Tripleurin XIIc: Peptide Folding Dynamics in Aqueous and Hydrophobic Environment Mimic Using Accelerated Molecular Dynamics

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
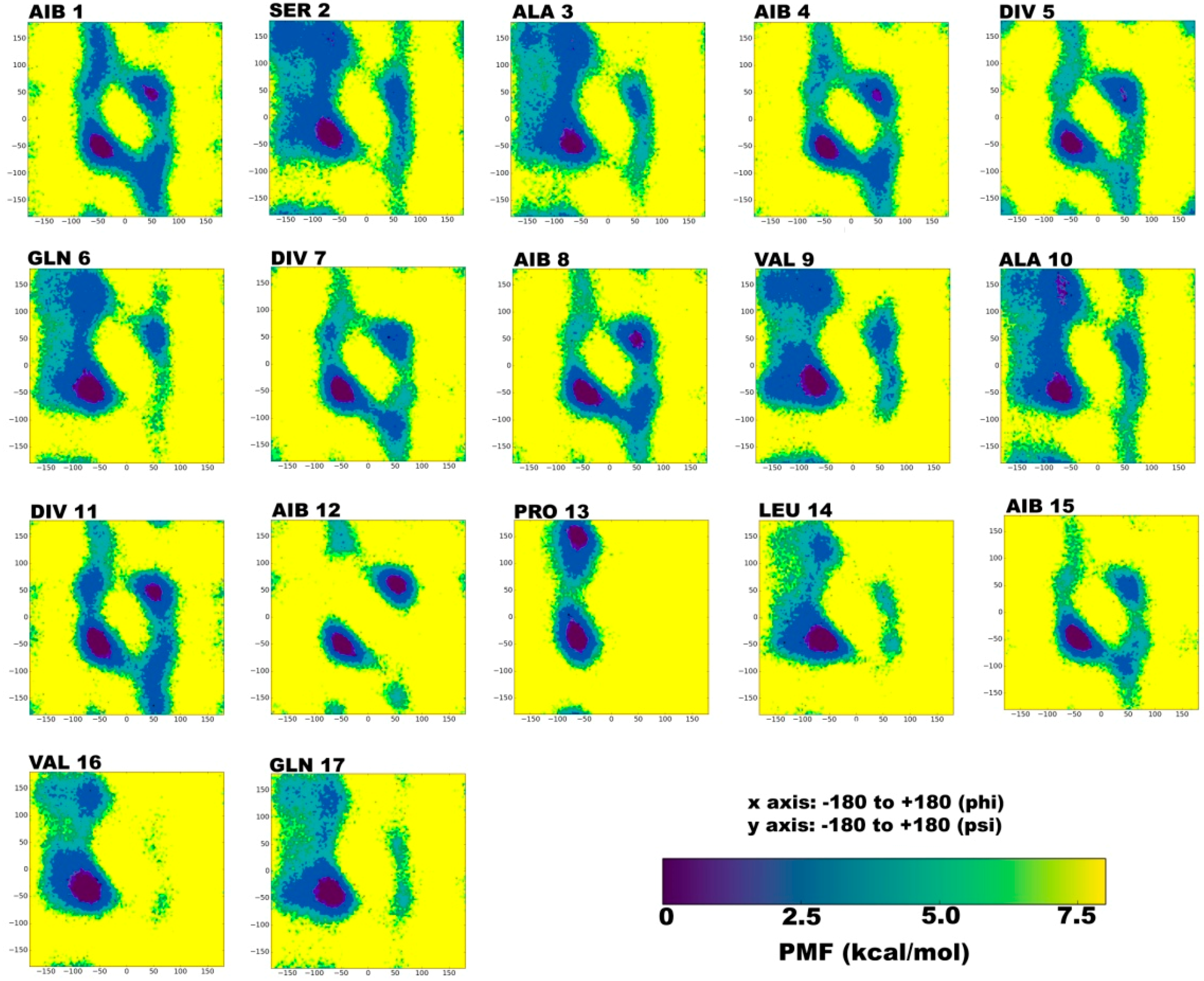
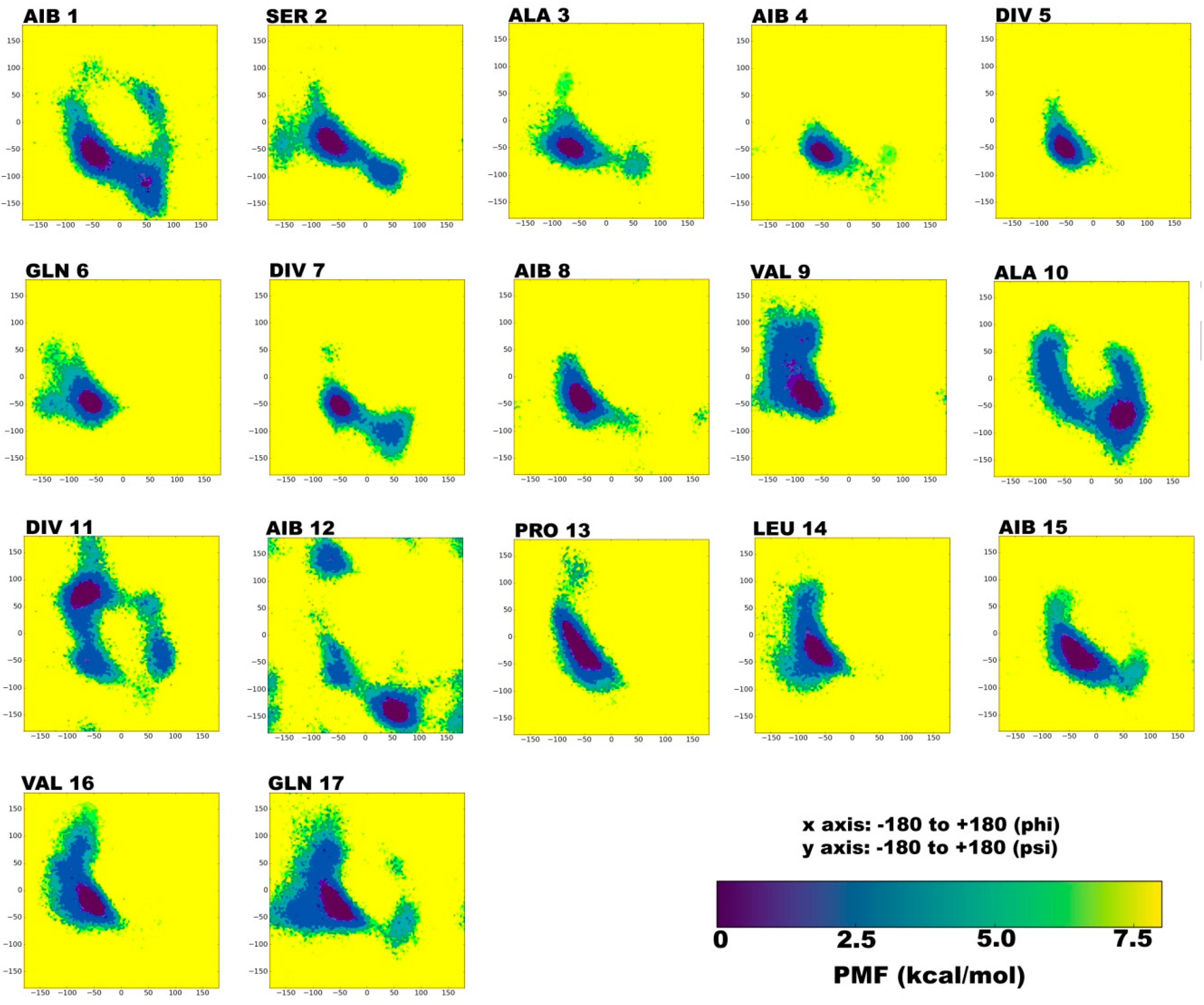
2.1. Secondary Structural Populations in Water and Chloroform Solvents
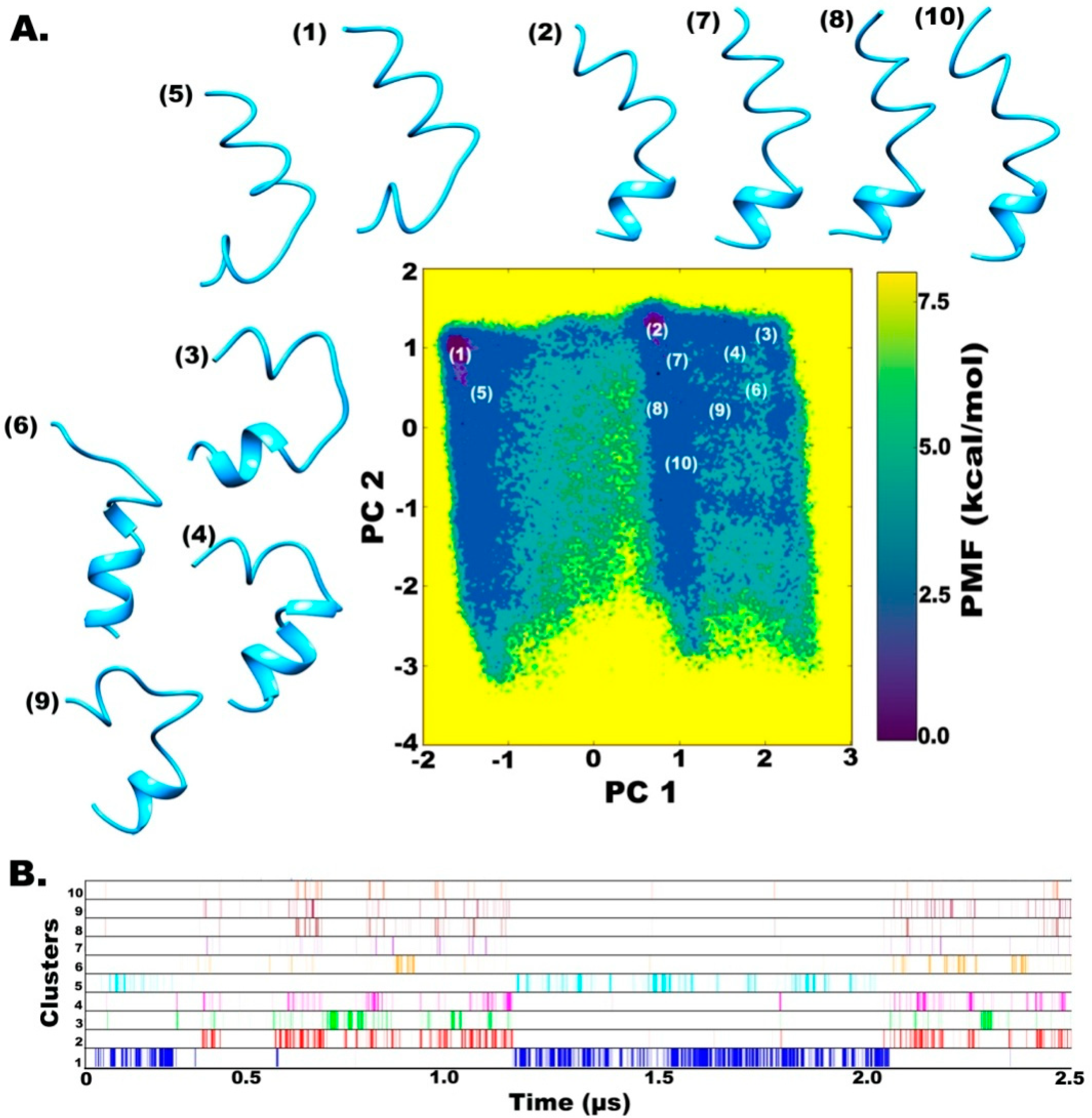
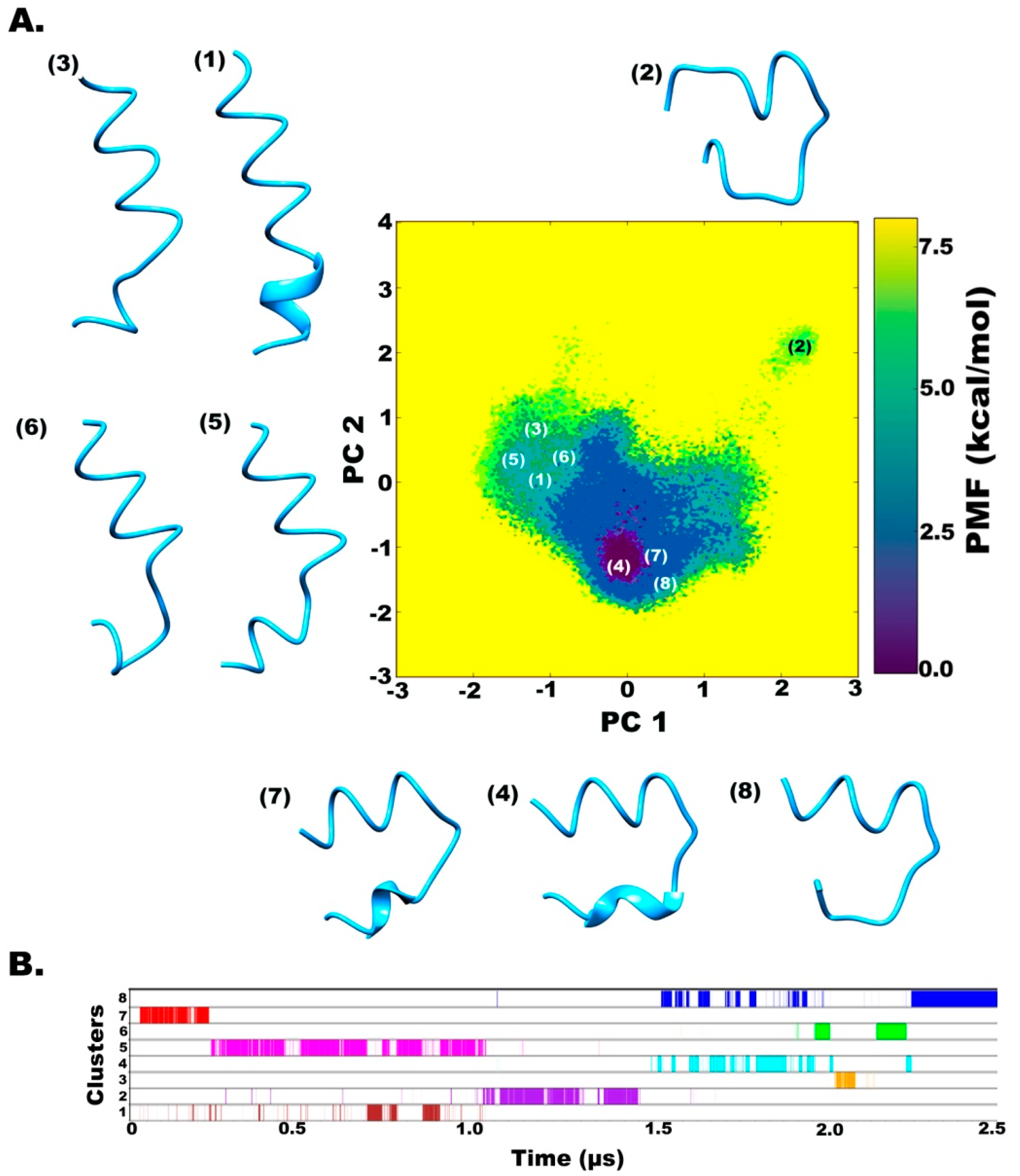
2.2. Clustering based on Free Energy Landscapes (FEL): Vision through Principal Component Analysis
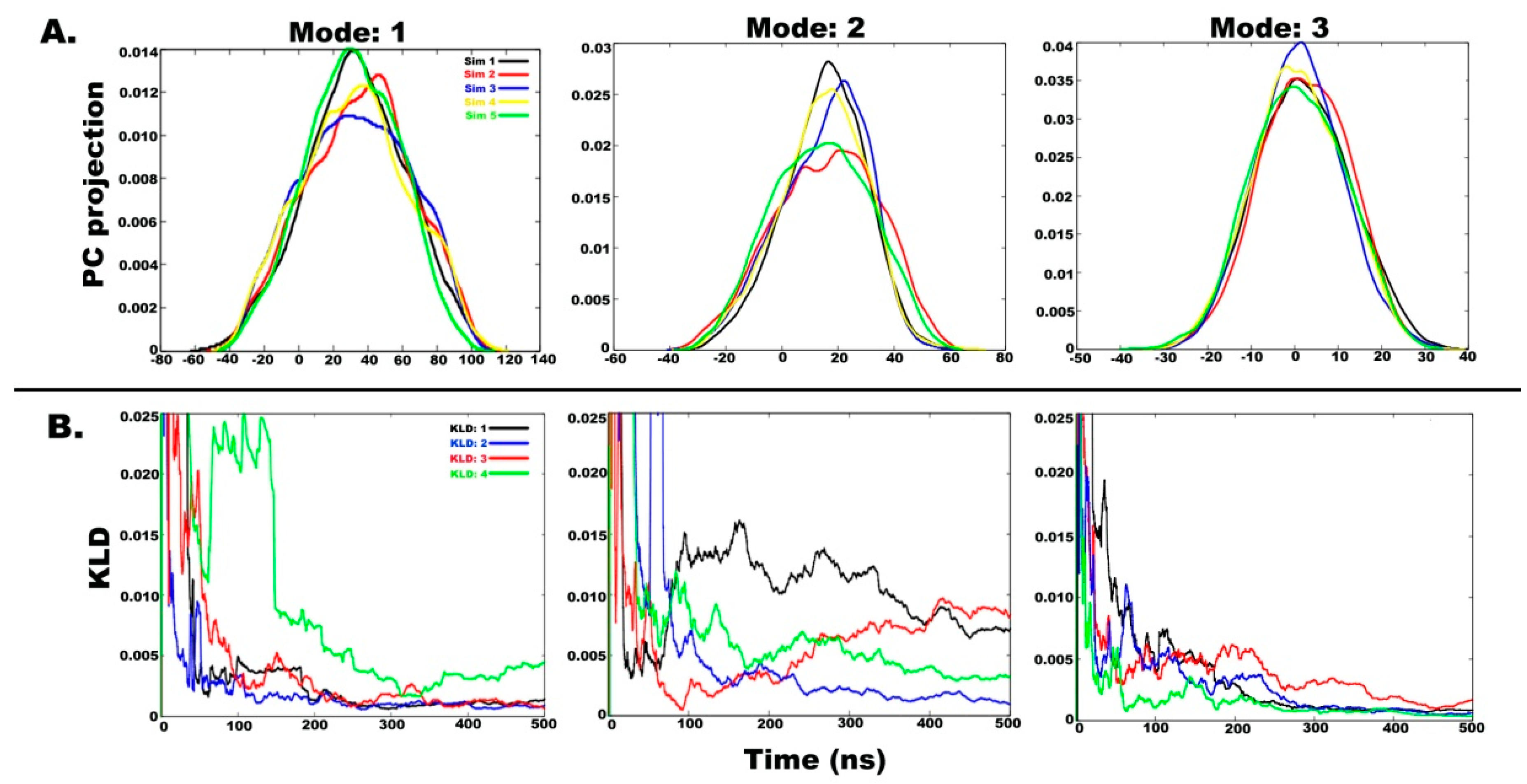
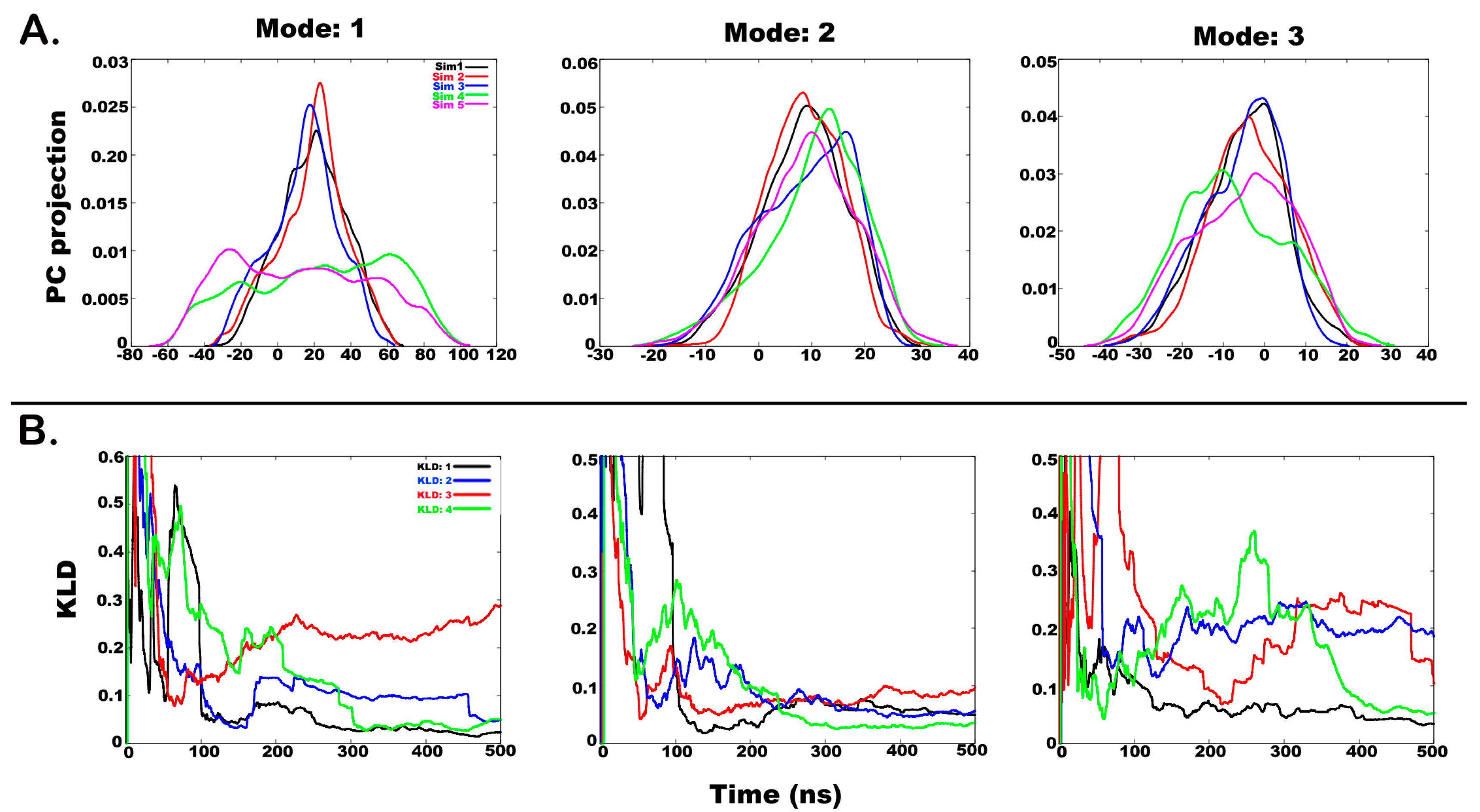
2.3. Understanding the Dynamics of TPN XIIc through Cartesian PCA
3. Materials and Methods
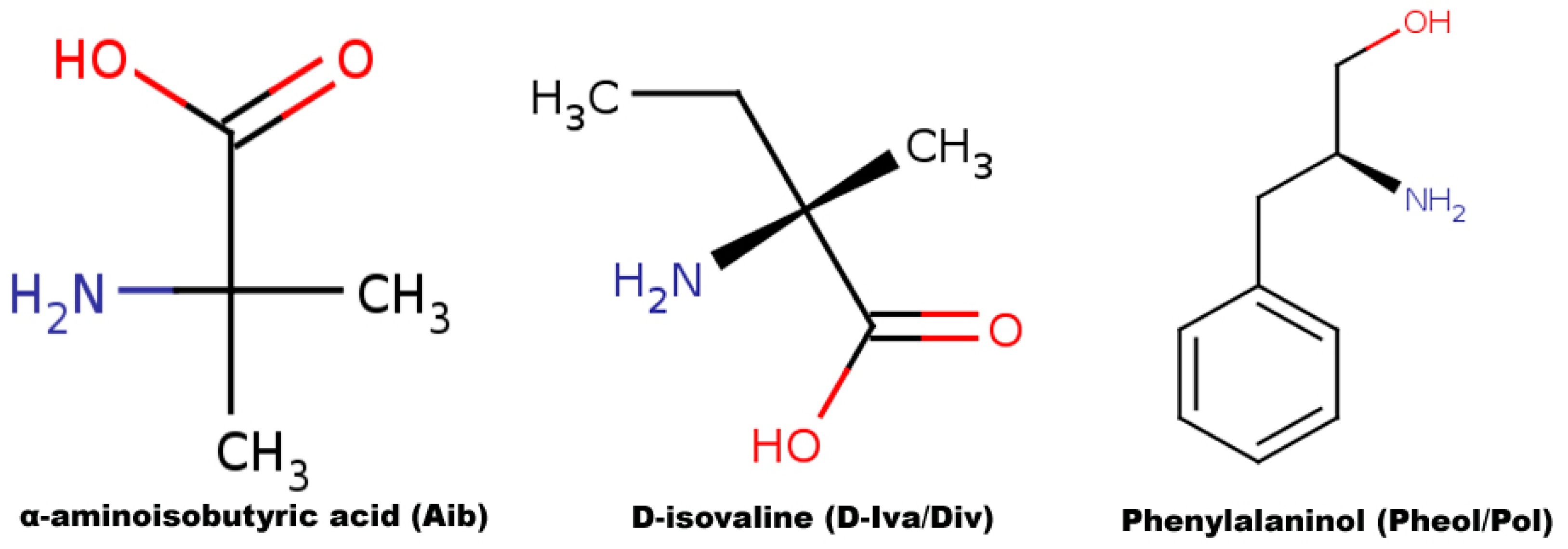
3.1. Sequence Selection
3.2. Partial charge Calculation and Force Field Library Generation for Non-Standard Residues
3.3. Accelerated Molecular Dynamics Simulations of TPN XIIc
Etotal = Vavg_total + b1 × Natoms, αtotal = b2 × Natoms
3.4. Tools for Simulation Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mohamed-Benkada, M.; Francois, P.Y.; Verite, P.; Pagniez, F.; Caroff, N.; Ruiz, N. Identification and biological activities of long-chain peptaibols produced by a marine-derived strain of Trichoderma longibrachiatum. Chem. Biodivers. 2016, 13, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.; Grigoriev, P.A.; Degenkolb, T.; Neuhof, T.; Hartl, A.; Schlegel, B.; Grafe, U. Isolation, structure elucidation and biological activities of trichofumins A, B, C and D, new 11 and 13mer peptaibols from Trichoderma sp. HKI 0276W. J. Pept. Sci. 2003, 9, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Leitgeb, B.; Szekeres, A.; Manczinger, L.; Vagvolgyi, C.; Kredics, L. The history of alamethicin: A review of the most extensively studied peptaibol. Chem. Biodivers. 2007, 4, 1027–1051. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.-S.; Yoo, I.-D.; Kim, Y.H.; Kim, Y.-S.; Lee, S.-J.; Kim, K.-S.; Yeo, W.-H. Peptaivirins A and B, two new antiviral peptaibols against TMV infection. Tetrahedron Lett. 2000, 41, 1429–1431. [Google Scholar] [CrossRef]
- Schiell, M.; Hofmann, J.; Kurz, M.; Schmidt, F.R.; Vertesy, L.; Vogel, M.; Wink, J.; Seibert, G. Cephaibols, new peptaibol antibiotics with anthelmintic properties from Acremonium tubakii DSM 12774. J. Antibiot. 2001, 54, 220–233. [Google Scholar] [CrossRef]
- Viterbo, A.; Wiest, A.; Brotman, Y.; Chet, I.; Kenerley, C. The 18mer peptaibols from Trichoderma virens elicit plant defence responses. Mol. Plant Pathol. 2007, 8, 737–746. [Google Scholar] [CrossRef]
- Engelberth, J.; Koch, T.; Kuhnemann, F.; Boland, W. Channel-Forming Peptaibols Are Potent Elicitors of Plant Secondary Metabolism and Tendril Coiling. Angew. Chem. Int. Ed. Engl. 2000, 39, 1860–1862. [Google Scholar] [CrossRef]
- Miller, B.R.; Gulick, A.M. Structural biology of nonribosomal peptide synthetases. In Nonribosomal Peptide and Polyketide Biosynthesis; Evans, B., Ed.; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1401, pp. 3–29. [Google Scholar]
- Raap, J.; Erkelens, K.; Ogrel, A.; Skladnev, D.A.; Brückner, H. Fungal biosynthesis of non-ribosomal peptide antibiotics and α, α-dialkylated amino acid constituents. J. Pept. Sci. 2005, 11, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Shenkarev, Z.O.; Balashova, T.A.; Efremov, R.G.; Yakimenko, Z.A.; Ovchinnikova, T.V.; Raap, J.; Arseniev, A.S. Spatial structure of zervamicin IIB bound to DPC micelles: Implications for voltage-gating. Biophys J 2002, 82, 762–771. [Google Scholar] [CrossRef]
- Karle, I.L.; Flippen-Anderson, J.L.; Agarwalla, S.; Balaram, P. Crystal structure of [Leu1]zervamicin, a membrane ion-channel peptide: Implications for gating mechanisms. Proc. Natl. Acad. Sci. USA 1991, 88, 5307–5311. [Google Scholar] [CrossRef]
- Fox, R.O., Jr.; Richards, F.M. A voltage-gated ion channel model inferred from the crystal structure of alamethicin at 1.5-A resolution. Nature 1982, 300, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Balashova, T.A.; Shenkarev, Z.O.; Tagaev, A.A.; Ovchinnikova, T.V.; Raap, J.; Arseniev, A.S. NMR structure of the channel-former zervamicin IIB in isotropic solvents. FEBS Lett. 2000, 466, 333–336. [Google Scholar] [CrossRef] [Green Version]
- Condamine, E.; Rebuffat, S.; Prigent, Y.; Ségalas, I.; Bodo, B.; Davoust, D. Three-dimensional structure of the ion-chanel forming peptide trichorzianin TA VII bound to sodium dodecyl sulfate micelles. Biopolymers 1998, 46, 75–88. [Google Scholar] [CrossRef]
- Snook, C.F.; Woolley, G.A.; Oliva, G.; Pattabhi, V.; Wood, S.F.; Blundell, T.L.; Wallace, B.A. The structure and function of antiamoebin I, a proline-rich membrane-active polypeptide. Structure 1998, 6, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Kronen, M.; Gorls, H.; Nguyen, H.H.; Reissmann, S.; Bohl, M.; Suhnel, J.; Grafe, U. Crystal structure and conformational analysis of ampullosporin A. J. Pept. Sci. 2003, 9, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Nagao, T.; Mishima, D.; Javkhlantugs, N.; Wang, J.; Ishioka, D.; Yokota, K.; Norisada, K.; Kawamura, I.; Ueda, K.; Naito, A. Structure and orientation of antibiotic peptide alamethicin in phospholipid bilayers as revealed by chemical shift oscillation analysis of solid state nuclear magnetic resonance and molecular dynamics simulation. Biochim. Biophys. Acta 2015, 1848, 2789–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, S.E.; Roberts, K.; Vaidehi, N. Position of helical kinks in membrane protein crystal structures and the accuracy of computational prediction. J. Mol. Graph. Model. 2009, 27, 944–950. [Google Scholar] [CrossRef]
- Marik, T.; Urban, P.; Tyagi, C.; Szekeres, A.; Leitgeb, B.; Vagvolgyi, M.; Manczinger, L.; Druzhinina, I.S.; Vagvolgyi, C.; Kredics, L. Diversity profile and dynamics of peptaibols produced by green mould trichoderma species in interactions with their hosts Agaricus bisporus and Pleurotus ostreatus. Chem. Biodivers. 2017, 14, e1700033. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Karplus, P.A. A fresh look at the Ramachandran plot and the occurrence of standard structures in proteins. Biomol. Concepts 2010, 1, 271–283. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef]
- Hamelberg, D.; de Oliveira, C.A.; McCammon, J.A. Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys. 2007, 127, 155102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, L.C.; Salomon-Ferrer, R.; Augusto, F.d.O.C.; McCammon, J.A.; Walker, R.C. Routine access to millisecond time scale events with accelerated molecular dynamics. J. Chem. Theory Comput. 2012, 8, 2997–3002. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Feixas, F.; Eun, C.; McCammon, J.A. Accelerated molecular dynamics simulations of protein folding. J. Comput. Chem. 2015, 36, 1536–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kullback, S.; Leibler, R.A. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Banerjee, R.; Basu, G. A short Aib/Ala-based peptide helix is as stable as an Ala-based peptide helix double its length. ChemBioChem 2002, 3, 1263–1266. [Google Scholar] [CrossRef]
- Banerjee, R.; Basu, G.; Roy, S.; Chène, P. Aib-based peptide backbone as scaffolds for helical peptide mimics. J. Pept. Res. 2002, 60, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, G.; Misawa, T.; Doi, M.; Demizu, Y. Extent of helical induction caused by introducing α-aminoisobutyric acid into an oligovaline sequence. ACS Omega 2018, 3, 6395–6399. [Google Scholar] [CrossRef]
- Mondal, S.; Adler-Abramovich, L.; Lampel, A.; Bram, Y.; Lipstman, S.; Gazit, E. Formation of functional super-helical assemblies by constrained single heptad repeat. Nat. Commun. 2015, 6, 8615. [Google Scholar] [CrossRef] [Green Version]
- Wada, S.-i.; Tsuda, H.; Okada, T.; Urata, H. Cellular uptake of Aib-containing amphipathic helix peptide. Bioorganic. Med. Chem. Lett. 2011, 21, 5688–5691. [Google Scholar] [CrossRef]
- Li, H.; Anuwongcharoen, N.; Malik, A.A.; Prachayasittikul, V.; Wikberg, J.E.; Nantasenamat, C. Roles of d-amino acids on the bioactivity of host defense peptides. Int. J. Mol. Sci. 2016, 17, 1023. [Google Scholar] [CrossRef]
- Carmona, G.; Rodriguez, A.; Juarez, D.; Corzo, G.; Villegas, E. Improved protease stability of the antimicrobial peptide Pin2 substituted with d-amino acids. Protein J. 2013, 32, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Tugyi, R.; Uray, K.; Ivan, D.; Fellinger, E.; Perkins, A.; Hudecz, F. Partial D-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 413–418. [Google Scholar] [CrossRef] [Green Version]
- De Zotti, M.; Biondi, B.; Crisma, M.; Hjorringgaard, C.U.; Berg, A.; Bruckner, H.; Toniolo, C. Isovaline in naturally occurring peptides: A nondestructive methodology for configurational assignment. Biopolymers 2012, 98, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Formaggio, F.; Crisma, M.; Bonora, G.M.; Pantano, M.; Valle, G.; Toniolo, C.; Aubry, A.; Bayeul, D.; Kamphuis, J. (R)-isovaline homo-peptides adopt the left-handed 3(10)-helical structure. Pept. Res. 1995, 8, 6–15. [Google Scholar] [PubMed]
- Shenkarev, Z.O.; Paramonov, A.S.; Lyukmanova, E.N.; Gizatullina, A.K.; Zhuravleva, A.V.; Tagaev, A.A.; Yakimenko, Z.A.; Telezhinskaya, I.N.; Kirpichnikov, M.P.; Ovchinnikova, T.V. Peptaibol antiamoebin I: Spatial structure, backbone dynamics, interaction with bicelles and lipid-protein nanodiscs, and pore formation in context of barrel-stave model. Chem. Biodivers. 2013, 10, 838–863. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.M.; Zondlo, N.J. A propensity scale for type II polyproline helices (PPII): Aromatic amino acids in proline-rich sequences strongly disfavor PPII due to proline-aromatic interactions. Biochemistry 2012, 51, 5041–5051. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Martinez, J.; Tomas, M.S.; Perez, J.J. Effect of the solvent on the conformational behavior of the alanine dipeptide deduced from MD simulations. J. Mol. Graph. Model. 2017, 78, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Principal component analysis for protein folding dynamics. J. Mol. Biol. 2009, 385, 312–329. [Google Scholar] [CrossRef]
- Sittel, F.; Jain, A.; Stock, G. Principal component analysis of molecular dynamics: On the use of Cartesian vs. internal coordinates. J. Chem. Phys. 2014, 141, 014111. [Google Scholar] [CrossRef]
- Altis, A.; Nguyen, P.H.; Hegger, R.; Stock, G. Dihedral angle principal component analysis of molecular dynamics simulations. J. Chem. Phys. 2007, 126, 244111. [Google Scholar] [CrossRef]
- Mu, Y.; Nguyen, P.H.; Stock, G. Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins 2005, 58, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Murillo, R.; Roe, D.R.; Cheatham, T.E., III. Convergence and reproducibility in molecular dynamics simulations of the DNA duplex d (GCACGAACGAACGAACGC). Biochim. Biophys. Acta (BBA) 2015, 1850, 1041–1058. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Bergonzo, C.; Cheatham, T.E., 3rd. Evaluation of enhanced sampling provided by accelerated molecular dynamics with Hamiltonian replica exchange methods. J. Phys. Chem. B 2014, 118, 3543–3552. [Google Scholar] [CrossRef] [PubMed]
- Koukos, P.I.; Glykos, N.M. On the application of Good-Turing statistics to quantify convergence of biomolecular simulations. J. Chem. Inf. Model. 2014, 54, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Serafeim, A.P.; Salamanos, G.; Patapati, K.K.; Glykos, N.M. Sensitivity of folding molecular dynamics simulations to even minor force field changes. J. Chem. Inf. Model. 2016, 56, 2035–2041. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering molecular dynamics trajectories: 1. characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Jortner, J.; Becker, O.M. Solvent effects on the energy landscapes and folding kinetics of polyalanine. Proc. Natl. Acad. Sci. USA 2001, 98, 2188–2193. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Medema, M.H.; Kottmann, R.; Lee, S.Y.; Weber, T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2017, 45, D555–D559. [Google Scholar] [CrossRef]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.Y. R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011, 39, W511–W517. [Google Scholar] [CrossRef]
- Dupradeau, F.Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The REd. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Gaussian Inc.: Wallingford, CT, USA, 2009.
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Cieplak, P.; Cornell, W.D.; Bayly, C.; Kollman, P.A. Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem. 1995, 16, 1357–1377. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.C.; Cheatham, T.E.I.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Wereszczynski, J.; McCammon, J.A. Using selectively applied accelerated molecular dynamics to enhance free energy calculations. J. Chem. Theory Comput. 2010, 6, 3285–3292. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Glykos, N.M. Software news and updates. Carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Koukos, P.I.; Glykos, N.M. Grcarma: A fully automated task-oriented interface for the analysis of molecular dynamics trajectories. J. Comput. Chem. 2013, 34, 2310–2312. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Sinko, W.; Pierce, L.; Bucher, D.; Walker, R.C.; McCammon, J.A. Improved reweighting of accelerated molecular dynamics simulations for free energy calculation. J. Chem. Theory Comput. 2014, 10, 2677–2689. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.; Sun, H. A comment on the reweighting method for accelerated molecular dynamics simulations. J. Chem. Theory Comput. 2015, 11, 2395–2397. [Google Scholar] [CrossRef]
Sample Availability: The producer of tripleurins, strain T. pleuroti SZMC 12454 is available from the Szeged Micrbiology Collection (www.szmc.hu). |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | ϕ, ψ Angles |
|---|---|
| α (alpha helix) | −63, −43 |
| β (beta region) | ‒157.2, 161.9 |
| PII-spirals | −65, +145 |
| γ-turns | +80, −80 |
| γ′-turns | −80, +80 |
| δ regions | Extending at 45° angle to the left of α-helix region |
| δ′ regions | Mirror image of δ region |
| ζ (pre-proline region) | −130, +80 |
| Acceptor | Donor | Fraction | Average Distance | Average Angle |
|---|---|---|---|---|
| Aib1 | Div5 | 0.0560 | 2.89 | 162.38 |
| Ser2 | Gln6 | 0.0843 | 2.88 | 160.03 |
| Ala3 | Gln6 | 0.1911 | 2.87 | 154.95 |
| Ala3 | Div7 | 0.1478 | 2.88 | 161.59 |
| Aib4 | Div7 | 0.0296 | 2.90 | 155.38 |
| Aib4 | Aib8 | 0.0929 | 2.89 | 161.68 |
| Div5 | Aib8 | 0.0242 | 2.88 | 154.44 |
| Div5 | Val9 | 0.0271 | 2.89 | 161.79 |
| Gln6 | Val9 | 0.1597 | 2.87 | 154.92 |
| Gln6 | Aib8 | 0.0545 | 2.78 | 149.02 |
| Aib8 | Aib12 | 0.1254 | 2.89 | 159.16 |
| Gln6 | Ala10 | 0.1417 | 2.87 | 160.11 |
| Div7 | Val9 | 0.0611 | 2.79 | 147.89 |
| Div7 | Ala10 | 0.1154 | 2.88 | 152.30 |
| Div7 | Div11 | 0.1484 | 2.89 | 161.04 |
| Val9 | Aib12 | 0.0456 | 2.88 | 159.08 |
| Ala10 | Aib12 | 0.0434 | 2.80 | 148.52 |
| Ala10 | Leu14 | 0.0351 | 2.88 | 159.89 |
| Div11 | Leu14 | 0.1235 | 2.86 | 151.96 |
| Div11 | Aib15 | 0.1890 | 2.88 | 162.72 |
| Pro13 | Val16 | 0.1974 | 2.87 | 151.15 |
| Pro13 | Gln17 | 0.2110 | 2.87 | 159.68 |
| Leu14 | Gln17 | 0.1082 | 2.88 | 152.47 |
| Leu14 | Pol18 | 0.4430 | 2.86 | 160.43 |
| Acceptor | Donor | Fraction | Average Distance | Average Angle |
|---|---|---|---|---|
| Aib1 | Div5 | 0.1538 | 2.90 | 162.68 |
| Ser2 | Aib4 | 0.0902 | 2.82 | 148.54 |
| Ser2 | Gln6 | 0.2475 | 2.88 | 160.23 |
| Ala3 | Div5 | 0.1023 | 2.80 | 149.61 |
| Ala3 | Div7 | 0.2394 | 2.90 | 162.90 |
| Ala3 | Gln6 | 0.0746 | 2.88 | 152.31 |
| Aib4 | Aib8 | 0.0904 | 2.90 | 163.41 |
| Gln6 | Val9 | 0.1050 | 2.89 | 154.32 |
| Gln6 | Div11 | 0.0767 | 2.90 | 152.28 |
| Gln6 | Ala10 | 0.1334 | 2.87 | 156.93 |
| Div7 | Val9 | 0.0451 | 2.91 | 162.20 |
| Div7 | Ala10 | 0.2448 | 2.88 | 152.99 |
| Aib8 | Ala10 | 0.0254 | 2.88 | 146.67 |
| Val9 | Div11 | 0.1822 | 2.83 | 147.37 |
| Val9 | Aib12 | 0.0186 | 2.88 | 161.66 |
| Ala10 | Aib12 | 0.4465 | 2.79 | 149.19 |
| Div11 | Leu14 | 0.1384 | 2.87 | 155.91 |
| Div11 | Aib15 | 0.0833 | 2.88 | 163.24 |
| Aib12 | Leu14 | 0.2099 | 2.85 | 151.67 |
| Aib12 | Aib15 | 0.0855 | 2.89 | 160.15 |
| Pro13 | Aib15 | 0.1378 | 2.84 | 148.97 |
| Pro13 | Val16 | 0.1997 | 2.88 | 158.57 |
| Aib15 | Pol18 | 0.2063 | 2.88 | 158.80 |
| Leu14 | Val16 | 0.1747 | 2.81 | 147.09 |
| Leu14 | Gln17 | 0.2091 | 2.88 | 159.58 |
| Leu14 | Pol18 | 0.1474 | 2.87 | 161.57 |
| Val16 | Pol18 | 0.0851 | 2.84 | 147.39 |
| Simulations | Time (ns) | Boost Option | Vavg_dihed (kcal mol−1) | Vavg_total (kcal mol−1) |
|---|---|---|---|---|
| In water | 2500 (500 × 3 + 1000 ns) | iamd = 3 | 210 | −25429 |
| In chloroform | 2500 (500 × 3 + 1000 ns) | iamd = 2 | 206 | −7535 |
| Water Simulation | Chloroform Simulation | ||||
|---|---|---|---|---|---|
| a1, a2 | b1, b2 | Avg. boost (kcal mol−1) | a1, a2 | b1, b2 | Avg. boost (kcal mol−1) |
| 4.0 | 0.16 | 5 | 4.0 | ------ | 6.5 |
| 3.5 | 0.30 | 45 | 4.5 | ------ | 10 |
| 3.5 | 0.20 | 15 | 6.0 | ------ | 30 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyagi, C.; Marik, T.; Szekeres, A.; Vágvölgyi, C.; Kredics, L.; Ötvös, F. Tripleurin XIIc: Peptide Folding Dynamics in Aqueous and Hydrophobic Environment Mimic Using Accelerated Molecular Dynamics. Molecules 2019, 24, 358. https://doi.org/10.3390/molecules24020358
Tyagi C, Marik T, Szekeres A, Vágvölgyi C, Kredics L, Ötvös F. Tripleurin XIIc: Peptide Folding Dynamics in Aqueous and Hydrophobic Environment Mimic Using Accelerated Molecular Dynamics. Molecules. 2019; 24(2):358. https://doi.org/10.3390/molecules24020358
Chicago/Turabian StyleTyagi, Chetna, Tamás Marik, András Szekeres, Csaba Vágvölgyi, László Kredics, and Ferenc Ötvös. 2019. "Tripleurin XIIc: Peptide Folding Dynamics in Aqueous and Hydrophobic Environment Mimic Using Accelerated Molecular Dynamics" Molecules 24, no. 2: 358. https://doi.org/10.3390/molecules24020358
APA StyleTyagi, C., Marik, T., Szekeres, A., Vágvölgyi, C., Kredics, L., & Ötvös, F. (2019). Tripleurin XIIc: Peptide Folding Dynamics in Aqueous and Hydrophobic Environment Mimic Using Accelerated Molecular Dynamics. Molecules, 24(2), 358. https://doi.org/10.3390/molecules24020358