Globular and Fibrous Proteins Modified with Deep Eutectic Solvents: Materials for Drug Delivery


Abstract
:
1. Introduction
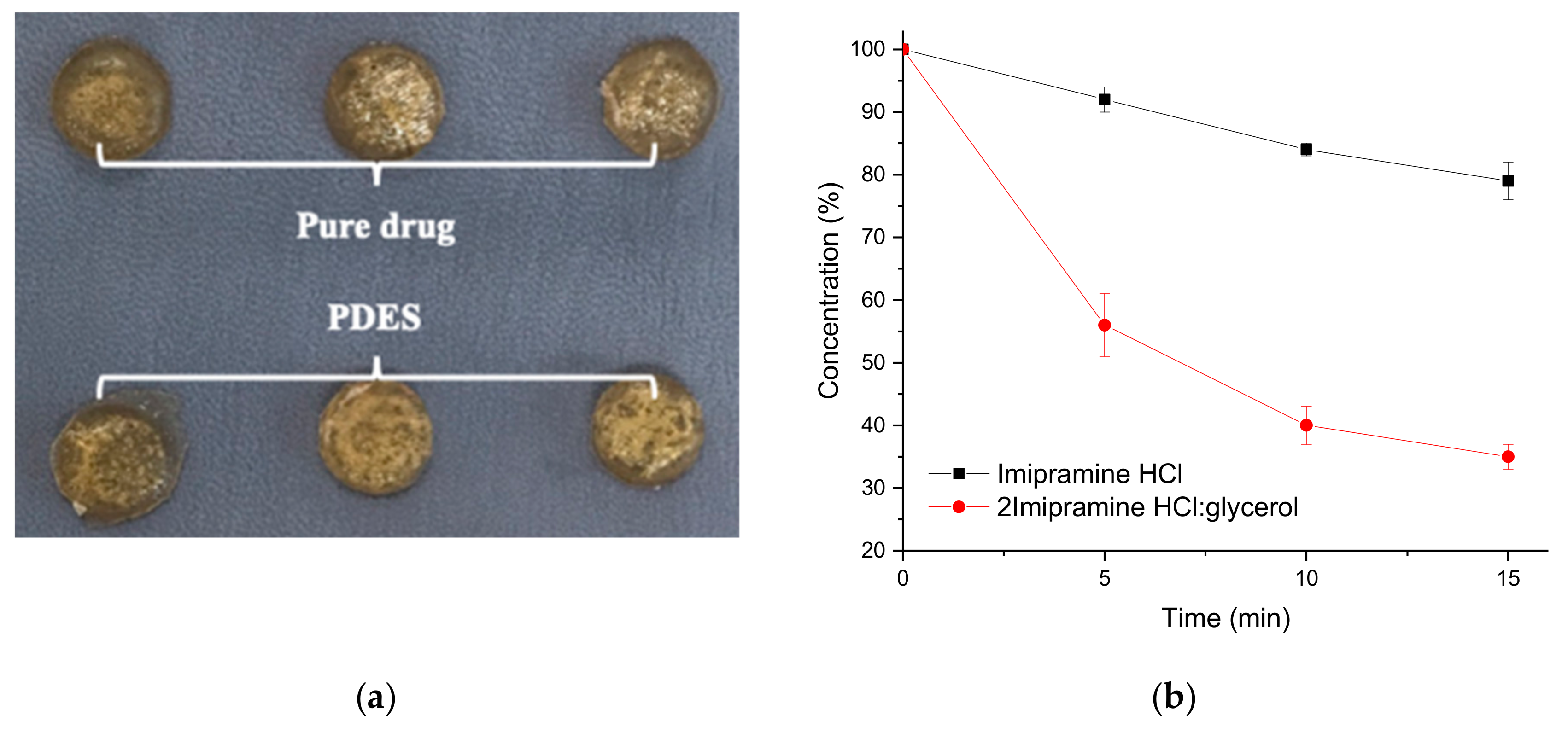
2. Results and Discussion
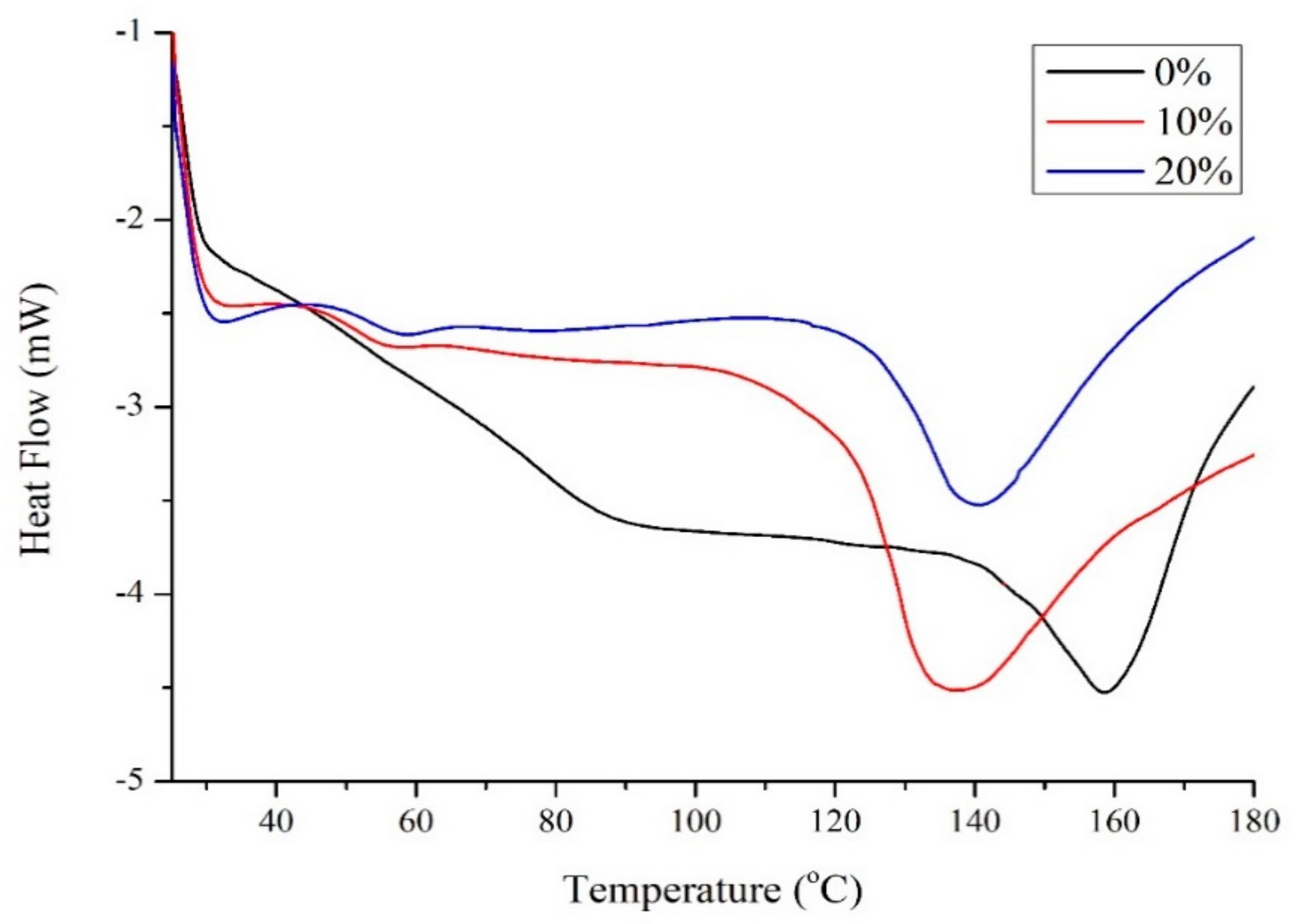
2.1. Plasticisation of Zein
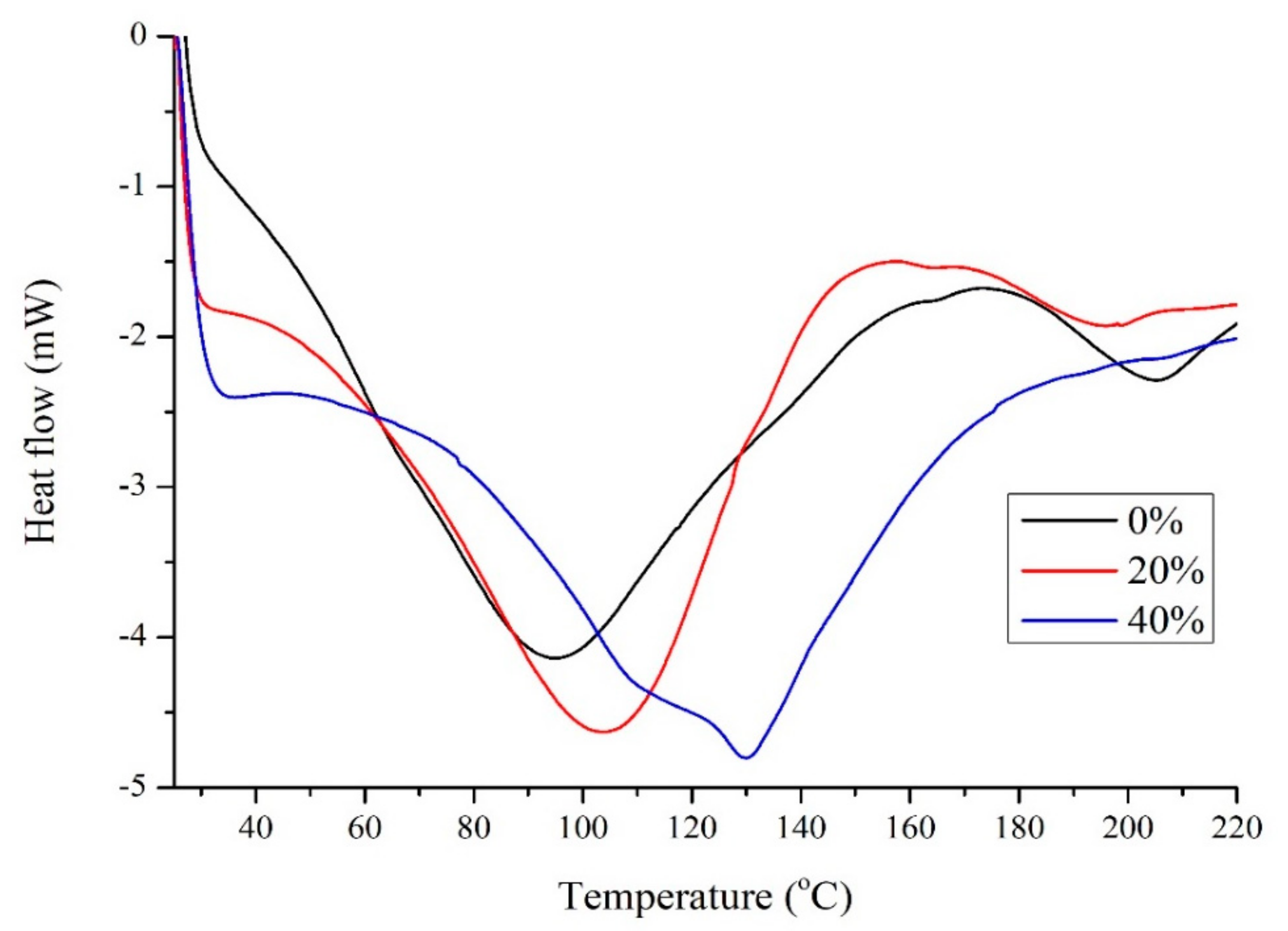
2.2. Plasticization of Soy Protein with Glyceline
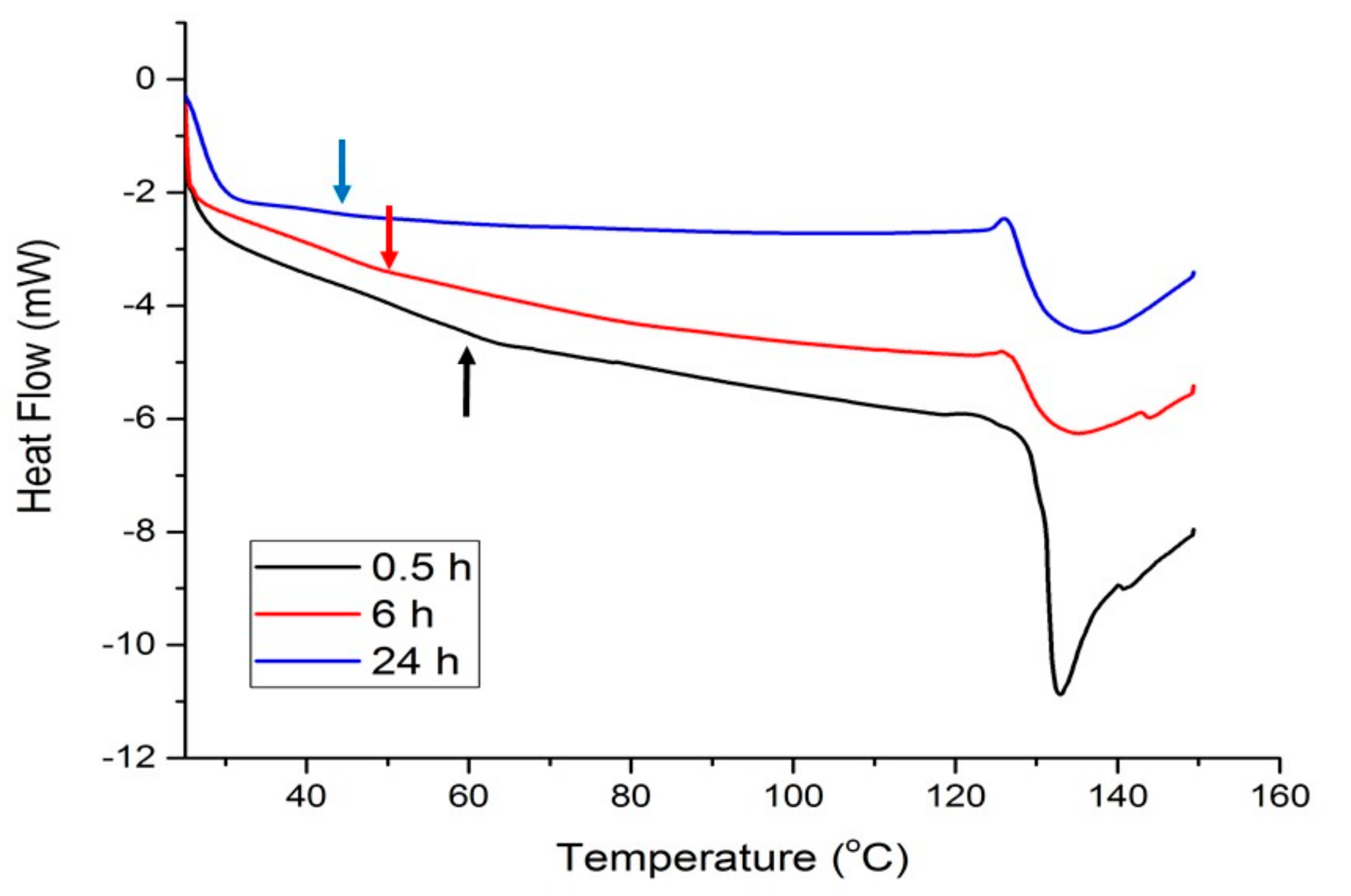
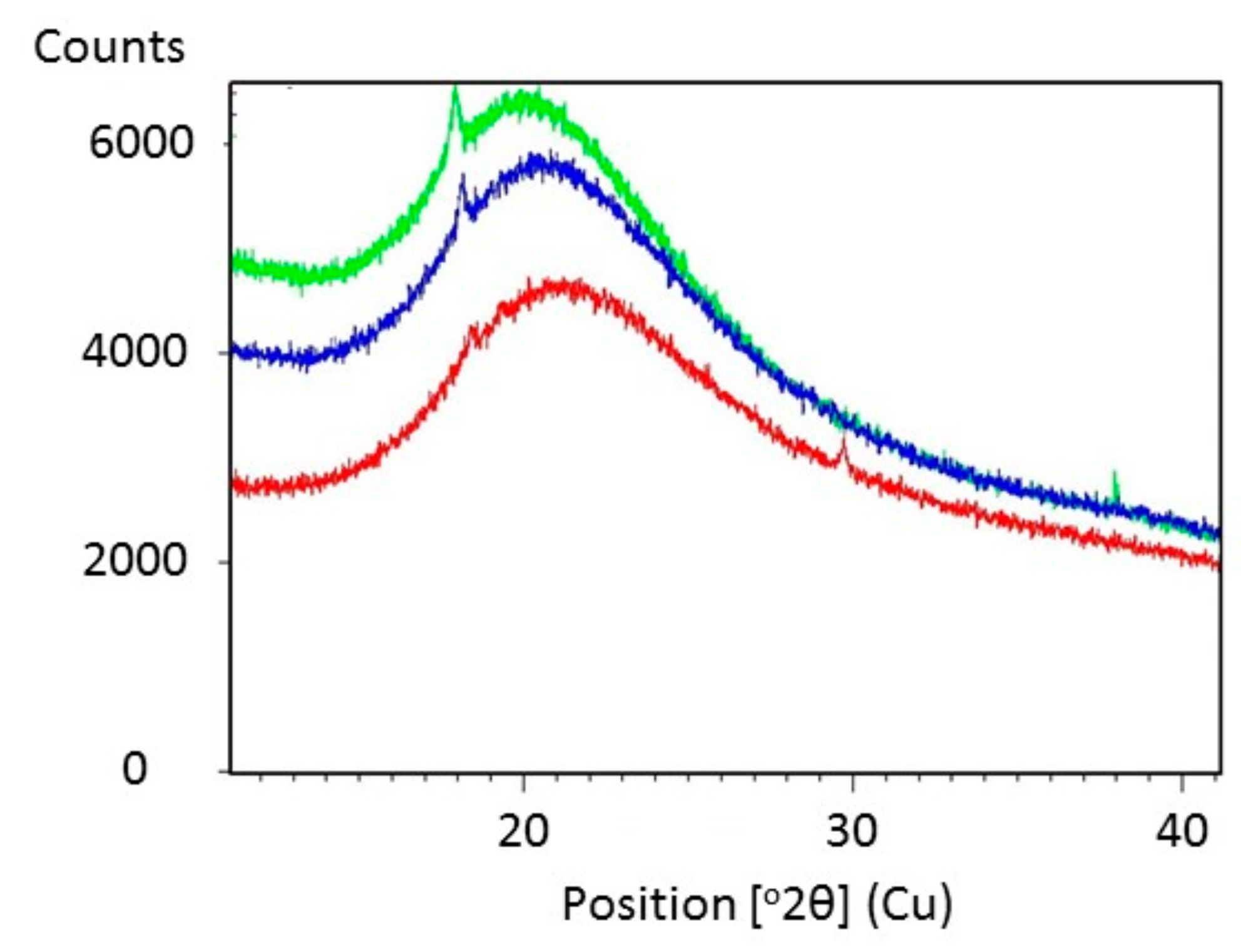
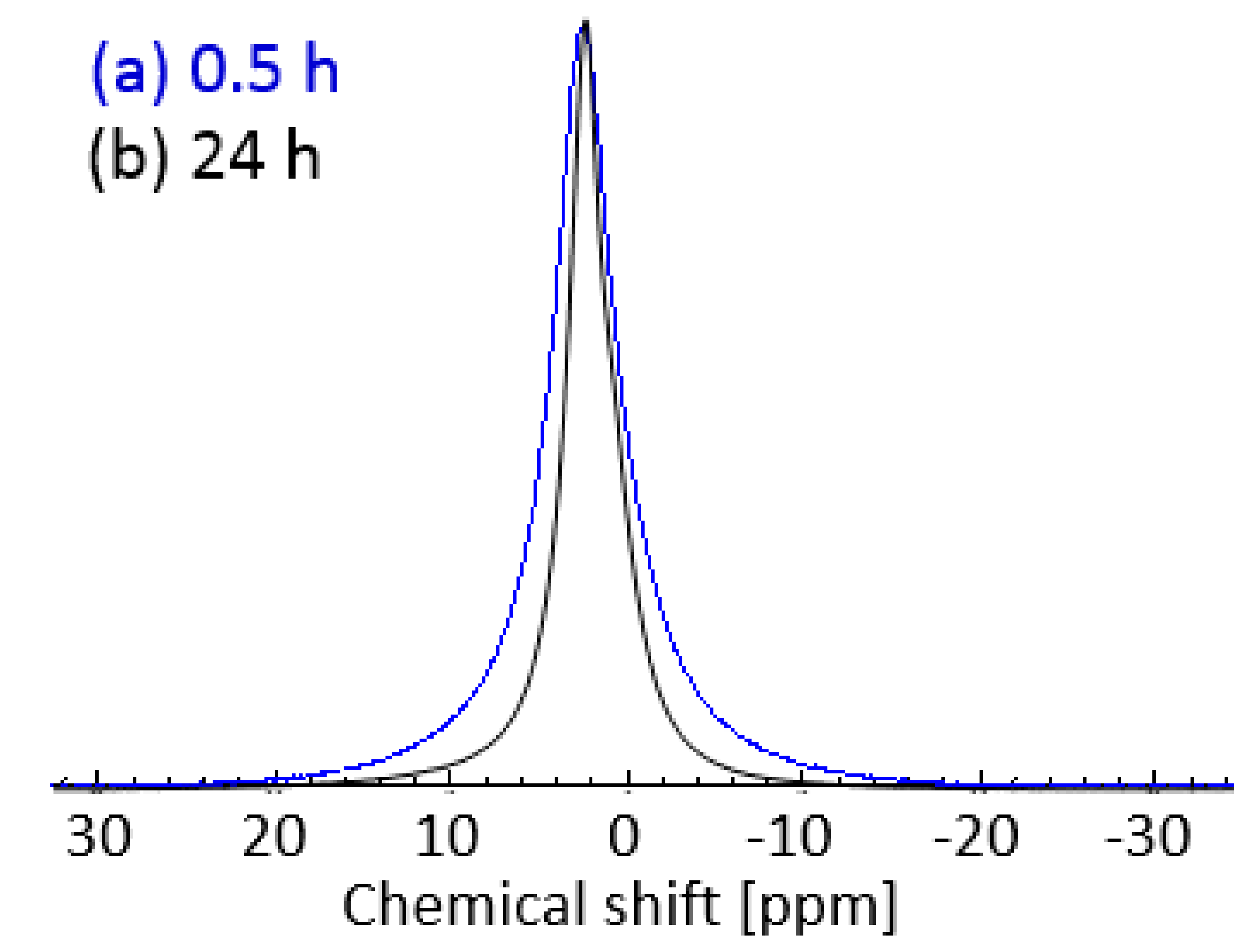
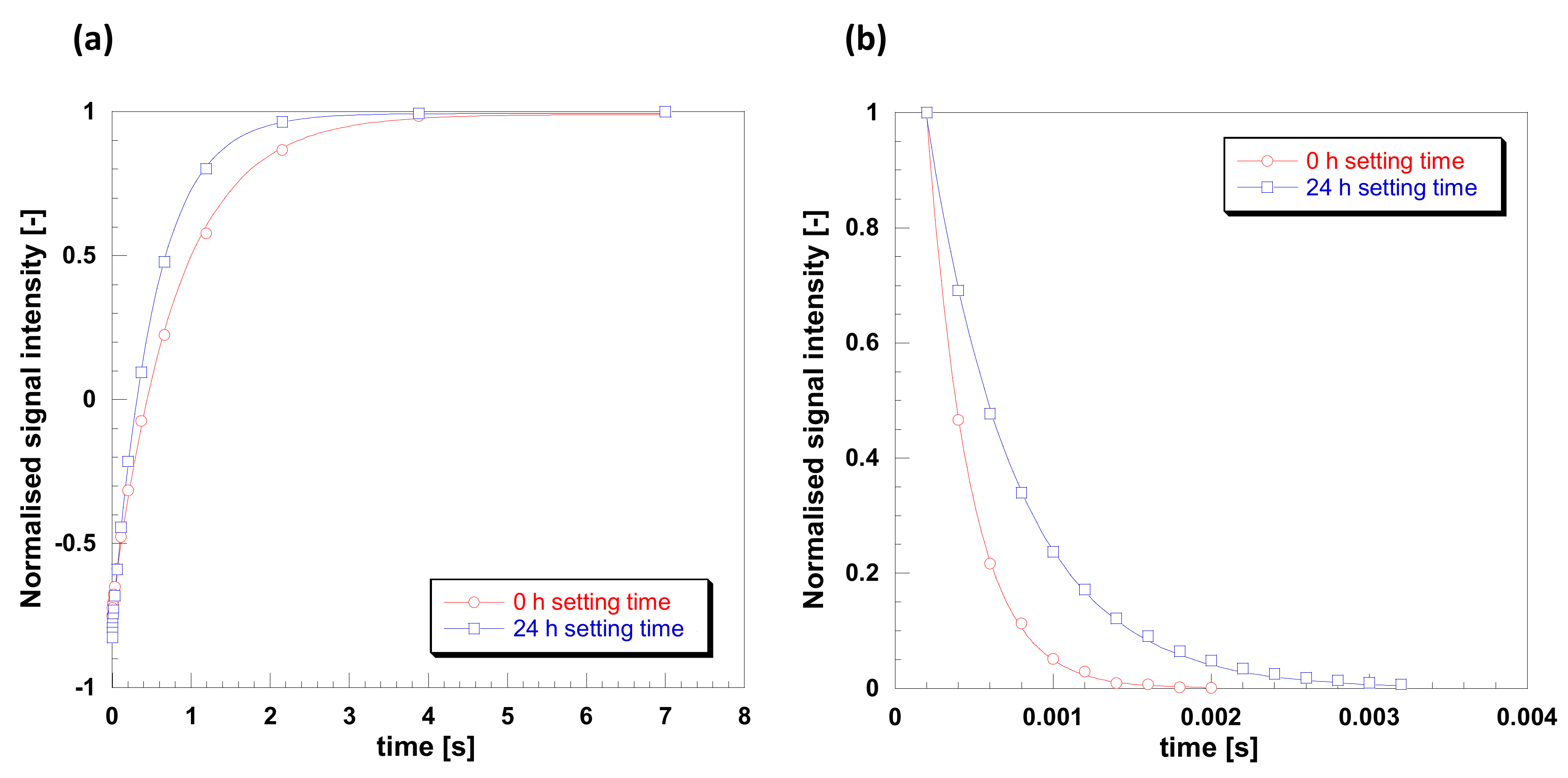
2.3. Plasticization of Gelatin with Glyceline
3. Material and Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Song, J.H.; Murphy, R.J.; Narayan, R.; Davies, G.B.H. Biodegradable and compostable alternatives to conventional plastics. Phil. Trans. Royal Soc. B. 2009, 364, 2127–2139. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Madbouly, S.A.; Schrader, J.A.; Srinivasan, G.D.; Grewell, K.G.; McCabe, M.R.; Kessler, W.; Graves, R. Characterization and biodegradation behavior of bio-based poly(lactic acid) and soy protein blends for sustainable horticultural applications. Green Chem. 2015, 17, 380–393. [Google Scholar]
- Kabasci, E.S. Bio-Based Plastics: Materials and Applications. John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2014. [Google Scholar]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [Green Version]
- Abbott, A.P.; Ballantyne, A.D.; Conde, J.P.; Ryder, K.S.; Wise, W.R. Salt modified starch: Sustainable, recyclable plastic. Green Chem. 2012, 14, 1302. [Google Scholar] [CrossRef]
- Abbott, A.P.; Abolibda, T.Z.; Davis, S.J.; Emmerling, F.; Lourdin, D.; Leroy, E.; Wise, W.R. Glycol based plasticisers for salt modified starch. RSC Adv. 2014, 4, 40421–40427. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Owyeung, R.E.; Sonkusale, S.R.; Panzer, M.J. Highly Stretchable and Nonvolatile Gelatin-Supported Deep Eutectic Solvent Gel Electrolyte-Based Ionic Skins for Strain and Pressure Sensing. J. Mater. Chem. C. 2019, 7, 601–608. [Google Scholar] [CrossRef]
- Biswas, A.; Shogren, R.L.; Stevenson, D.G.; Willett, J.L.; Bhowmik, P.K. Ionic Liquids as Solvents for Biopolymers: Acylation of Starch and Zein Protein. Carbohydr. Polym. 2006, 66, 546–550. [Google Scholar] [CrossRef]
- Esquembre, R.; Sanz, J.M.; Wall, J.G.; del Monte, F.; Mateo, C.R.; Ferrer, M.L. Thermal Unfolding and Refolding of Lysozyme in Deep Eutectic Solvents and Their Aqueous Dilutions. Phys. Chem. Chem. Phys. 2013, 15, 11248. [Google Scholar] [CrossRef]
- Sanchez-Fernandez, A.; Edler, K.J.; Arnold, T.; Alba Venero, D.; Jackson, A.J. Protein Conformation in Pure and Hydrated Deep Eutectic Solvents. Phys. Chem. Chem. Phys. 2017, 19, 8667–8670. [Google Scholar] [CrossRef]
- Abbott, A.P.; Ahmed, E.I.; Prasad, K.I.; Qader, B.; Ryder, K.S. Liquid pharmaceuticals formulation by eutectic formation. Fluid Phase Equil. 2017, 448, 2–8. [Google Scholar] [CrossRef]
- Leroy, E.; Decaen, P.; Jacquet, P.; Coativy, G.; Pontoire, B.; Reguerre, A.-L.; Lourdin, D. Deep Eutectic Solvents as Functional Additives for Starch Based Plastics. Green Chem. 2012, 14, 3063. [Google Scholar] [CrossRef]
- Lawton, J.W. Zein: A history of processing and use. Cereal Chem. 2002, 79, 1–18. [Google Scholar] [CrossRef]
- Abbott, A.P.; Abolibda, T.Z.; Qu, W.; Wise, W.R.; Wright, L.A. Thermoplastic starch–polyethylene blends homogenised using deep eutectic solvents. RSC Adv. 2017, 7, 7268–7273. [Google Scholar]
- Kokini, J.; Cocero, A.; Madeka, H. State diagrams help predict rheology of cereal proteins. Food Tech. 1995, 49, 74–82. [Google Scholar]
- Di Gioia, L.; Guilbert, S. Corn Protein-Based Thermoplastic Resins: Effect of Some Polar and Amphiphilic Plasticizers. J. Agri. Food Chem. 1999, 47, 1254–1261. [Google Scholar] [CrossRef]
- Lawton, J. Plasticizers for zein: Their effect on tensile properties and water absorption of zein films. Cereal Chem. 2004, 81, 1–5. [Google Scholar] [CrossRef]
- Chang, Y.; Karim, A.A.; Seow, C. Interactive plasticizing–antiplasticizing effects of water and glycerol on the tensile properties of tapioca starch films. Food 2006, 20, 1–8. [Google Scholar] [CrossRef]
- Lourdin, D.; Coignard, L.; Bizot, H.; Colonna, P. Influence of equilibrium relative humidity and plasticizer concentration on the water content and glass transition of starch materials. Polymer 1997, 38, 5401–5406. [Google Scholar]
- Liang, J.; Xia, Q.; Wang, S.; Li, J.; Huang, Q.; Ludescher, R.D. Influence of glycerol on the molecular mobility, oxygen permeability and microstructure of amorphous zein films. Food Hydrocoll. 2015, 44, 94–100. [Google Scholar] [CrossRef]
- Thakur, V.K. Soy-Based Bioplastics; Smithers Rapra: Shawbury, UK, 2017. [Google Scholar]
- Schmidt, V.; Soldi, V. Influence of polycaprolactone-triol addition on thermal stability of soy protein isolate based films. Polymer Deg. Stab. 2006, 91, 3124–3130. [Google Scholar] [CrossRef]
- Cuadri, A.A.; Romero, A.; Bengoechea, C.; Guerrero, A. Natural superabsorbent plastic materials based on a functionalized soy protein. Poly. Test. 2017, 58, 126. [Google Scholar]
- Zhang, J.; Mungara, P.; Jane, J. Mechanical and thermal properties of extruded soy protein sheets. Polymer 2001, 42, 2569–2578. [Google Scholar] [CrossRef]
- Ortiz, S.M.; Añón, M. Enzymatic hydrolysis of soy protein isolates. DSC study. J. Therm. Anal. Cal. 2001, 66, 489–499. [Google Scholar] [CrossRef]
- Fakhoury, F.M.; Martelli, S.M.; Bertan, L.C.; Yamashita, F.; Mei, L.H.I.; Queiroz, F.P.C. Edible films made from blends of manioc starch and gelatin–Influence of different types of plasticizer and different levels of macromolecules on their properties. LWT-Food Sci. Tech. 2012, 49, 149–154. [Google Scholar] [CrossRef]
- Al-Hassan, A.; Norziah, M. Starch-gelatin edible films: Water vapor permeability and mechanical properties as affected by plasticizers. Food Hydrocoll. 2012, 26, 108–117. [Google Scholar] [CrossRef]
- Cao, N.; Yang, X.; Fu, Y. Effects of various plasticizers on mechanical and water vapor barrier properties of gelatin films. Food Hydrocoll. 2009, 23, 729–735. [Google Scholar] [CrossRef]
- Singh, T.; Boral, S.; Bohidar, H.; Kumar, A. Interaction of gelatin with room temperature ionic liquids: A detailed physicochemical study. J. Phys. Chem. B. 2010, 114, 8441–8448. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A.P.; Alaysuy, O.; Antunes, A.P.M.; Douglas, A.C.; Guthrie-Strachan, J.; Wise, W.R. Processing of leather using deep eutectic solvents. ACS Sus. Chem. Eng. 2015, 3, 1241–1247. [Google Scholar]
- Alasuy, O. Processing leather using deep eutectic solvents. PhD Thesis, University of Leicester, Leicester, UK, 2019. [Google Scholar]
- Adamus, J.; Spychaj, T.; Zdanowicz, M.; Jędrzejewski, R. Thermoplastic Starch with Deep Eutectic Solvents and Montmorillonite as a Base for Composite Materials. Ind. Crops Prod. 2018, 123, 278–284. [Google Scholar] [CrossRef]
- Gómez-Estaca, J.; Montero, P.F.; Alemán, F.-M.A.; Gómez-Guillén, M. Physical and chemical properties of tuna-skin and bovine-hide gelatin films with added aqueous oregano and rosemary extracts. Food Hydrocoll. 2009, 23, 1334–1341. [Google Scholar] [Green Version]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 1948, 73, 679–746. [Google Scholar]
- Abd, E.; Yousef, S.A..; Pastore, M.N.; Telaprolu, K.; Mohammed, Y.H.; Namjoshi, S.; Grice, J.E.; Roberts, M.S. Skin models for the testing of transdermal drugs. Clin. Pharmacol. 2016, 8, 163–176. [Google Scholar]
- Fukushima, E.; Roeder, S.B.W. Experimental Pulse NMR: A Nuts and Bolts Approach. Addison-Wesley Publishing Company Inc.: Reading, MA, USA, 1981. [Google Scholar]
- Carr, H.Y.; Purcell, E.M. Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Phys. Rev. 1954, 94, 630–638. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glyceline Contents (wt%) | Ultimate Tensile Strength (MPa) | Elongation (%) | Ts (°C) |
|---|---|---|---|
| 0% | 10.2 ± 1.4 | 1.38 ± 0.28 | 159 |
| 10% | 6.5 ± 1.7 | 0.84 ± 0.09 | 139 |
| 20% | 5.8 ± 0.78 | 0.80 ± 0.08 | 140 |
| Glyceline Contents (wt%) | UTS (MPa) | Elongation (%) |
|---|---|---|
| 10% | 2.0± 0.73 | 0.84 ± 0.28 |
| 20% | 3.9 ± 0.74 | 17.2 ± 1.5 |
| 30% | 2.1 ± 0.68 | 20.0 ± 4.6 |
| 40% | 1.6 ± 0.55 | 18.6 ± 3.5 |
| 50% | 0.37 ± 0.05 | 5.8 ± 1.8 |
| Glyceline Contents (wt%) | Ultimate Tensile Strength (MPa) | Elongation (%) | 24 h Setting | Glyceline Contents (wt%) | Ultimate Tensile Strength (MPa) | Elongation (%) |
|---|---|---|---|---|---|---|
| 10% | 14.6 ± 2.8 | 3.5 ± 0.83 | 10% | 0.84 ± 0.13 | 23.9 ± 6.8 | |
| 20% | 23.8 ± 5.7 | 3.3 ± 0.48 | 20% | 0.05 ± 0.01 | 442± 80 | |
| 30% | 22.0 ± 6.0 | 22.6 ± 6.8 | 30% | 0.01 ± 0.01 | 1265 ± 264 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, W.; Häkkinen, R.; Allen, J.; D’Agostino, C.; Abbott, A.P. Globular and Fibrous Proteins Modified with Deep Eutectic Solvents: Materials for Drug Delivery. Molecules 2019, 24, 3583. https://doi.org/10.3390/molecules24193583
Qu W, Häkkinen R, Allen J, D’Agostino C, Abbott AP. Globular and Fibrous Proteins Modified with Deep Eutectic Solvents: Materials for Drug Delivery. Molecules. 2019; 24(19):3583. https://doi.org/10.3390/molecules24193583
Chicago/Turabian StyleQu, Wanwan, Riina Häkkinen, Jack Allen, Carmine D’Agostino, and Andrew P. Abbott. 2019. "Globular and Fibrous Proteins Modified with Deep Eutectic Solvents: Materials for Drug Delivery" Molecules 24, no. 19: 3583. https://doi.org/10.3390/molecules24193583
APA StyleQu, W., Häkkinen, R., Allen, J., D’Agostino, C., & Abbott, A. P. (2019). Globular and Fibrous Proteins Modified with Deep Eutectic Solvents: Materials for Drug Delivery. Molecules, 24(19), 3583. https://doi.org/10.3390/molecules24193583