Characterization of New Oligosaccharides Obtained by An Enzymatic Cleavage of the Exopolysaccharide Produced by the Deep-Sea Bacterium Alteromonas infernus Using its Cell Extract

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results and Discussion
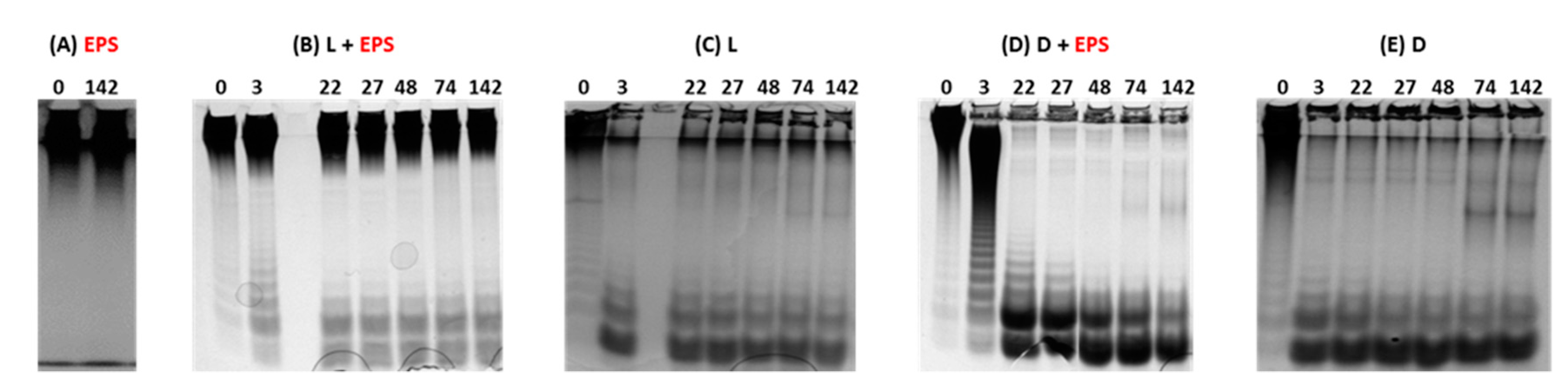
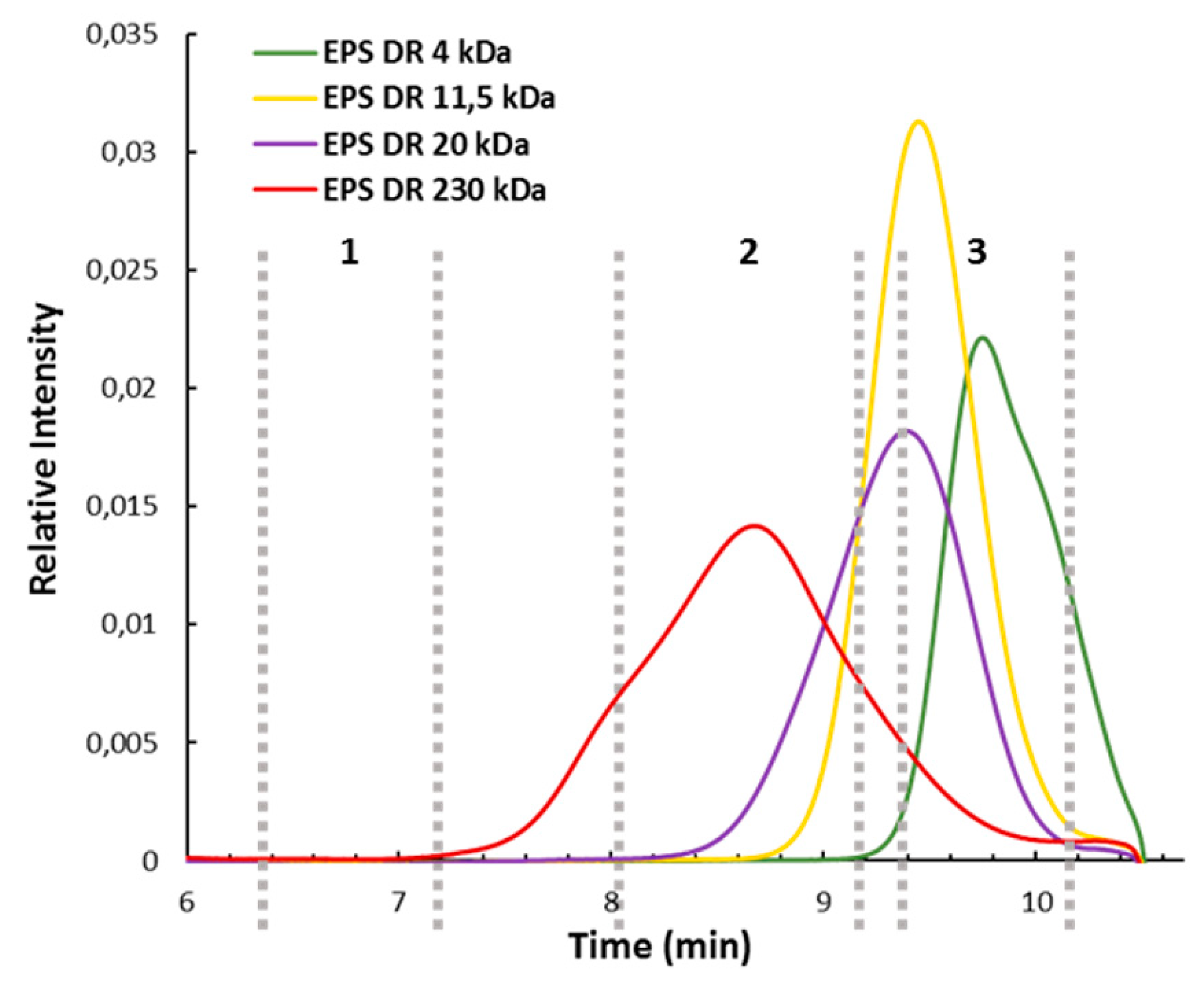
2.1. Evaluation of the Depolymerization by Gel Electrophoresis and High Pressure Size Exclusion Chromatography with Multi Angle Light Scattering (HPSEC-MALS)
2.2. Characterization of Oligosaccharide Fractions Generated by Insoluble Cell Debris
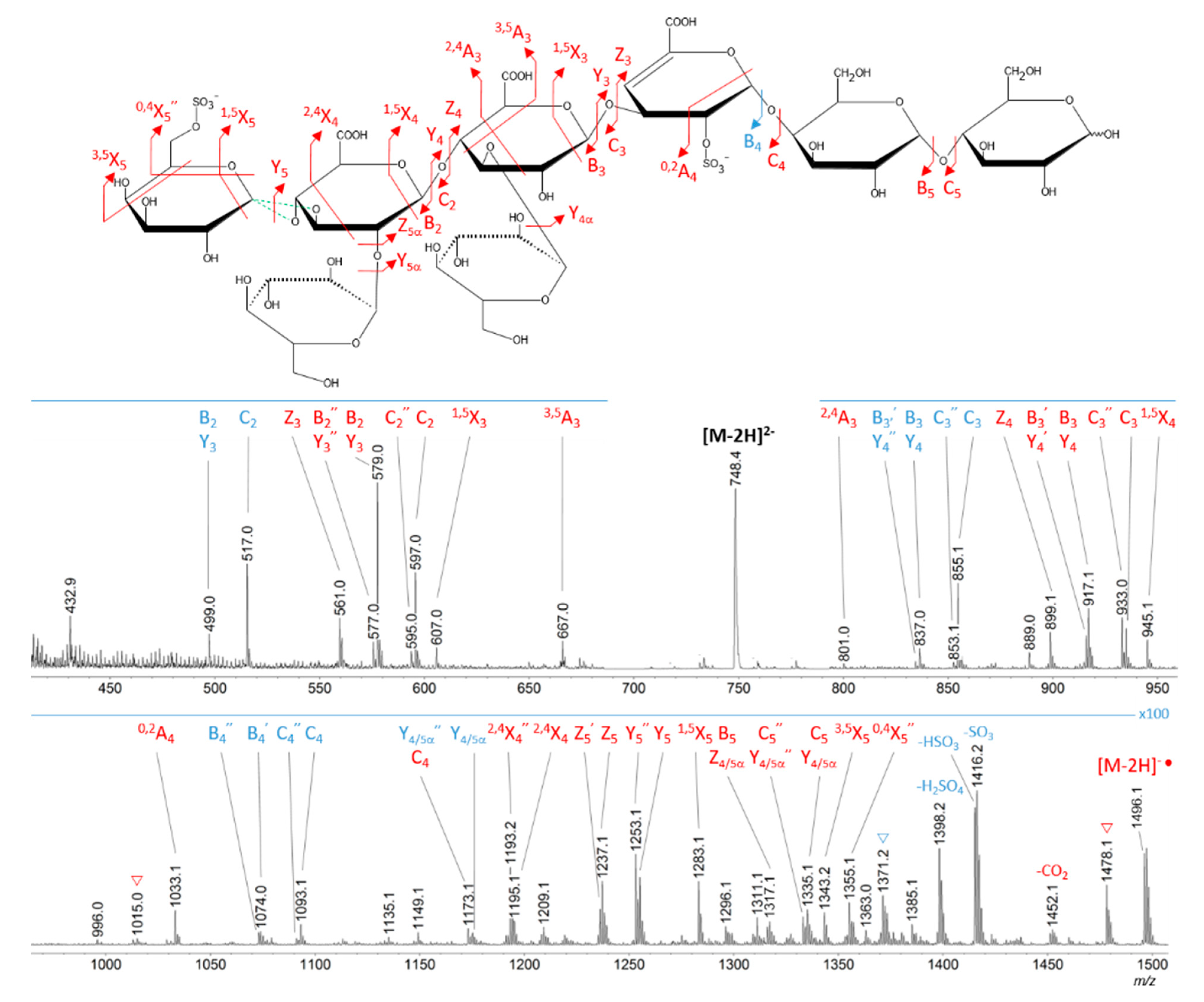
2.3. Oligosaccharide Structure Characterization by Mass Spectrometry
3. Conclusions
4. Materials and Methods
4.1. Production of the Native GY785 EPS
4.2. Enzymatic Assays Using A. infernus Protein Extracts
4.3. Batch Depolymerization Using A. infernus Cell Debris
4.4. Electrophoresis in Polyacrylamide Gels (PAGE)
4.5. HPSEC-MALS
4.6. Monosaccharide Composition
4.7. Protein Content
4.8. UHPLC-MS
4.9. He-CTD-MS/MS
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Held, M.A.; Jiang, N.; Basu, D.; Showalter, A.M.; Faik, A. Plant Cell Wall Polysaccharides: Structure and Biosynthesis; Springer Science and Business Media LLC: Berlin, Germany, 2015; pp. 3–54. [Google Scholar]
- Raposo, M.F.D.J.; De Morais, R.M.S.C.; De Morais, A.B.; De Morais, A.M.M.B. Bioactivity and Applications of Sulphated Polysaccharides from Marine Microalgae. Mar. Drugs 2013, 11, 233–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, G.; Yu, G.; Zhang, J.; Ewart, H.S. Chemical Structures and Bioactivities of Sulfated Polysaccharides from Marine Algae. Mar. Drugs 2011, 9, 196–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez, J.; Rodríguez-Amado, I.; Montemayor, M.; Fraguas, J.; González, M.; Murado, M. Chondroitin Sulfate, Hyaluronic Acid and Chitin/Chitosan Production Using Marine Waste Sources: Characteristics, Applications and Eco-Friendly Processes: A Review. Mar. Drugs 2013, 11, 747–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öner, E.T. Microbial Production of Extracellular Polysaccharides from Biomass. In Waste Energy for Life Cycle Assessment; Springer Science and Business Media LLC: Berlin, Germany, 2013; pp. 35–56. [Google Scholar]
- Delbarre-Ladrat, C.; Sinquin, C.; Lebellenger, L.; Zykwinska, A.; Colliec-Jouault, S. Exopolysaccharides produced by marine bacteria and their applications as glycosaminoglycan-like molecules. Front. Chem. 2014, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Sworn, G.; Sanderson, G.; Gibson, W. Gellan gum fluid gels. Food Hydrocoll. 1995, 9, 265–271. [Google Scholar] [CrossRef]
- Yu, Y.; Shen, M.; Song, Q.; Xie, J. Biological activities and pharmaceutical applications of polysaccharide from natural resources: A review. Carbohydr. Polym. 2018, 183, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.L.; Busch, S.J.; Cardin, A.D. Glycosaminoglycans: Molecular properties, protein interactions, and role in physiological processes. Physiol. Rev. 1991, 71, 481–539. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.S.; Mancera, R.L. The Structure of Glycosaminoglycans and their Interactions with Proteins. Chem. Boil. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef]
- Pomin, V.H. A Dilemma in the Glycosaminoglycan-Based Therapy: Synthetic or Naturally Unique Molecules? Med. Res. Rev. 2015, 35, 1195–1219. [Google Scholar] [CrossRef]
- Kishimoto, T.K.; Viswanathan, K.; Ganguly, T.; Elankumaran, S.; Smith, S.; Pelzer, K.; Lansing, J.C.; Sriranganathan, N.; Zhao, G.; Galcheva-Gargova, Z.; et al. Contaminated Heparin Associated with Adverse Clinical Events and Activation of the Contact System. N. Engl. J. Med. 2008, 358, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Mende, M.; Bednarek, C.; Wawryszyn, M.; Sauter, P.; Biskup, M.B.; Schepers, U.; Bräse, S.; Braese, S. Chemical Synthesis of Glycosaminoglycans. Chem. Rev. 2016, 116, 8193–8255. [Google Scholar] [CrossRef] [PubMed]
- Petitou, M.; Duchaussoy, P.; Lederman, I.; Choay, J.; Sinaÿ, P.; Jacquinet, J.-C.; Torri, G. Synthesis of heparin fragments. A chemical synthesis of the pentasaccharide O-(2-deoxy-2-sulfamido-6-O-sulfo-α-d-glucopyranosyl)-(1→4)-O-(β-d-glucopyranosyluronic acid)-(1→4)-O-(2-deoxy-2-sulfamido-3,6-di-O-sulfo-α-d-glucopyranosyl)-(1→4)-O-(2-O-sulfo-α-l-idopyranosyluronic acid)-(1→4)-2-deoxy-2-sulfamido-6-O-sulfo-d-glucopyranose decasodium salt, a heparin fragment having high affinity for antithrombin III. Carbohydr. Res. 1986, 147, 221–236. [Google Scholar] [PubMed]
- Van Boeckel, C.A.; Beetz, T.; Vos, J.N.; De Jong, A.J.; Van Aelst, S.F.; Bosch, R.H.V.D.; Mertens, J.M.R.; Van Der Vlugt, F.A. Synthesis of a Pentasaccharide Corresponding to the Antithrombin III Binding Fragment of Heparin. J. Carbohydr. Chem. 1985, 4, 293–321. [Google Scholar] [CrossRef]
- Petitou, M.; Van Boeckel, C.A.A. A Synthetic Antithrombin III Binding Pentasaccharide Is Now a Drug! What Comes Next? Angew. Chem. Int. Ed. 2004, 43, 3118–3133. [Google Scholar] [CrossRef] [PubMed]
- Kadokawa, J.-I. Precision Polysaccharide Synthesis Catalyzed by Enzymes. Chem. Rev. 2011, 111, 4308–4345. [Google Scholar] [CrossRef]
- Xu, Y.; Chandarajoti, K.; Zhang, X.; Pagadala, V.; Dou, W.; Hoppensteadt, D.M.; Sparkenbaugh, E.M.; Cooley, B.; Daily, S.; Key, N.S.; et al. Synthetic oligosaccharides can replace animal-sourced low–molecular weight heparins. Sci. Transl. Med. 2017, 9, eaan5954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pagadala, V.; Jester, H.M.; Lim, A.M.; Pham, T.Q.; Goulas, A.M.P.; Liu, J.; Linhardt, R.J. Chemoenzymatic Synthesis of Heparan Sulfate and Heparin Oligosaccharides and NMR Analysis: Paving the Way to a Diverse Library for Glycobiologists. Chem. Sci. 2017, 8, 7932–7940. [Google Scholar] [CrossRef]
- Vann, W.F.; Schmidt, M.A.; Jann, B.; Jann, K. The Structure of the Capsular Polysaccharide (K5 Antigenn) of Urinary-Tract-Infective Escherichia Coli 010:K5:H4. Eur. J. Biochem. 1981, 116, 359–364. [Google Scholar] [CrossRef]
- DeAngelis, P.L.; White, C.L. Identification of a Distinct, Cryptic Heparosan Synthase from Pasteurella multocida Types A, D, and F. J. Bacteriol. 2004, 186, 8529–8532. [Google Scholar] [CrossRef]
- Restaino, O.F.; Cimini, D.; De Rosa, M.; Catapano, A.; De Rosa, M.; Schiraldi, C. High cell density cultivation of Escherichia coli K4 in a microfiltration bioreactor: A step towards improvement of chondroitin precursor production. Microb. Cell Factories 2011, 10, 10–19. [Google Scholar] [CrossRef]
- Pan, N.C.; Pereira, H.C.B.; da Silva, M.D.L.C.; Vasconcelos, A.F.D.; Celligoi, M.A.P.C. Improvement Production of Hyaluronic Acid by Streptococcus Zooepidemicus in Sugarcane Molasses. Appl. Biochem. Biotechnol. 2017, 182, 276–293. [Google Scholar] [CrossRef] [PubMed]
- Im, J.-H.; Song, J.-M.; Kang, J.-H.; Kang, D.-J. Optimization of Medium Components for High-Molecular-Weight Hyaluronic Acid Production by Streptococcus sp. ID9102 via a Statistical Approach. J. Ind. Microbiol. Biotechnol. 2009, 36, 1337. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I. Bacterial Exopolysaccharides. In Advances in Bacterial Electron Transport Systems and Their Regulation; Elsevier: Amsterdam, The Netherlands, 1972; Volume 8, pp. 143–213. [Google Scholar]
- Guezennec, J. Deep-sea hydrothermal vents: A new source of innovative bacterial exopolysaccharides of biotechnological interest? J. Ind. Microbiol. Biotechnol. 2002, 29, 204–208. [Google Scholar] [CrossRef] [PubMed]
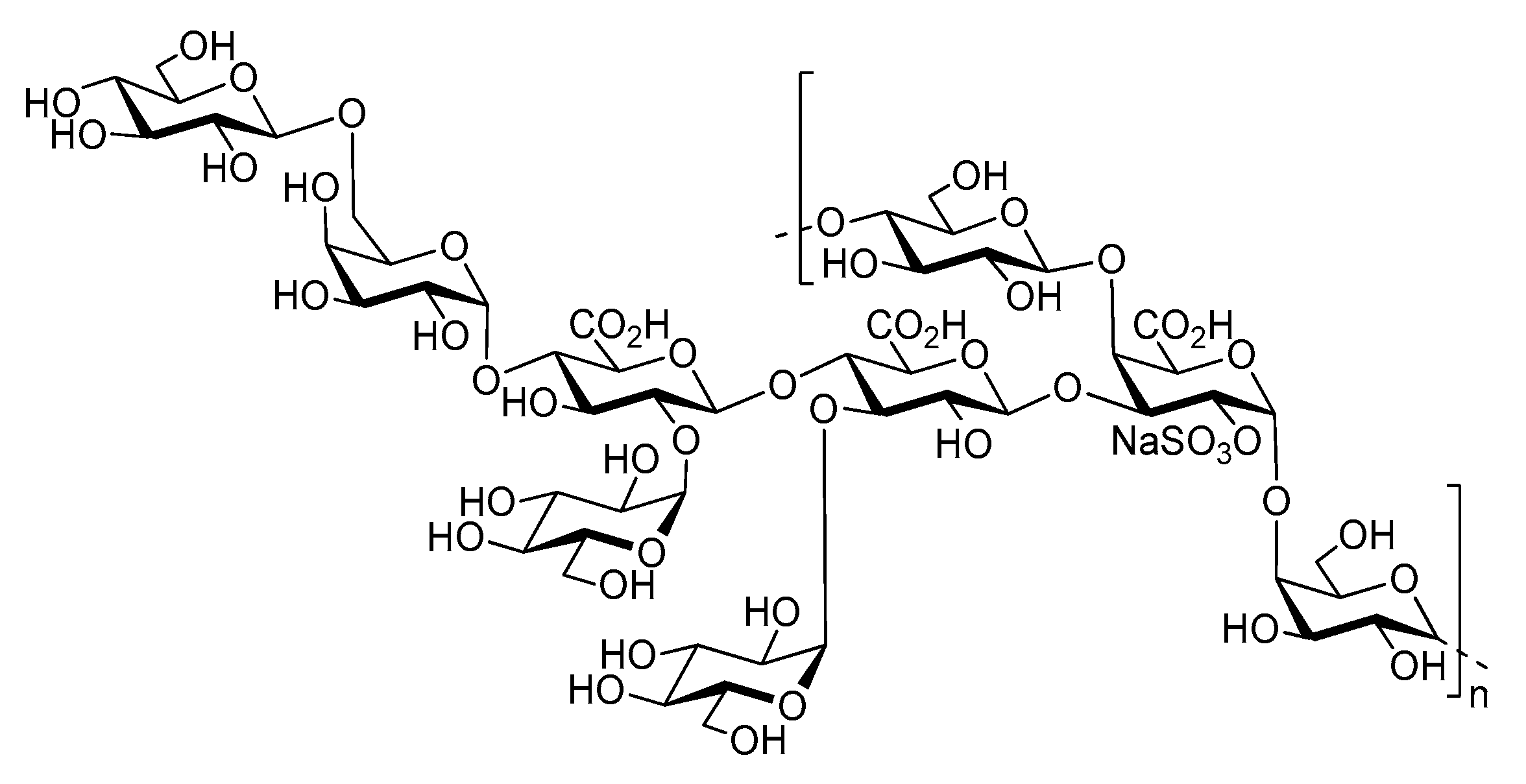
- Roger, O.; Kervarec, N.; Ratiskol, J.; Colliec-Jouault, S.; Chevolot, L. Structural studies of the main exopolysaccharide produced by the deep-sea bacterium Alteromonas infernus. Carbohydr. Res. 2004, 339, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Heymann, D.; Ruiz-Velasco, C.; Chesneau, J.; Ratiskol, J.; Sinquin, C.; Colliec-Jouault, S. Anti-Metastatic Properties of a Marine Bacterial Exopolysaccharide-Based Derivative Designed to Mimic Glycosaminoglycans. Molecules 2016, 21, 309. [Google Scholar] [CrossRef] [PubMed]
- Merceron, C.; Portron, S.; Vignes-Colombeix, C.; Rederstorff, E.; Masson, M.; Lesoeur, J.; Sourice, S.; Sinquin, C.; Colliec-Jouault, S.; Weiss, P.; et al. Pharmacological Modulation of Human Mesenchymal Stem Cell Chondrogenesis by a Chemically Oversulfated Polysaccharide of Marine Origin: Potential Application to Cartilage Regenerative Medicine. Stem Cells 2012, 30, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Petit, A.-C.; Noiret, N.; Sinquin, C.; Ratiskol, J.; Guezennec, J.; Colliec-Jouault, S. Free-radical depolymerization with metallic catalysts of an exopolysaccharide produced by a bacterium isolated from a deep-sea hydrothermal vent polychaete annelid. Carbohydr. Polym. 2006, 64, 597–602. [Google Scholar] [CrossRef] [Green Version]
- Shively, J.E.; Conrad, H.E. Formation of anhydrosugars in the chemical depolymerization of heparin. Biochemistry 1976, 15, 3932–3942. [Google Scholar] [CrossRef] [PubMed]
- Ruijssenaars, H.; Hartmans, S. Plate screening methods for the detection of polysaccharase-producing microorganisms. Appl. Microbiol. Biotechnol. 2001, 55, 143–149. [Google Scholar] [CrossRef]
- Levasseur, A.; Drula, E.; Lombard, V.; Coutinho, P.M.; Henrissat, B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 2013, 6, 41. [Google Scholar] [CrossRef]
- Zykwinska, A.; Berre, L.T.-L.; Sinquin, C.; Ropartz, D.; Rogniaux, H.; Colliec-Jouault, S.; Delbarre-Ladrat, C. Enzymatic depolymerization of the GY785 exopolysaccharide produced by the deep-sea hydrothermal bacterium Alteromonas infernus: Structural study and enzyme activity assessment. Carbohydr. Polym. 2018, 188, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Aebi, M.; Packer, N.H.; Seeberger, P.H.; Esko, J.D.; Stanley, P.; Hart, G.; Darvill, A.; Kinoshita, T.; et al. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 2015, 25, 1323–1324. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W.D.; Jackson, G.P. Charge Transfer Dissociation (CTD) Mass Spectrometry of Peptide Cations Using Kiloelectronvolt Helium Cations. J. Am. Soc. Mass Spectrom. 2014, 25, 1939–1943. [Google Scholar] [CrossRef]
- Ropartz, D.; Li, P.; Jackson, G.P.; Rogniaux, H. Negative Polarity Helium Charge Transfer Dissociation Tandem Mass Spectrometry: Radical-Initiated Fragmentation of Complex Polysulfated Anions. Anal. Chem. 2017, 89, 3824–3828. [Google Scholar] [CrossRef] [PubMed]
- Domon, B.; Costello, C.E. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 1988, 5, 397–409. [Google Scholar] [CrossRef]
- Kreuger, J.; Kjellén, L. Heparan Sulfate Biosynthesis: Regulation and Variability. J. Histochem. Cytochem. 2012, 60, 898–907. [Google Scholar] [CrossRef]
- Raguénès, G.; Peres, A.; Ruimy, R.; Pignet, P.; Christen, R.; Loäec, M.; Rougeaux, H.; Barbier, G.; Guezennec, J. Alteromonas infernus sp. nov., a new polysaccharide-producing bacterium isolated from a deep-sea hydrothermal vent. J. Appl. Microbiol. 1997, 82, 422–430. [Google Scholar] [CrossRef]
- Lee, H.; Cowman, M. An Agarose Gel Electrophoretic Method for Analysis of Hyaluronan Molecular Weight Distribution. Anal. Biochem. 1994, 219, 278–287. [Google Scholar] [CrossRef]
- Kamerling, J.P.; Gerwig, G.J.; Vliegenthart, J.F.G.; Clamp, J.R. Characterization by gas-liquid chromatography-mass spectrometry and proton-magnetic-resonance spectroscopy of pertrimethylsilyl methyl glycosides obtained in the methanolysis of glycoproteins and glycopeptides. Biochem. J. 1975, 151, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Montreuil, J.; Bouguelet, S.; Debray, H.; Fournet, B.; Spik, G.; Strecker, G. Glycoproteins. In Carbohydrate Analysis. A Practical Approach; Chaplin, M.F., Kennedy, J.F., Eds.; IRL Press: Oxford, UK, 1986; pp. 143–204. [Google Scholar]
- Niedermeyer, T.H.J.; Strohalm, M. mMass as a Software Tool for the Annotation of Cyclic Peptide Tandem Mass Spectra. PLoS ONE 2012, 7, 44913. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fraction | F1 | F2 | F3 | F4 | F5 |
|---|---|---|---|---|---|
| Amount (mg) | 34.6 | 2.4 | 1.4 | 1.5 | 0.5 |
| Osidic Composition (Molar Ratio) | |||||||
|---|---|---|---|---|---|---|---|
| Rha | Fuc | Man | Gal | Glc | GalA | GlcA | |
| GY785 EPS | 0.2 | 0.1 | 0.4 | 3.6 | 4.7 | 1.0 | 2.0 |
| F1 | 1.0 | 0.4 | 0.5 | 0.3 | 1.0 | 0 | 1.6 |
| F2 | 0 | 0 | 0 | 0.1 | 1.1 | 0 | 1.0 |
| F3 | 0 | 0 | 0 | 1.0 | 1.6 | 0.2 | 1.0 |
| F4 | 0 | 0 | 0 | 0.7 | 1.1 | 0.1 | 1.0 |
| Cell debris (D) | 0.3 | 0.1 | 0 | 0 | 1.0 | 0 | 0 |
| Structure | Monoisotopic Mass (Theoretical, Da) | RetentionTime (min) | Measured m/z | Adduct | ||
|---|---|---|---|---|---|---|
| F5 | A |  | 1498.27 | 2.73 | 748.13 | [M − 2H]2− |
| Supp. Species: A—(1⇨3 hexoses) | 1336.22 ⇨ 1012.12 | 2.76 ⇨ 2.88 | 667.10 ⇨ 505.05 | [M − 2H]2− | ||
| F4 | B |  | 2996.55 | 7.00 | 1031.55 | [M − 4H + 1IPR]3− |
| Supp. Species: B—(1⇨4 hexoses) | 2834.5 ⇨ 2348.34 | 7.05 ⇨ 7.21 | 977.52 ⇨ 815.47 | [M − 4H + 1IPR]3− | ||
| F3 | C |  | 4494.82 | 8.56 | 1173.26 | [M − 6H + 2IPR]4− |
| Supp. Species: C—(1⇨5 hexoses) | 4332.77 ⇨ 3684.56 | 8.59 ⇨ 8.84 | 1132.75 ⇨ 970.69 | [M − 6H + 2IPR]4− | ||
| F2 | D |  | 5993.10 | 9.69 | 1598.38 | [M − 8H + 4IPR]4− |
| Supp. Species: D—(1⇨5 hexoses) | 5831.04 ⇨ 5182.83 | 9.69 ⇨ 9.79 | 1557.88 ⇨ 1395.82 | [M − 8H + 4IPR]4- | ||
| E |  | 7491.37 | 10.27 | 1598.40 | [M − 10H + 5IPR]5− | |
| Supp. Species: E—(1⇨7 hexoses) | 7329.32 ⇨ 6357.00 | 10.29 ⇨ 10.43 | 1565.98 ⇨ 1371.51 | [M − 10H + 5IPR]5− | ||
| F |  | 8989.64 | 10.77 | 1598.40 | [M − 12H + 6IPR]6− | |
| Supp. Species: F—(1⇨2 hexoses) | 8827.59 ⇨ 8665.54 | 10.79 ⇨ 10.80 | 1571.39 ⇨ 1544.36 | [M − 12H + 6IPR]6− | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akoumany, K.; Zykwinska, A.; Sinquin, C.; Marchand, L.; Fanuel, M.; Ropartz, D.; Rogniaux, H.; Pipelier, M.; Delbarre-Ladrat, C.; Colliec-Jouault, S. Characterization of New Oligosaccharides Obtained by An Enzymatic Cleavage of the Exopolysaccharide Produced by the Deep-Sea Bacterium Alteromonas infernus Using its Cell Extract. Molecules 2019, 24, 3441. https://doi.org/10.3390/molecules24193441
Akoumany K, Zykwinska A, Sinquin C, Marchand L, Fanuel M, Ropartz D, Rogniaux H, Pipelier M, Delbarre-Ladrat C, Colliec-Jouault S. Characterization of New Oligosaccharides Obtained by An Enzymatic Cleavage of the Exopolysaccharide Produced by the Deep-Sea Bacterium Alteromonas infernus Using its Cell Extract. Molecules. 2019; 24(19):3441. https://doi.org/10.3390/molecules24193441
Chicago/Turabian StyleAkoumany, Katy, Agata Zykwinska, Corinne Sinquin, Laëtitia Marchand, Mathieu Fanuel, David Ropartz, Hélène Rogniaux, Muriel Pipelier, Christine Delbarre-Ladrat, and Sylvia Colliec-Jouault. 2019. "Characterization of New Oligosaccharides Obtained by An Enzymatic Cleavage of the Exopolysaccharide Produced by the Deep-Sea Bacterium Alteromonas infernus Using its Cell Extract" Molecules 24, no. 19: 3441. https://doi.org/10.3390/molecules24193441
APA StyleAkoumany, K., Zykwinska, A., Sinquin, C., Marchand, L., Fanuel, M., Ropartz, D., Rogniaux, H., Pipelier, M., Delbarre-Ladrat, C., & Colliec-Jouault, S. (2019). Characterization of New Oligosaccharides Obtained by An Enzymatic Cleavage of the Exopolysaccharide Produced by the Deep-Sea Bacterium Alteromonas infernus Using its Cell Extract. Molecules, 24(19), 3441. https://doi.org/10.3390/molecules24193441