSH) fused Cell-penetrating Peptide. Biomaterials 2014, 35, 3396–3405. [Google Scholar] [CrossRef]
- Iguchi, Y.; Michiue, H.; Kitamatsu, M.; Hayashi, Y.; Takenaka, F.; Nishiki, T.-i.; Matsui, H. Tumor-specific Delivery of BSH-3R for Boron Neutron Capture Therapy and Positron Emission Tomography Imaging in a Mouse Brain Tumor Model. Biomaterials 2015, 56, 10–17. [Google Scholar] [CrossRef]
- Doi, A.; Kawabata, S.; Iida, K.; Yokoyama, K.; Kajimoto, Y.; Kuroiwa, T.; Shirakawa, T.; Kirihata, M.; Kasaoka, S.; Maruyama, K.; et al. Tumor-specific Targeting of Sodium Borocaptate (BSH) to Malignant Glioma by Transferrin-PEG Liposomes: a Modality for Boron Neutron Capture Therapy. J. Neurooncol. 2008, 87, 287–294. [Google Scholar] [CrossRef]
- Romero-Canelón, I.; Phoenix, B.; Pitto-Barry, A.; Tran, J.; Soldevila-Barreda, J.J.; Kirby, N.; Green, S.; Sadler, P.J.; Barry, N.P.E. Arene Ruthenium Dithiolato–carborane Complexes for Boron Neutron Capture Therapy (BNCT). J. Organomet. Chem. 2015, 796, 17–25. [Google Scholar] [CrossRef]
- Kettenbach, K.; Schieferstein, H.; Grunewald, C.; Iffland, D.; Reffert, L.M.; Hampel, G.; Schütz, C.L.; Bings, N.H.; Ross, T.L. Synthesis and Evaluation of Boron Folates for Boron-Neutron-Capture-Therapy (BNCT). Radiochim. Acta 2015, 103, 799–809. [Google Scholar] [CrossRef]
- Mier, W.; Gabel, D.; Haberkorn, U.; Eisenhut, M. Conjugation of the closo-Borane Mercaptoundecahydrododecaborate (BSH) to a Tumour Selective Peptide. Z. Anorg. Allg. Chem. 2004, 630, 1258–1262. [Google Scholar] [CrossRef]
- Lai, C.-H.; Lin, Y.-C.; Chou, F.-I.; Liang, C.-F.; Lin, E.-W.; Chuang, Y.-J.; Lin, C.-C. Design of Multivalent Galactosyl Carborane as a Targeting Specific Agent for Potential Application to Boron Neutron Capture Therapy. Chem. Commun. 2012, 48, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Otero, R.; Seoane, S.; Sigüeiro, R.; Belorusova, A.Y.; Maestro, M.A.; Pérez-Fernández, R.; Rochel, N.; Mouriño, A. Carborane-based Design of a Potent Vitamin D Receptor Agonist. Chem. Sci. 2016, 7, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Worm, D.J.; Els-Heindl, S.; Kellert, M.; Kuhnert, R.; Saretz, S.; Koebberling, J.; Riedl, B.; Hey-Hawkins, E.; Beck-Sickinger, A.G. A Stable meta-Carborane Enables the Generation of Boron-rich Peptide Agonists Targeting the Ghrelin Receptor. J. Pept. Sci. 2018, 32, e3119. [Google Scholar] [CrossRef]
- Ciofani, G.; Raffa, V.; Menciassi, A.; Cuschieri, A. Folate Functionalized Boron Nitride Nanotubes and their Selective Uptake by Glioblastoma Multiforme Cells: Implications for their Use as Boron Carriers in Clinical Boron Neutron Capture Therapy. Nanoscale Res. Lett. 2008, 4, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kueffer, P.J.; Maitz, C.A.; Khan, A.A.; Schuster, S.A.; Shlyakhtina, N.I.; Jalisatgi, S.S.; Brockman, J.D.; Nigg, D.W.; Hawthorne, M.F. Boron Neutron Capture Therapy Demonstrated in Mice Bearing EMT6 Tumors Following Selective Delivery of Boron by Rationally Designed Liposomes. Proc. Natl. Acad. Sci. USA 2013, 110, 6512–6517. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, J.; Goldhaber, M. Disintegration by Slow Neutrons. Nature 1935, 135, 65. [Google Scholar] [CrossRef]
- Taylor, H.J.; Goldhaber, M. Detection of Nuclear Disintegration in a Photographic Emulsion. Nature 1935, 135, 341. [Google Scholar] [CrossRef]
- Hattori, Y.; Kusaka, S.; Mukumoto, M.; Uehara, K.; Asano, T.; Suzuki, M.; Masunaga, S.-I.; Ono, K.; Tanimori, S.; Kirihata, M. Biological Evaluation of Dodecaborate-Containing L-Amino Acids for Boron Neutron Capture Therapy. J. Med. Chem. 2012, 55, 6980–6984. [Google Scholar] [CrossRef]
- Hartman, T.; Carlsson, J. Radiation Dose Heterogeneity in Receptor and Antigen-mediated Boron Neutron Capture Therapy. Radiother. Oncol. 1994, 31, 61–75. [Google Scholar] [CrossRef]
- Ahrens, V.M.; Frank, R.; Stadlbauer, S.; Beck-Sickinger, A.G.; Hey-Hawkins, E. Incorporation of ortho-Carbaboranyl-Nε-Modified L-lysine into Neuropeptide Y Receptor Y1- and Y2-Selective Analogues. J. Med. Chem. 2011, 54, 2368–2377. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Saravanakumar, G.; Park, J.H.; Park, K. Hyaluronic Acid-based Nanocarriers for Intracellular Targeting: Interfacial Interactions with Proteins in Cancer. Colloid Surface B 2012, 99, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Agosteo, S.; Colautti, P.; Esposito, J.; Fazzi, A.; Introini, M.V.; Pola, A. Characterization of the Energy Distribution of Neutrons generated by 5 MeV Protons on a Thick Beryllium Target at Different Emission Angles. Appl. Radiat. Isotopes 2011, 69, 1664–1667. [Google Scholar] [CrossRef] [PubMed]
- Kumada, H.; Matsumura, A.; Sakurai, H.; Sakae, T.; Yoshioka, M.; Kobayashi, H.; Matsumoto, H.; Kiyanagi, Y.; Shibata, T.; Nakashima, H. Project for the Development of the Linac-based NCT Facility in University of Tsukuba. Appl. Radiat. Isotopes 2014, 88, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Durisi, E.; Alikaniotis, K.; Borla, O.; Bragato, F.; Costa, M.; Giannini, G.; Monti, V.; Visca, L.; Vivaldo, G.; Zanini, A. Design and Simulation of an Optimized e-Linac-based Neutron Source for BNCT Research. Appl. Radiat. Isotopes 2015, 106, 63–67. [Google Scholar] [CrossRef]
- Onishi, T.; Kumada, H.; Takada, K.; Naito, F.; Kurihara, T.; Sakae, T. Investigation of the Neutron Spectrum Measurement Method for Dose Evaluation in Boron Neutron Capture Therapy. Appl. Radiat. Isotopes 2018, 140, 5–11. [Google Scholar] [CrossRef]
- Lerouge, F.; Viñas, C.; Teixidor, F.; Núñez, R.; Abreu, A.; Xochitiotzi, E.; Santillan, R.; Farfán, N. High Boron Content Carboranyl-functionalized Aryl Ether Derivatives Displaying Photoluminescent Properties. Dalton Trans. 2007, 92, 1898–1903. [Google Scholar] [CrossRef]
- Parrott, M.C.; Marchington, E.B.; Valliant, J.F.; Adronov, A. Synthesis and Properties of Carborane-Functionalized Aliphatic Polyester Dendrimers. J. Am. Chem. Soc. 2005, 127, 12081–12089. [Google Scholar] [CrossRef]
- Kawasaki, R.; Sasaki, Y.; Akiyoshi, K. Intracellular Delivery and Passive Tumor Targeting of a Self-assembled Nanogel Containing Carborane Clusters for Boron Neutron Capture Therapy. Biochem. Biophys. Res. Commun. 2017, 483, 147–152. [Google Scholar] [CrossRef]
- Mollard, A.; Zharov, I. Tricarboranyl Pentaerythritol-Based Building Block. Inorg. Chem. 2006, 45, 10172–10179. [Google Scholar] [CrossRef] [PubMed]
- Nar, I.; Bortolussi, S.; Postuma, I.; Atsay, A.; Berksun, E.; Viola, E.; Ferrari, C.; Cansolino, L.; Ricciardi, G.; Donzello, M.P.; et al. A Phthalocyanine-ortho-Carborane Conjugate for Boron Neutron Capture Therapy: Synthesis, Physicochemical Properties, and in Vitro Tests. ChemPlusChem 2019, 84, 345–351. [Google Scholar] [CrossRef]
- Bregadze, V.I. Dicarba-closo-dodecaboranes C2B10H12 and Their Derivatives. Chem. Rev. 1992, 92, 209–223. [Google Scholar] [CrossRef]
- Fox, M.A.; Gill, W.R.; Herbertson, P.L.; MacBride, J.A.H.; Wade, K. Deboronation of C-Substituted ortho- and meta-closo-Carboranes Using “wet” Fluoride Ion Solutions. Polyhedron 1996, 15, 565–571. [Google Scholar] [CrossRef]
- Kellert, M.; Worm, D.J.; Hoppenz, P.; Sárosi, M.B.; Lönnecke, P.; Riedl, B.; Koebberling, J.; Beck-Sickinger, A.G.; Hey-Hawkins, E. Modular Triazine-based Carborane-containing Carboxylic Acids - Synthesis and Characterisation of Potential Boron Neutron Capture Therapy Agents Made of Readily Accessible Building Blocks. Dalton Trans. 2019, 48, 10834–10844. [Google Scholar] [CrossRef] [PubMed]
- Kellert, M. s-Triazine-based Boron-rich Carboxylic Acids and Amines. The Journey of Modular Boron Neutron Capture Therapy Precursors with an Enhanced Boron Load. Ph.D. Thesis, Leipzig University, Leipzig, Germany, 2019. [Google Scholar]
- Hey-Hawkins, E.; Beck-Sickinger, A.G.; Kellert, M.; Kuhnert, R.; Saretz, S.; Riedl, B.; Bierer, D.; Koebberling, J.; Griebenow, N. Novel 1,7-Dicarba-closo-dodecaborane(12) (meta-Carborane)-derived Carboxylic Acids and Amines Suitable for Peptide Modification for Application in Boron Neutron Capture Therapy (BNCT). WO/2019/115617, 20 June 2019. [Google Scholar]
- Timofeev, S.V.; Bregadze, V.I.; Osipov, S.N.; Titanyuk, I.D.; Petrovskii, P.V.; Starikova, Z.A.; Glukhov, I.V.; Beletskaya, I.P. New Carborane-containing Amino Acids and Their Derivatives. Crystal Structures of N-Protected Carboranylalaninates. Russ. Chem. Bull. 2007, 56, 791–797. [Google Scholar] [CrossRef]
- Timofeev, S.V.; Prikaznova, E.A.; Starikova, Z.A.; Osipov, S.N.; Bregadze, V.I. Synthesis and Structure of Diethyl (1-benzyloxycarbonylamino-1-carboranyl-3,3,3-trifluoropropyl)phosphonate. Russ. Chem. Bull. 2013, 62, 1934–1937. [Google Scholar] [CrossRef]
- Smith, H.D.; Obenland, C.O.; Papetti, S. A New Series of Organoboranes. IX. The Preparation and Some Reactions of Sulfur-Carborane Derivatives. Inorg. Chem. 1966, 5, 1013–1015. [Google Scholar] [CrossRef]
- Reiner, J.; Alexander, R.P.; Schröder, H. A New Series of Organoboranes. VIII. The Reaction of Phosgene with the Dilithium Salts of o- and m-Carborane. Inorg. Chem. 1966, 5, 1460–1462. [Google Scholar] [CrossRef]
- Viñas, C.; Benakki, R.; Teixidor, F.; Casabó, J. Dimethoxyethane as a Solvent for the Synthesis of C-Monosubstituted o-Carborane Derivatives. Inorg. Chem. 1995, 34, 3844–3845. [Google Scholar] [CrossRef]
- Bauer, S.; Tschirschwitz, S.; Lönnecke, P.; Frank, R.; Kirchner, B.; Clarke, M.L.; Hey-Hawkins, E. Enantiomerically Pure Bis(phosphanyl)carbaborane(12) Compounds. Eur. J. Inorg. Chem. 2009, 2776–2788. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective Carbon-Carbon Bond Formation by Cross-Coupling of Grignard Reagents with Organic Halides. Catalysis by Nickel-Phosphine Complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar] [CrossRef]
- Zhao, J.; Huang, P.; Chen, G.; Zhan, M. Copper (I)-catalyzed Cross-coupling Reaction for the Synthesis of 1,2-Bis(3-butenyl)-1,2-dicarba-closo-dedocaborane. Inorg. Chem. Commun. 2012, 15, 321–323. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kiso, Y.; Zembayashi, M.; Fujioka, A.; Kodama, S.-i.; Nakajima, I.; Minato, A.; Kumada, M. Nickel-Phosphine Complex-Catalyzed Grignard Coupling. I. Cross-Coupling of Alkyl, Aryl, and Alkenyl Grignard Reagents with Aryl and Alkenyl Halides: General Scope and Limitations. Bull. Chem. Soc. Jpn. 1976, 49, 1958–1969. [Google Scholar] [CrossRef]
- Himmelspach, A.; Finze, M. Dicarba-closo-dodecaboranes with One and Two Ethynyl Groups Bonded to Boron. Eur. J. Inorg. Chem. 2010, 2012–2024. [Google Scholar] [CrossRef]
- Teixidor, F.; Sillanpää, R.; Pepiol, A.; Lupu, M.; Viñas, C. Synthesis of Globular Precursors. Chem. Eur. J. 2015, 21, 12778–12786. [Google Scholar] [CrossRef] [PubMed]
- Puga, A.V.; Teixidor, F.; Sillanpää, R.; Kivekäs, R.; Viñas, C. Synthesis of Quadruped-Shaped Polyfunctionalized o-Carborane Synthons. Chem. Commun. 2011, 47, 2252–2254. [Google Scholar] [CrossRef] [PubMed]
- Vaca, A.; Teixidor, F.; Kivekäs, R.; Sillanpää, R.; Viñas, C. A Solvent-free Regioselective Iodination Route of ortho-Carboranes. Dalton Trans. 2006, 4884–4885. [Google Scholar] [CrossRef] [PubMed]
- Lupu, M.; Zaulet, A.; Teixidor, F.; Sillanpää, R.; Viñas, C. Poly-iodinated-closo-1,2-C2B10 and nido-[7,8-C2B9]- Carborane Frameworks: Synthesis and Consequences. J. Organomet. Chem. 2015, 798, 171–181. [Google Scholar] [CrossRef]
- Zheng, Z.; Jiang, W.; Zinn, A.A.; Knobler, C.B.; Hawthorne, M.F. Facile Electrophilic Iodination of Icosahedral Carboranes. Synthesis of Carborane Derivatives with Boron-Carbon Bonds via the Palladium-Catalyzed Reaction of Diiodocarboranes with Grignard Reagents. Inorg. Chem. 1995, 34, 2095–2100. [Google Scholar] [CrossRef]
- Andrews, J.S.; Zayas, J.; Jones, M., Jr. 9-Iodo-o-carborane. Inorg. Chem. 1985, 24, 3715–3716. [Google Scholar] [CrossRef]
- Wingen, L.M.; Scholz, M.S. B-Cyanodicarba-closo-dodecaboranes: Facile Synthesis and Spectroscopic Features. Inorg. Chem. 2016, 55, 8274–8276. [Google Scholar] [CrossRef] [PubMed]
- Zakharkin, L.I. Synthesis and Some Reactions of 1-Halomethyl-m-carboranes. Zh. Obshch. Khim. 1981, 51, 357–361. [Google Scholar]
- Li, N.; Zeng, F.; Qu, D.; Zhang, J.; Shao, L.; Bai, Y. Synthesis and Characterization of Carborane-containing Polyester with Excellent Thermal and Ultrahigh Char Yield. J. Appl. Polym. Sci. 2016, 133, 44202. [Google Scholar] [CrossRef]
- Goto, T.; Ohta, K.; Suzuki, T.; Ohta, S.; Endo, Y. Design and Synthesis of Novel Androgen Receptor Antagonists with Sterically Bulky Icosahedral Carboranes. Bioorgan. Med. Chem. 2005, 13, 6414–6424. [Google Scholar] [CrossRef] [PubMed]
- Zakharkin, L.I. Synthesis of B-organyl-o-and-m-Carboranes by the Cross-Coupling of B-Iodo-o-and-m-Carboranes with Organozinc Compounds Catalyzed by Palladium Complexes. Russ. J. Gen. Chem. 1998, 68, 925–927. [Google Scholar]
- Zakharkin, L.I.; Ol’shevskaya, V.A.; Nesmeyanov’s, A.N. Synthesis of 9-Organyl-1,2 and 1,7-Dicarba-closo-dodecaboranes(12) via the Cross-Coupling Reactions Between Organozinc Compounds and 9-Iodo-1,2- or 1,7-Dicarba-closo-dodecaboranes. Syn. React. Inorg. Met.-Org. Chem. 1991, 21, 1041–1046. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Pisareva, I.V. A New Simple Method for the Production and Some Conversions of B-S Bond-containing o- and m-Carboranyl. Phosphorus Sulfur 1984, 20, 357–370. [Google Scholar] [CrossRef]
- Oae, S.; Takata, T.; Kim, Y.H. Oxidation of Unsymmetrical Disulfide and Thiosuldinic S-Esters with Peroxy Acids. Search for Formation of α-Disulfoxide as an Intermediate in the Electrophilic Oxidation of Thiosulfinic S-Ester. Bull. Chem. Soc. Jpn. 1982, 55, 2484–2494. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR Nomenclature: Nuclear Spin Properties and Conventions for Chemical Shifts. IUPAC Recommendations 2001. Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- MestReNova; v12.0.0-20080; Mestrelab Research S. L.: Santiago de Compostela, Spain, 2017.
- CrysAlis Pro: Data Collection and Data Reduction Software Package; Rigaku Oxford Diffraction: Tokyo, Japan, 2015.
- SCALE3 ABSPACK: Empirical Absorption Correction Using Sperical Harmonics, Implemented in SCALE3 ABSPACK Scaling Algorithm of CrysAlis Pro.; Rigaku Oxford Diffraction: Tokyo, Japan, 2015.
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. Diamond; v4.5.3; Crystal Impact GbR: Bonn, Germany, 1997–2019.
Sample Availability: Samples of the compounds are not available from the authors. |








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kellert, M.; Lönnecke, P.; Riedl, B.; Koebberling, J.; Hey-Hawkins, E. Enlargement of a Modular System—Synthesis and Characterization of an s-Triazine-Based Carboxylic Acid Ester Bearing a Galactopyranosyl Moiety and an Enormous Boron Load. Molecules 2019, 24, 3288. https://doi.org/10.3390/molecules24183288
Kellert M, Lönnecke P, Riedl B, Koebberling J, Hey-Hawkins E. Enlargement of a Modular System—Synthesis and Characterization of an s-Triazine-Based Carboxylic Acid Ester Bearing a Galactopyranosyl Moiety and an Enormous Boron Load. Molecules. 2019; 24(18):3288. https://doi.org/10.3390/molecules24183288
Chicago/Turabian StyleKellert, Martin, Peter Lönnecke, Bernd Riedl, Johannes Koebberling, and Evamarie Hey-Hawkins. 2019. "Enlargement of a Modular System—Synthesis and Characterization of an s-Triazine-Based Carboxylic Acid Ester Bearing a Galactopyranosyl Moiety and an Enormous Boron Load" Molecules 24, no. 18: 3288. https://doi.org/10.3390/molecules24183288
APA StyleKellert, M., Lönnecke, P., Riedl, B., Koebberling, J., & Hey-Hawkins, E. (2019). Enlargement of a Modular System—Synthesis and Characterization of an s-Triazine-Based Carboxylic Acid Ester Bearing a Galactopyranosyl Moiety and an Enormous Boron Load. Molecules, 24(18), 3288. https://doi.org/10.3390/molecules24183288