
TiO2 Photocatalysis for Transfer Hydrogenation




Abstract
:1. Introduction
2. Transfer Hydrogenation of C=C and C≡C Bonds
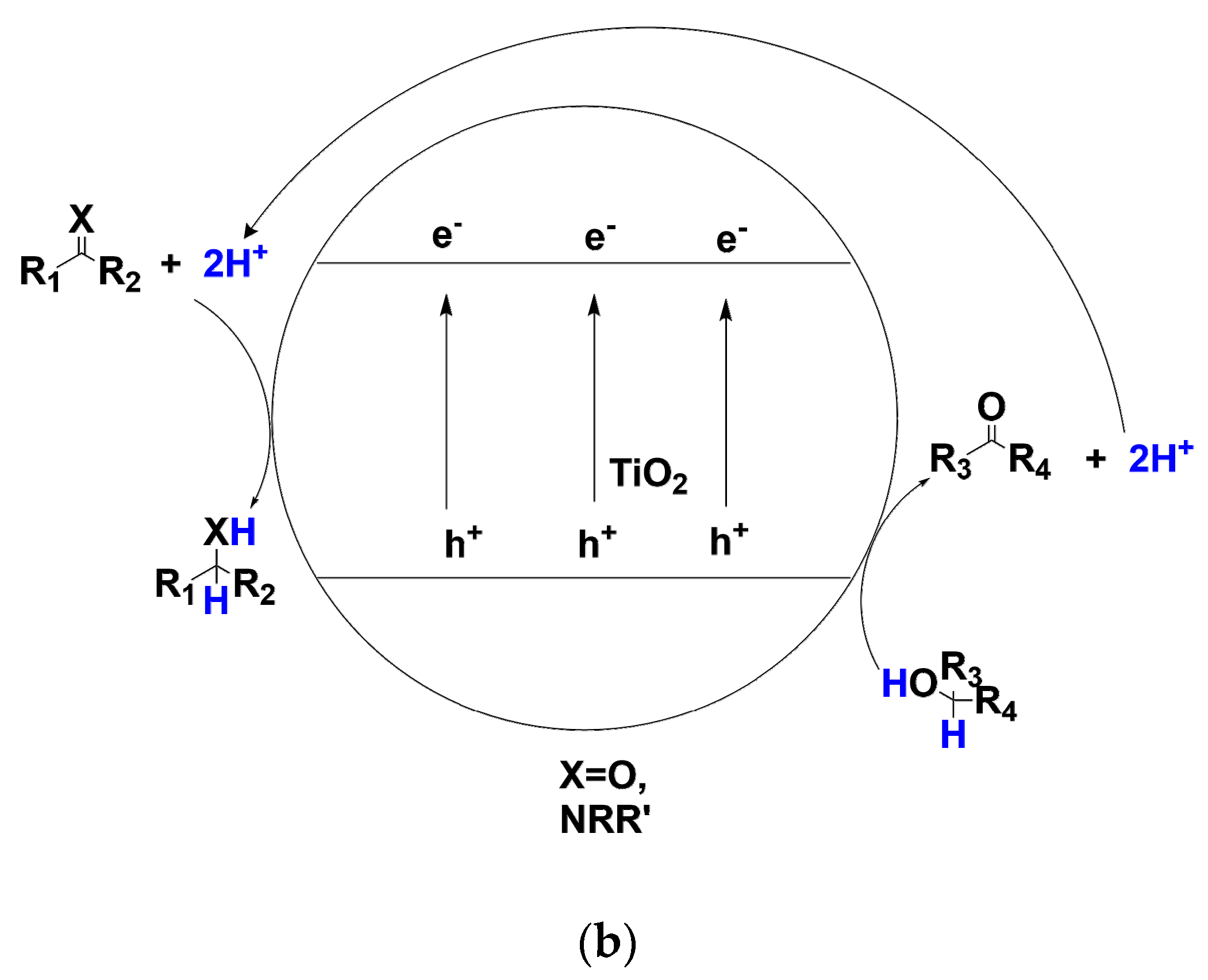
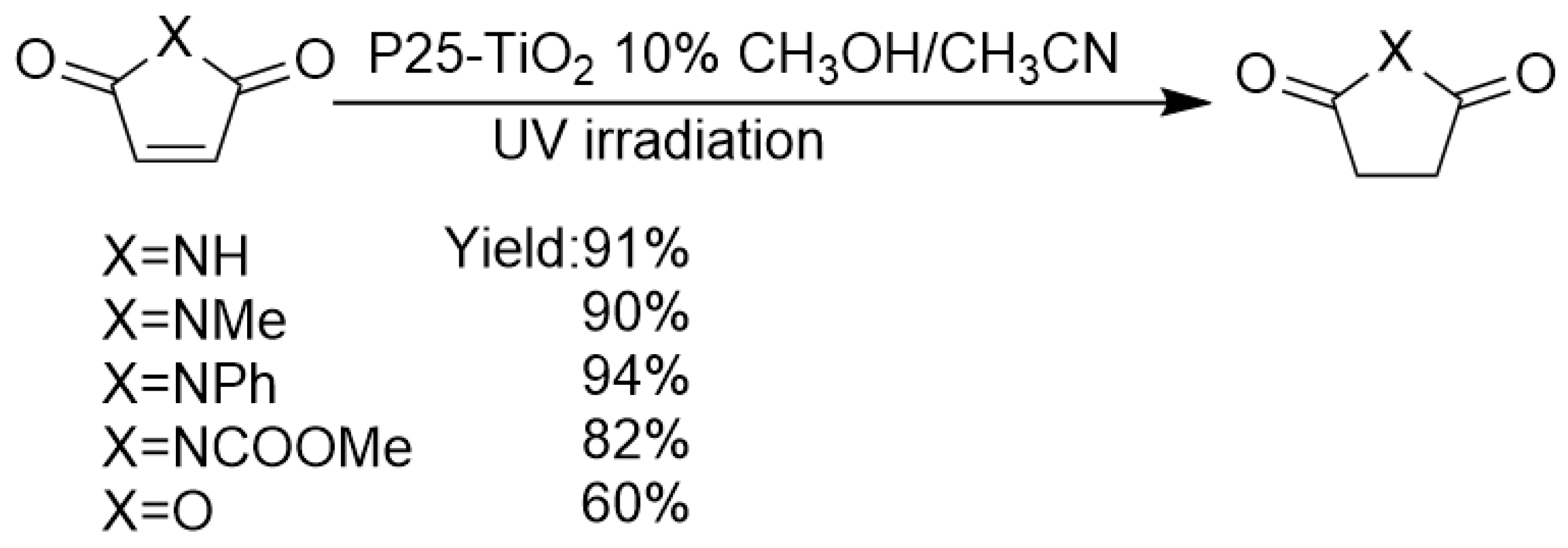
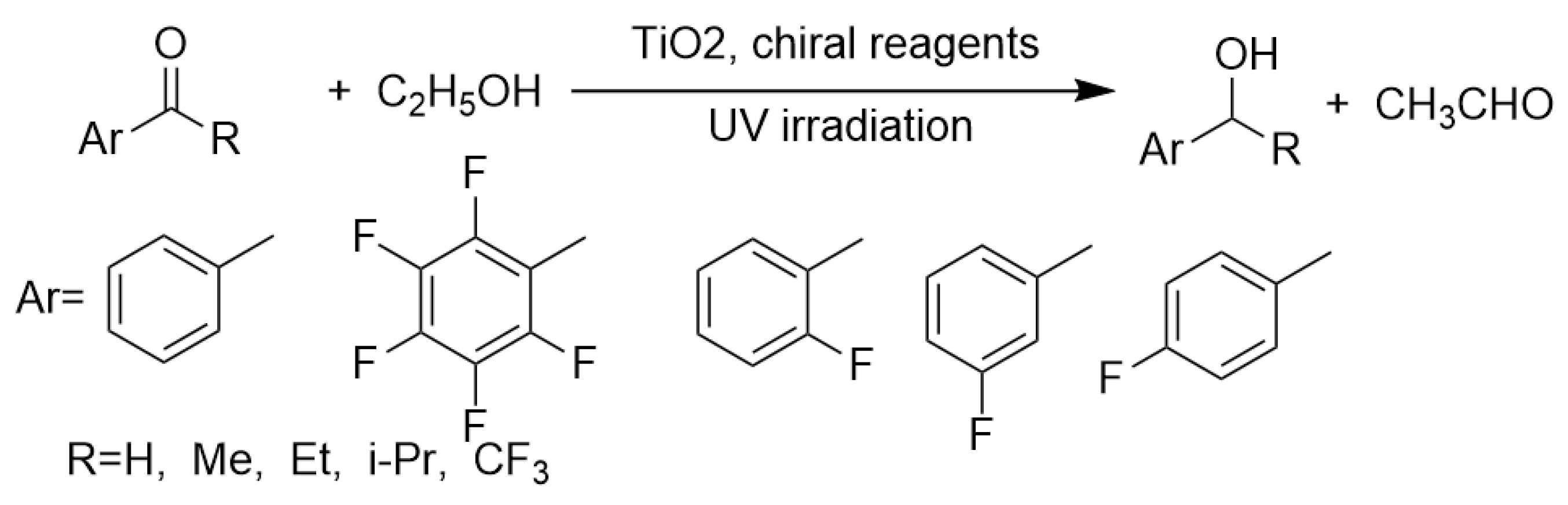
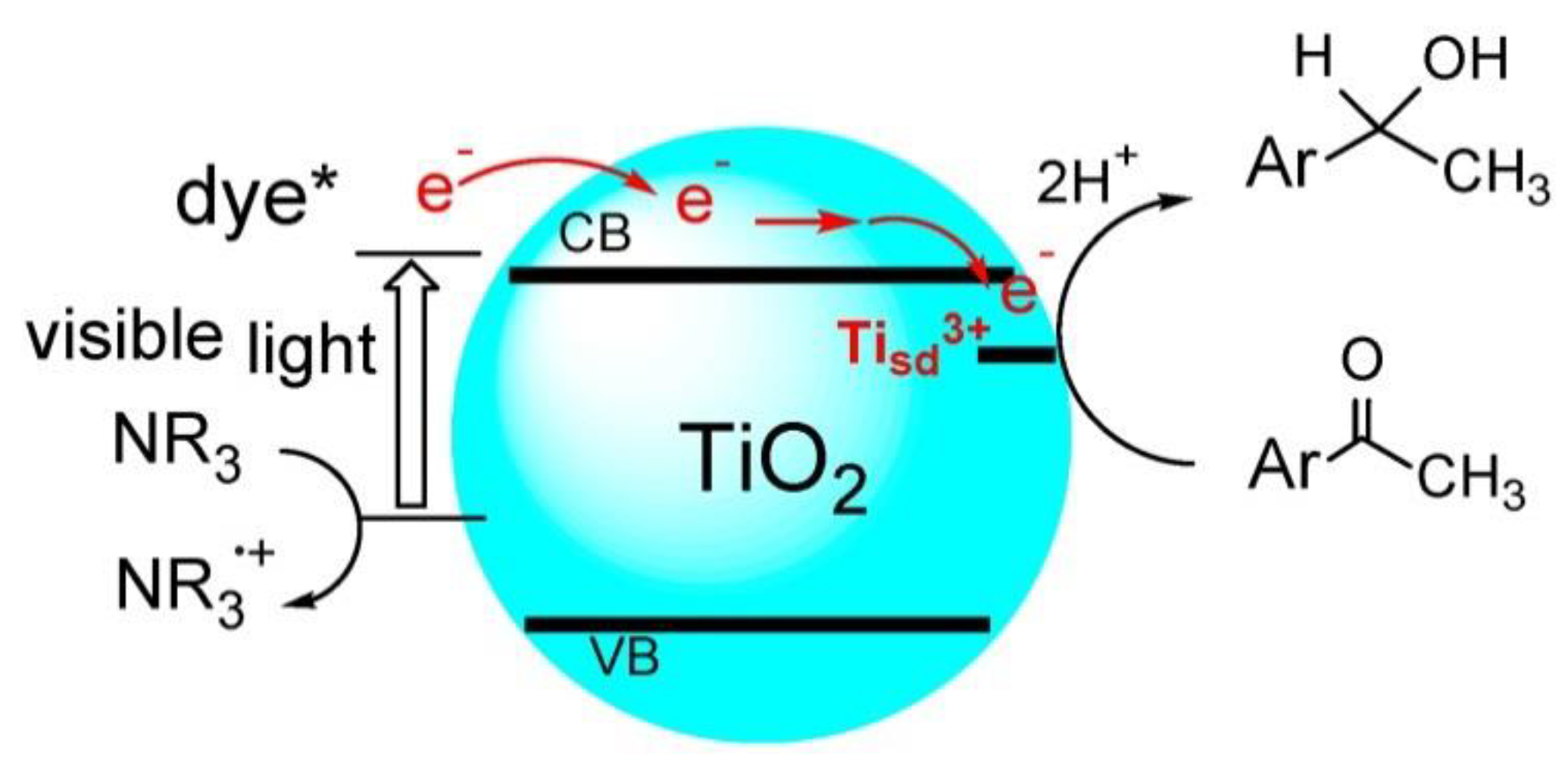
3. Transfer Hydrogenation of C=O and C=N Bonds
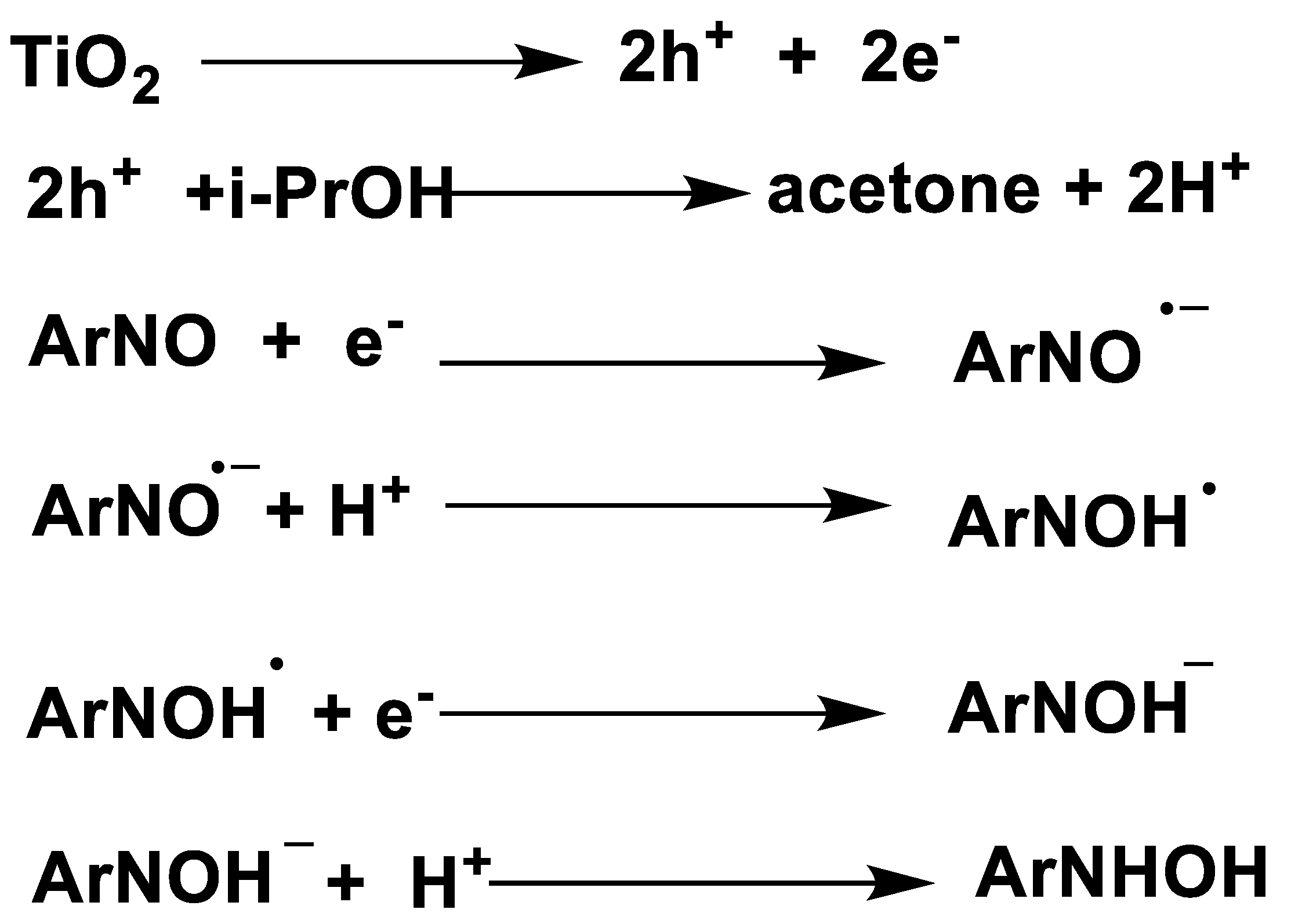
4. Transfer Hydrogenation of N=O Bonds
5. Conclusions
Funding
Conflicts of Interest
References
- Johnstone, R.A.W.; Wilby, A.H.; Entwistle, I.D. Heterogeneous catalytic transfer hydrogenation and its relation to other methods for reduction of organic compounds. Chem. Rev. 1985, 85, 129–170. [Google Scholar] [CrossRef]
- Brieger, G.; Nestrick, T.J. Catalytic transfer hydrogenation. Chem. Rev. 1974, 74, 567–580. [Google Scholar] [CrossRef]
- Tsuji, J. Catalytic hydrogenation, transfer hydrogenation and hydrosilylation. In Transition Metal Reagents and Catalysts: Innovations in Organic Synthesis; John Wiley & Sons, Ltd.: Weinheim, Germany, 2000. [Google Scholar]
- Stefane, B.; Pozgan, F. Advances in catalyst systems for the asymmetric hydrogenation and transfer hydrogenation of ketones. Cat. Rev. Sci. Eng. 2014, 56, 82–174. [Google Scholar] [CrossRef]
- Stefane, B.; Pozgan, F. Metal-catalysed transfer hydrogenation of ketones. Top. Curr. Chem. 2016, 374, 18. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Dufour, J.; Schoepke, F.R. Advances in catalytic metal-free reductions: From bio-inspired concepts to applications in the organocatalytic synthesis of pharmaceuticals and natural products. Green Chem. 2011, 13, 1084–1105. [Google Scholar] [CrossRef]
- Robertson, A.; Matsumoto, T.; Ogo, S. The development of aqueous transfer hydrogenation catalysts. Dalton Trans. 2011, 40, 10304–10310. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Astruc, D. The golden age of transfer hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Clapham, S.E.; Hadzovic, A.; Morris, R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004, 248, 2201–2237. [Google Scholar] [CrossRef]
- Bäckvall, J.-E. Transition metal hydrides as active intermediates in hydrogen transfer reactions. J. Organomet. Chem. 2002, 652, 105–111. [Google Scholar] [CrossRef]
- Meerwein, H.; Schmidt, R. Ein neues verfahren zur reduktion von aldehyden und ketonen. Justus Liebigs Ann. Chem. 1925, 444, 221–238. [Google Scholar] [CrossRef]
- Ponndorf, W. Der reversible austausch der oxydationsstufen zwischen aldehyden oder ketonen einerseits und primären oder sekundären alkoholen anderseits. Angew. Chem. 1926, 39, 138–143. [Google Scholar] [CrossRef]
- Verley, A. Exchange of functional groups between two molecules. Exchange of alcohol and aldehyde groups. Bull. Soc. Chim. Fr. 1925, 37, 537–542. [Google Scholar]
- Oppenauer, R.V. Eine methode der dehydrierung von sekundären alkoholen zu ketonen. I. Zur herstellung von sterinketonen und sexualhormonen. Recl. Trav. Chim. Pays-Bas 1937, 56, 137–144. [Google Scholar] [CrossRef]
- Aranyos, A.; Csjernyik, G.; Szabó, K.J.; Bäckvall, J.-E. Evidence for a ruthenium dihydride species as the active catalyst in the RuCl2(PPh3)-catalyzed hydrogen transfer reaction in the presence of base. Chem. Commun. 1999, 4, 351–352. [Google Scholar] [CrossRef]
- Sues, P.E.; Demmans, K.Z.; Morris, R.H. Rational development of iron catalysts for asymmetric transfer hydrogenation. Dalton Trans. 2014, 43, 7650–7667. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wang, Y.; Liu, A.; Li, S.; Lu, C.; Chen, C. Covalent organic frameworks: Promising materials as heterogeneous catalysts for C-C bond formations. Catalysts 2018, 8, 404. [Google Scholar] [CrossRef]
- Liu, M.; Huang, Q.; Wang, S.; Li, Z.; Li, B.; Tan, B.; Jin, S. Crystalline covalent triazine frameworks by in-situ oxidation of alcohols to aldehyde monomers. Angew. Chem. Int. Ed. 2018, 57, 11968–11972. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.-F.; Qi, M.-Z.; Wang, Z.-P.; Ding, S.-Y.; Yu, W.; Liu, Q.; Wang, L.-K.; Wang, H.-Z.; An, W.-K.; Wang, W. Benzoxazole-linked ultrastable covalent organic frameworks for photocatalysis. J. Am. Chem. Soc. 2018, 140, 4623–4631. [Google Scholar] [CrossRef] [PubMed]
- Santoro, S.; Kozhushkov, S.I.; Ackermann, L.; Vaccaro, L. Heterogeneous catalytic approaches in C-H activation reactions. Green Chem. 2016, 18, 3471–3493. [Google Scholar] [CrossRef]
- Su, F.; Mathew, S.C.; Lipner, G.; Fu, X.; Antonietti, M.; Blechert, S.; Wang, X. Mpg-C3N4-catalyzed selective oxidation of alcohols using O2 and visible light. J. Am. Chem. Soc. 2010, 132, 16299–16301. [Google Scholar] [CrossRef]
- Gilkey, M.J.; Xu, B. Heterogeneous catalytic transfer hydrogenation as an effective pathway in biomass upgrading. ACS Catal. 2016, 6, 1420–1436. [Google Scholar] [CrossRef]
- Takanabe, K.; Domen, K. Preparation of inorganic photocatalytic materials for overall water splitting. ChemCatChem 2012, 4, 1485–1497. [Google Scholar] [CrossRef]
- Chen, X.; Mao, S.S. Titanium dioxide nanomaterials: Synthesis, properties, modifications, and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.; Liu, P.; Hu, X.; Stucky, G.; Sun, S.-G.; Chen, L.; Han, W.; Whang, D.R.; Chen, G.; Zhan, C.; et al. Solar cells and photocatalytic systems: General discussion. Faraday Discuss. 2014, 176, 313–331. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, M.D.; Lee, J.S. Current progress and scientific challenges in the advancement of organic–inorganic lead halide perovskite solar cells. New J. Chem. 2017, 41, 10508–10527. [Google Scholar] [CrossRef]
- Chen, C.C.; Ma, W.H.; Zhao, J.C. Semiconductor-mediated photodegradation of pollutants under visible-light irradiation. Chem. Soc. Rev. 2010, 39, 4206–4219. [Google Scholar] [CrossRef]
- Fujishima, A.; Zhang, X.T.; Tryk, D.A. TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 2008, 63, 515–582. [Google Scholar] [CrossRef]
- Palmisano, G.; Augugliaro, V.; Pagliaro, M.; Palmisano, L. Photocatalysis: A promising route for 21st century organic chemistry. Chem. Commun. 2007, 3425–3437. [Google Scholar] [CrossRef]
- Palmisano, G.; Garcia-Lopez, E.; Marci, G.; Loddo, V.; Yurdakal, S.; Augugliaro, V.; Palmisano, L. Advances in selective conversions by heterogeneous photocatalysis. Chem. Commun. 2010, 46, 7074–7089. [Google Scholar] [CrossRef]
- Gambarotti, C.; Punta, C.; Recupero, F.; Caronna, T.; Palmisano, L. TiO2 in organic photosynthesis: Sunlight induced functionalization of heterocyclic bases. Curr. Org. Chem. 2010, 14, 1153–1169. [Google Scholar] [CrossRef]
- Ma, D.; Liu, A.; Li, S.; Lu, C.; Chen, C. TiO2 photocatalysis for C-C bond formation. Catal. Sci. Technol. 2018, 8, 2030–2045. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, A.; Ma, D.; Li, S.; Lu, C.; Li, T.; Chen, C. TiO2 photocatalyzed C-H bond transformation for C-C coupling reactions. Catalysts 2018, 8, 355. [Google Scholar] [CrossRef]
- Yurdakal, S.; Palmisano, G.; Loddo, V.; Augugliaro, V.; Palmisano, L. Nanostructured rutile TiO2 for selective photocatalytic oxidation of aromatic alcohols to aldehydes in water. J. Am. Chem. Soc. 2008, 130, 1568–1569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, C.C.; Ma, W.H.; Zhao, J.C. Visible-light-induced aerobic oxidation of alcohols in a coupled photocatalytic system of dye-sensitized TiO2 and TEMPO. Angew. Chem. Int. Ed. 2008, 47, 9730–9733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Q.; Chen, C.C.; Zang, L.; Ma, W.H.; Zhao, J.C. Oxygen atom transfer in the photocatalytic oxidation of alcohols by TiO2: Oxygen isotope studies. Angew. Chem. Int. Ed. 2009, 48, 6081–6084. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Liu, A.; Lu, C.; Chen, C. Photocatalytic dehydrogenation of primary alcohols: Selectivity goes against adsorptivity. ACS Omega 2017, 2, 4161–4172. [Google Scholar] [CrossRef]
- Lang, X.J.; Ji, H.W.; Chen, C.C.; Ma, W.H.; Zhao, J.C. Selective formation of imines by aerobic photocatalytic oxidation of amines on TiO2. Angew. Chem. Int. Ed. 2011, 50, 3934–3937. [Google Scholar] [CrossRef]
- Lang, X.J.; Ma, W.H.; Zhao, Y.B.; Chen, C.C.; Ji, H.W.; Zhao, J.C. Visible-light-induced selective photocatalytic aerobic oxidation of amines into imines on TiO2. Chem. Eur. J. 2012, 18, 2624–2631. [Google Scholar] [CrossRef]
- Wang, Z.; Lang, X. Visible light photocatalysis of dye-sensitized TiO2: The selective aerobic oxidation of amines to imines. Appl. Catal. B Environ. 2018, 224, 404–409. [Google Scholar] [CrossRef]
- Li, X.; Xu, H.; Shi, J.-L.; Hao, H.; Yuan, H.; Lang, X. Salicylic acid complexed with TiO2 for visible light-driven selective oxidation of amines into imines with air. Appl. Catal. B Environ. 2019, 244, 758–766. [Google Scholar] [CrossRef]
- Lang, X.J.; Leow, W.R.; Zhao, J.C.; Chen, X.D. Synergistic photocatalytic aerobic oxidation of sulfides and amines on TiO2 under visible-light irradiation. Chem. Sci. 2015, 6, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Lang, X.J.; Hao, W.; Leow, W.R.; Li, S.Z.; Zhao, J.C.; Chen, X.D. Tertiary amine mediated aerobic oxidation of sulfides into sulfoxides by visible-light photoredox catalysis on TiO2. Chem. Sci. 2015, 6, 5000–5005. [Google Scholar] [CrossRef] [PubMed]
- Fagnoni, M.; Dondi, D.; Ravelli, D.; Albini, A. Photocatalysis for the formation of the C-C bond. Chem. Rev. 2007, 107, 2725–2756. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, M.; Tung, C.-H.; Wang, Y. TiO2 photocatalytic cyclization reactions for the syntheses of aryltetralones. ACS Catal. 2016, 6, 8389–8394. [Google Scholar] [CrossRef]
- Qiao, X.; Biswas, S.; Wu, W.; Zhu, F.; Tung, C.-H.; Wang, Y. Selective endoperoxide formation by heterogeneous TiO2 photocatalysis with dioxygen. Tetrahedron 2018, 74, 2421–2427. [Google Scholar] [CrossRef]
- Cermenati, L.; Richter, C.; Albini, A. Solar light induced carbon-carbon bond formation via TiO2 photocatalysis. Chem. Commun. 1998, 805–806. [Google Scholar] [CrossRef]
- Manley, D.W.; McBurney, R.T.; Miller, P.; Howe, R.F.; Rhydderch, S.; Walton, J.C. Unconventional titania photocatalysis: Direct deployment of carboxylic acids in alkylations and annulations. J. Am. Chem. Soc. 2012, 134, 13580–13583. [Google Scholar] [CrossRef] [PubMed]
- Manley, D.W.; McBurney, R.T.; Miller, P.; Walton, J.C. Titania-promoted carboxylic acid alkylations of alkenes and cascade addition-cyclizations. J. Org. Chem. 2014, 79, 1386–1398. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, S.; Hoffmann, N. Efficient radical addition of tertiary amines to electron-deficient alkenes using semiconductors as photochemical sensitisers. Chem. Commun. 2001, 10, 1576–1577. [Google Scholar] [CrossRef]
- Marinkovic, S.; Hoffmann, N. Semiconductors as sensitisers for the radical addition of tertiary amines to electron deficient alkenes. Int. J. Photoenergy 2003, 5, 175–182. [Google Scholar] [CrossRef]
- Cherevatskaya, M.; Neumann, M.; Fueldner, S.; Harlander, C.; Kuemmel, S.; Dankesreiter, S.; Pfitzner, A.; Zeitler, K.; Koenig, B. Visible-light-promoted stereoselective alkylation by combining heterogeneous photocatalysis with organocatalysis. Angew. Chem. Int. Ed. 2012, 51, 4062–4066. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Zoller, J.; Fabry, D.C.; Poscharny, K.; Koenigs, R.M.; Weirich, T.E.; Mayer, J. Light-mediated heterogeneous cross dehydrogenative coupling reactions: Metal oxides as efficient, recyclable, photoredox catalysts in C-C bond-forming reactions. Chem. Eur. J. 2012, 18, 3478–3481. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.G.; Yan, Y.; Ji, H.W.; Chen, C.C.; Zhao, J.C. Photocatalytic activation of pyridine for addition reactions: An unconventional reaction feature between a photo-induced hole and electron on TiO2. Chem. Commun. 2015, 51, 17451–17454. [Google Scholar] [CrossRef] [PubMed]
- Kohtani, S.; Nishioka, S.; Yoshioka, E.; Miyabe, H. Dye-sensitized photo-hydrogenation of aromatic ketones on titanium dioxide under visible light irradiation. Catal. Commun. 2014, 43, 61–65. [Google Scholar] [CrossRef]
- Kohtani, S.; Kawashima, A.; Masuda, F.; Sumi, M.; Kitagawa, Y.; Yoshioka, E.; Hasegawa, Y.; Miyabe, H. Chiral α-hydroxy acid-coadsorbed TiO2 photocatalysts for asymmetric induction in hydrogenation of aromatic ketones. Chem. Commun. 2018, 54, 12610–12613. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, Z. Coupling plasmonic noble metal with TiO2 for efficient photocatalytic transfer hydrogenation: M/TiO2 (m = Au and Pt) for chemoselective transformation of cinnamaldehyde to cinnamyl alcohol under visible and 365 nm UV light. Appl. Surf. Sci. 2018, 452, 279–285. [Google Scholar] [CrossRef]
- Füldner, S.; Mild, R.; Siegmund, H.I.; Schroeder, J.A.; Gruber, M.; König, B. Green-light photocatalytic reduction using dye-sensitized TiO2 and transition metal nanoparticles. Green Chem. 2010, 12, 400–406. [Google Scholar] [CrossRef]
- Zhu, H.Y.; Ke, X.B.; Yang, X.Z.; Sarina, S.; Liu, H.W. Reduction of nitroaromatic compounds on supported gold nanoparticles by visible and ultraviolet light. Angew. Chem. Int. Ed. 2010, 49, 9657–9661. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, B.; Goto, Y.; Nishimoto, S.-I.; Inui, T. Photocatalytic transfer hydrogenation of Schiff bases with propan-2-ol by suspended semiconductor particles loaded with platinum deposits. J. Chem. Soc. Faraday Trans. 1996, 92, 4291–4295. [Google Scholar] [CrossRef]
- Brezova, V.; Blazkova, A.; Surina, I.; Havlinova, B. Solvent effect on the photocatalytic reduction of 4-nitrophenol in titanium dioxide suspensions. J. Photochem. Photobiol. A Chem. 1997, 107, 233–237. [Google Scholar] [CrossRef]
- Brezová, V.; Tarábek, P.; Dvoranová, D.; Staško, A.; Biskupič, S. EPR study of photoinduced reduction of nitroso compounds in titanium dioxide suspensions. J. Photochem. Photobiol. A Chem. 2003, 155, 179–198. [Google Scholar] [CrossRef]
- Ferry, J.L.; Glaze, W.H. Photocatalytic reduction of nitroorganics over illuminated titanium dioxide: Electron transfer between excited-state TiO2 and nitroaromatics. J. Phys. Chem. B 1998, 102, 2239–2244. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, Y.; Song, W.; Chen, C.; Zhao, J. Photocatalytic hydrodehalogenation for the removal of halogenated aromatic contaminants. ChemCatChem 2018. [Google Scholar] [CrossRef]
- Ouellet, S.G.; Tuttle, J.B.; MacMillan, D.W.C. Enantioselective organocatalytic hydride reduction. J. Am. Chem. Soc. 2005, 127, 32–33. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, J.B.; Ouellet, S.G.; MacMillan, D.W.C. Organocatalytic transfer hydrogenation of cyclic enones. J. Am. Chem. Soc. 2006, 128, 12662–12663. [Google Scholar] [CrossRef]
- Chau, A.; Paquin, J.-F.; Lautens, M. Diastereoselective palladium-catalyzed formate reduction of allylic carbonates en route to polypropionate systems. J. Org. Chem. 2006, 71, 1924–1933. [Google Scholar] [CrossRef]
- Ohshima, T.; Tadaoka, H.; Hori, K.; Sayo, N.; Mashima, K. Highly enantio- and s-trans C=C bond selective catalytic hydrogenation of cyclic enones: Alternative synthesis of (-)-menthol. Chem. Eur. J. 2008, 14, 2060–2066. [Google Scholar] [CrossRef]
- Blakemore, D.C.; Castro, L.; Churcher, I.; Rees, D.C.; Thomas, A.W.; Wilson, D.M.; Wood, A. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 2018, 10, 383–394. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37. [Google Scholar] [CrossRef]
- Boonstra, A.H.; Mutsaers, C.A.H.A. Photohydrogenation of ethyne and ethene on the surface of titanium dioxide. J. Phys. Chem. 1975, 79, 2025–2027. [Google Scholar] [CrossRef]
- Yun, C.; Ampo, M.; Kodama, S.; Kubokawa, Y. UV Irradiation-induced fission of a C=C or C≡C bond adsorbed on TiO2. J. Chem. Soc. Chem. Commun. 1980, 9, 609. [Google Scholar] [CrossRef]
- Anpo, M.; Aikawa, N.; Kodama, S.; Kubokawa, Y. Photocatalytic hydrogenation of alkynes and alkenes with water over titanium dioxide. Hydrogenation accompanied by bond fission. J. Phys. Chem. 1984, 88, 2569–2572. [Google Scholar] [CrossRef]
- Anpo, M.; Aikawa, N.; Kubokawa, Y. Photocatalytic hydrogenation of alkynes and alkenes with water over titanium dioxide. Platinum loading effect on the primary processes. J. Phys. Chem. 1984, 88, 3998–4000. [Google Scholar] [CrossRef]
- Anpo, M.; Shima, T.; Kodama, S.; Kubokawa, Y. Photocatalytic hydrogenation of propyne with water on small-particle titania: Size quantization effects and reaction intermediates. J. Phys. Chem. 1987, 91, 4305–4310. [Google Scholar] [CrossRef]
- Yamataka, H.; Seto, N.; Ichihara, J.; Hanafusa, T.; Teratani, S. Reduction of C-C multiple bonds using an illuminated semiconductor catalyst. J. Chem. Soc. Chem. Commun. 1985, 788–789. [Google Scholar] [CrossRef]
- Baba, R.; Nakabayashi, S.; Fujishima, A.; Honda, K. Photocatalytic hydrogenation of ethylene on bimetal-deposited semiconductor powders. J. Am. Chem. Soc. 1987, 109, 2273–2277. [Google Scholar] [CrossRef]
- Cai, Z.S.; Kuntz, R.R. Photohydrogenation of acetylene in titanium dioxide-based colloidal aqueous solutions. Langmuir 1988, 4, 830–836. [Google Scholar] [CrossRef]
- Al-Thabaiti, S.; Kuntz, R.R. Photocatalytic hydrogenation of acetylene by dimolybdenum and trimolybdenum oxo species in colloidal titania solutions. Langmuir 1990, 6, 782–786. [Google Scholar] [CrossRef]
- Lin, L.; Kuntz, R.R. Photocatalytic hydrogenation of acetylene by molybdenum-sulfur complexes supported on titania. Langmuir 1992, 8, 870–875. [Google Scholar] [CrossRef]
- Kuntz, R.R. Comparative study of Mo2OxSy(cys)22− complexes as catalysts for electron transfer from irradiated colloidal TiO2 to acetylene. Langmuir 1997, 13, 1571–1576. [Google Scholar] [CrossRef]
- Manley, D.; Buzzetti, L.; MacKessack-Leitch, A.; Walton, J. Hydrogenations without hydrogen: Titania photocatalyzed reductions of maleimides and aldehydes. Molecules 2014, 19, 15324. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Okubo, Y.; Ito, T.; Tanaka, A.; Hashimoto, K.; Kominami, H. Photocatalytic hydrogenation of alkenes to alkanes in alcoholic suspensions of palladium-loaded titanium(iv) oxide without the use of hydrogen gas. RSC Adv. 2014, 4, 19883–19886. [Google Scholar] [CrossRef]
- Kominami, H.; Kitagawa, S.-Y.; Okubo, Y.; Fukui, M.; Hashimoto, K.; Imamura, K. Organically modified titania having a metal catalyst: A new type of liquid-phase hydrogen-transfer photocatalyst working under visible light irradiation and H2-free conditions. Phys. Chem. Chem. Phys. 2016, 18, 16076–16079. [Google Scholar] [CrossRef] [PubMed]
- Kohtani, S.; Yoshioka, E.; Saito, K.; Kudo, A.; Miyabe, H. Photocatalytic hydrogenation of acetophenone derivatives and diaryl ketones on polycrystalline titanium dioxide. Catal. Commun. 2010, 11, 1049–1053. [Google Scholar] [CrossRef]
- Kohtani, S.; Yoshioka, E.; Saito, K.; Kudo, A.; Miyabe, H. Adsorptive and kinetic properties on photocatalytic hydrogenation of aromatic ketones upon uv irradiated polycrystalline titanium dioxide: Differences between acetophenone and its trifluoromethylated derivative. J. Phys. Chem. C 2012, 116, 17705–17713. [Google Scholar] [CrossRef]
- Kohtani, S.; Kamoi, Y.; Yoshioka, E.; Miyabe, H. Kinetic study on photocatalytic hydrogenation of acetophenone derivatives on titanium dioxide. Catal. Sci. Technol. 2014, 4, 1084–1091. [Google Scholar] [CrossRef]
- Kohtani, S.; Mori, M.; Yoshioka, E.; Miyabe, H. Photohydrogenation of acetophenone using coumarin dye-sensitized titanium dioxide under visible light irradiation. Catalysts 2015, 5, 1417–1424. [Google Scholar] [CrossRef]
- Kohtani, S.; Kurokawa, T.; Yoshioka, E.; Miyabe, H. Photoreductive transformation of fluorinated acetophenone derivatives on titanium dioxide: Defluorination vs. Reduction of carbonyl group. Appl. Catal. A Gen. 2016, 521, 68–74. [Google Scholar] [CrossRef]
- Kohtani, S.; Kawashima, A.; Miyabe, H. Reactivity of trapped and accumulated electrons in titanium dioxide photocatalysis. Catalysts 2017, 7, 303. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, M.; Sun, K.; Tang, Z.; Kotov, N.A. Similar topological origin of chiral centers in organic and nanoscale inorganic structures: Effect of stabilizer chirality on optical isomerism and growth of cdte nanocrystals. J. Am. Chem. Soc. 2010, 132, 6006–6013. [Google Scholar] [CrossRef]
- Jiang, S.; Chekini, M.; Qu, Z.-B.; Wang, Y.; Yeltik, A.; Liu, Y.; Kotlyar, A.; Zhang, T.; Li, B.; Demir, H.V.; et al. Chiral ceramic nanoparticles and peptide catalysis. J. Am. Chem. Soc. 2017, 139, 13701–13712. [Google Scholar] [CrossRef] [PubMed]
- Joycepruden, C.; Pross, J.K.; Li, Y.Z. Photoinduced reduction of aldehydes on titanium-dioxide. J. Org. Chem. 1992, 57, 5087–5091. [Google Scholar] [CrossRef]
- Ohtani, B.; Osaki, H.; Nishimoto, S.; Kagiya, T. A novel photocatalytic process of amine N-alkylation by platinized semiconductor particles suspended in alcohols. J. Am. Chem. Soc. 1986, 108, 308–310. [Google Scholar] [CrossRef]
- Ohtani, B.; Tsuru, S.; Nishimoto, S.; Kagiya, T.; Izawa, K. Photocatalytic one-step syntheses of cyclic imino acids by aqueous semiconductor suspensions. J. Org. Chem. 1990, 55, 5551–5553. [Google Scholar] [CrossRef]
- Nishimoto, S.; Ohtani, B.; Yoshikawa, T.; Kagiya, T. Photocatalytic conversion of primary amines to secondary amines and cyclization of polymethylene-α,ω-diamines by an aqueous suspension of titanium(iv) oxide/platinum. J. Am. Chem. Soc. 1983, 105, 7180–7182. [Google Scholar] [CrossRef]
- Fukui, M.; Tanaka, A.; Hashimoto, K.; Kominami, H. Visible light-induced heterogeneous Meerwein–Ponndorf–Verley-type reduction of an aldehyde group over an organically modified titanium dioxide photocatalyst. Chem. Commun. 2017, 53, 4215–4218. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Blaser, H.-U.; Steiner, H.; Studer, M. Selective catalytic hydrogenation of functionalized nitroarenes: An update. ChemCatChem 2009, 1, 210–221. [Google Scholar] [CrossRef]
- Blaser, H.U.; Malan, C.; Pugin, B.; Spindler, F.; Steiner, H.; Studer, M. Selective hydrogenation for fine chemicals: Recent trends and new developments. Adv. Synth. Catal. 2003, 345, 103–151. [Google Scholar] [CrossRef]
- Imamura, K.; Nakanishi, K.; Hashimoto, K.; Kominami, H. Chemoselective reduction of nitrobenzenes having other reducible groups over titanium(iv) oxide photocatalyst under protection-, gas-, and metal-free conditions. Tetrahedron 2014, 70, 6134–6139. [Google Scholar] [CrossRef]
- Mahdavi, F.; Bruton, T.C.; Li, Y.Z. Photoinduced reduction of nitro-compounds on semiconductor particles. J. Org. Chem. 1993, 58, 744–746. [Google Scholar] [CrossRef]
- Ferry, J.L.; Glaze, W.H. Photocatalytic reduction of nitro organics over illuminated titanium dioxide: Role of the TiO2 surface. Langmuir 1998, 14, 3551–3555. [Google Scholar] [CrossRef]
- Makarova, O.V.; Rajh, T.; Thurnauer, M.C.; Martin, A.; Kemme, P.A.; Cropek, D. Surface modification of TiO2 nanoparticles for photochemical reduction of nitrobenzene. Environ. Sci. Technol. 2000, 34, 4797–4803. [Google Scholar] [CrossRef]
- Rajh, T.; Makarova, O.V.; Thurnauer, M.C.; Kemme, P.A.; Cropek, D. Surface modification of TiO2 nanoparticles for selective adsorption of nitrobenzene. Abstr. Pap. Am. Chem. Soc. 2001, 222, U428. [Google Scholar]
- Cropek, D.; Kemme, P.A.; Makarova, O.V.; Chen, L.X.; Rajh, T. Selective photocatalytic decomposition of nitrobenzene using surface modified TiO2 nanoparticles. J. Phys. Chem. C 2008, 112, 8311–8318. [Google Scholar] [CrossRef]
- Tada, H.; Ishida, T.; Takao, A.; Ito, S. Drastic enhancement of TiO2-photocatalyzed reduction of nitrobenzene by loading Ag clusters. Langmuir 2004, 20, 7898–7900. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; You, L.; Zhang, Y. Photocatalytic reduction of p-chloronitrobenzene on illuminated nano-titanium dioxide particles. Dyes Pigments 2006, 68, 95–100. [Google Scholar] [CrossRef]
- Hiroshi, K.; Shin-ichi, I.; Tsuyoshi, M.; Kazuya, I.; Keiji, H.; Yoshiya, K.; Bunsho, O. Photocatalytic reduction of nitrobenzene to aniline in an aqueous suspension of titanium(iv) oxide particles in the presence of oxalic acid as a hole scavenger and promotive effect of dioxygen in the system. Chem. Lett. 2009, 38, 410–411. [Google Scholar]
- Shiraishi, Y.; Hirakawa, H.; Togawa, Y.; Sugano, Y.; Ichikawa, S.; Hirai, T. Rutile crystallites isolated from degussa (evonik) P25 TiO2: Highly efficient photocatalyst for chemoselective hydrogenation of nitroaromatics. ACS Catal. 2013, 3, 2318–2326. [Google Scholar] [CrossRef]
- Hoffmann, M.R.; Martin, S.T.; Choi, W.; Bahnemann, D.W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar] [CrossRef]
- Parrino, F.; Bellardita, M.; García-López, E.I.; Marcì, G.; Loddo, V.; Palmisano, L. Heterogeneous photocatalysis for selective formation of high-value-added molecules: Some chemical and engineering aspects. ACS Catal. 2018, 8, 11191–11225. [Google Scholar] [CrossRef]
Sample Availability: Not available. |



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
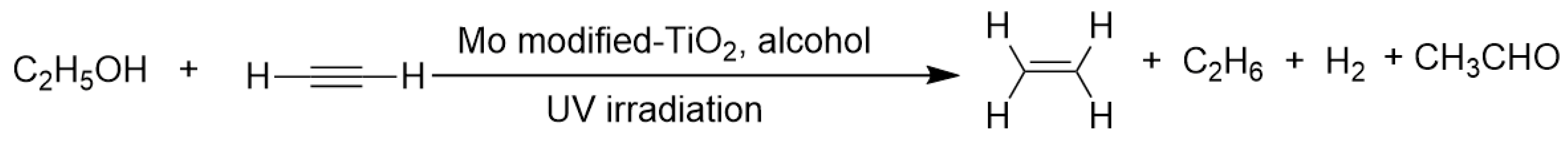{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Solvent | t/h | Conversion/% | Product | Yield/% |
|---|---|---|---|---|---|---|
| 1 |  | i-PrOH | 6 | >99 |  | >99 |
| 2 |  | i-PrOH/toluene (1/9) | 4 | >99 |  | 98 |
| 3 |  | i-PrOH/toluene (9/1) | 5 | >99 |  | 94 |
| 4 |  | i-PrOH/toluene (5/5) | 7 | >99 |  | 98 |
| 5 |  | i-PrOH/toluene (1/9) | 6 | >99 |  | 94 |
| 6 |  | i-PrOH/toluene (1/9) | 6 | >99 |  | 97 |
| 7 |  | i-PrOH/toluene (1/9) | 6 | >99 |  | 94 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, D.; Zhai, S.; Wang, Y.; Liu, A.; Chen, C. TiO2 Photocatalysis for Transfer Hydrogenation. Molecules 2019, 24, 330. https://doi.org/10.3390/molecules24020330
Ma D, Zhai S, Wang Y, Liu A, Chen C. TiO2 Photocatalysis for Transfer Hydrogenation. Molecules. 2019; 24(2):330. https://doi.org/10.3390/molecules24020330
Chicago/Turabian StyleMa, Dongge, Shan Zhai, Yi Wang, Anan Liu, and Chuncheng Chen. 2019. "TiO2 Photocatalysis for Transfer Hydrogenation" Molecules 24, no. 2: 330. https://doi.org/10.3390/molecules24020330
APA StyleMa, D., Zhai, S., Wang, Y., Liu, A., & Chen, C. (2019). TiO2 Photocatalysis for Transfer Hydrogenation. Molecules, 24(2), 330. https://doi.org/10.3390/molecules24020330