Discovery of Novel µ-Opioid Receptor Inverse Agonist from a Combinatorial Library of Tetrapeptides through Structure-Based Virtual Screening



 ,
,  , ,
, , 
Abstract
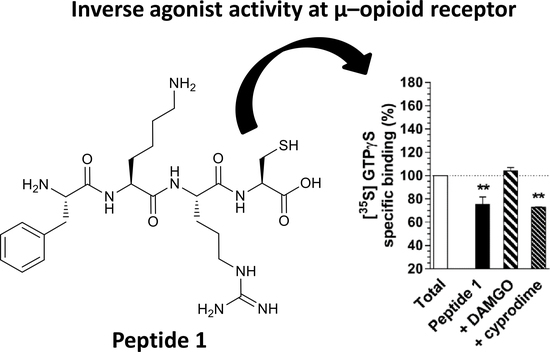
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Peptide Library Generation
2.2. Pharmacophore Modeling
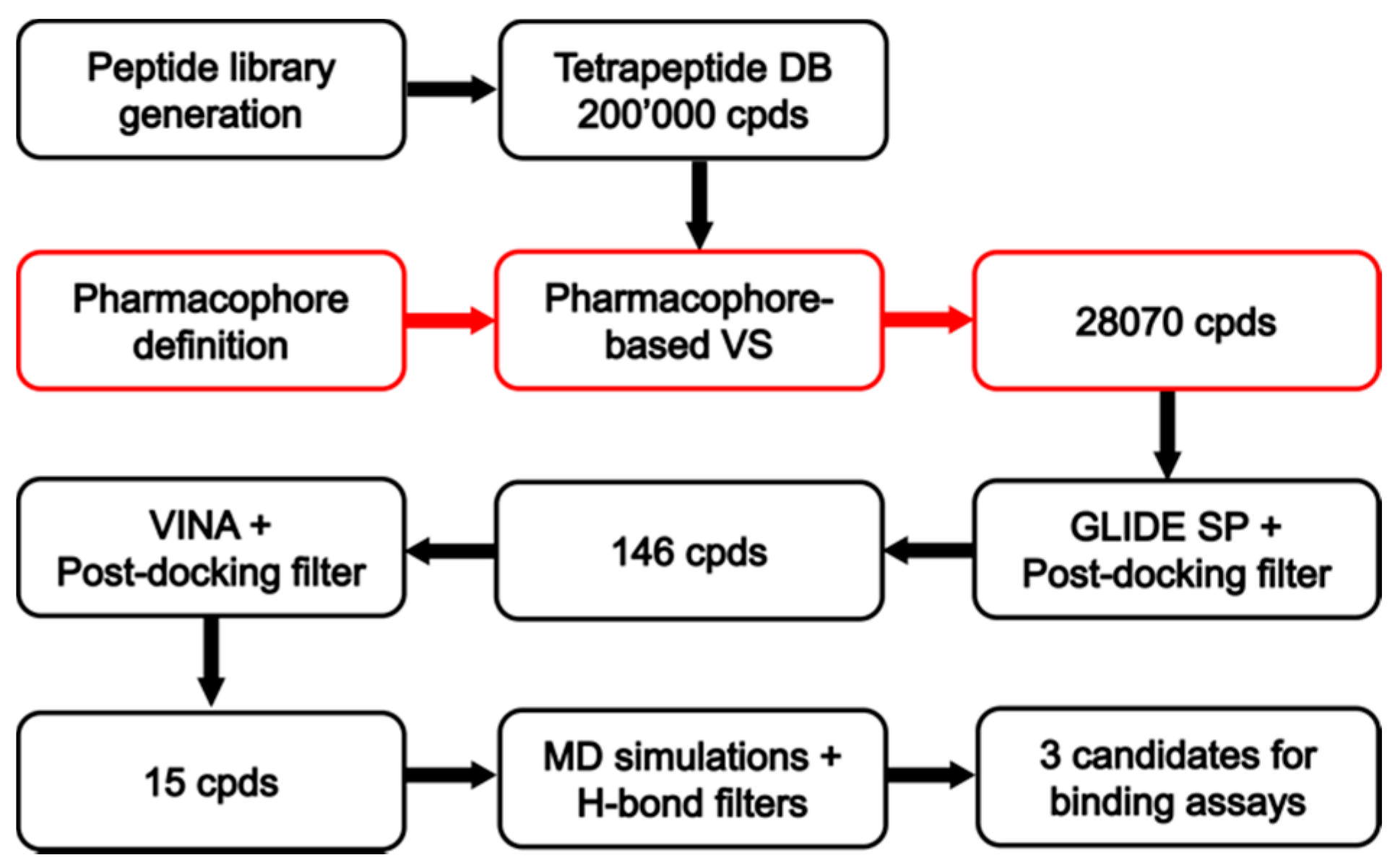
2.3. Database Generation and Pharmacophore Screening
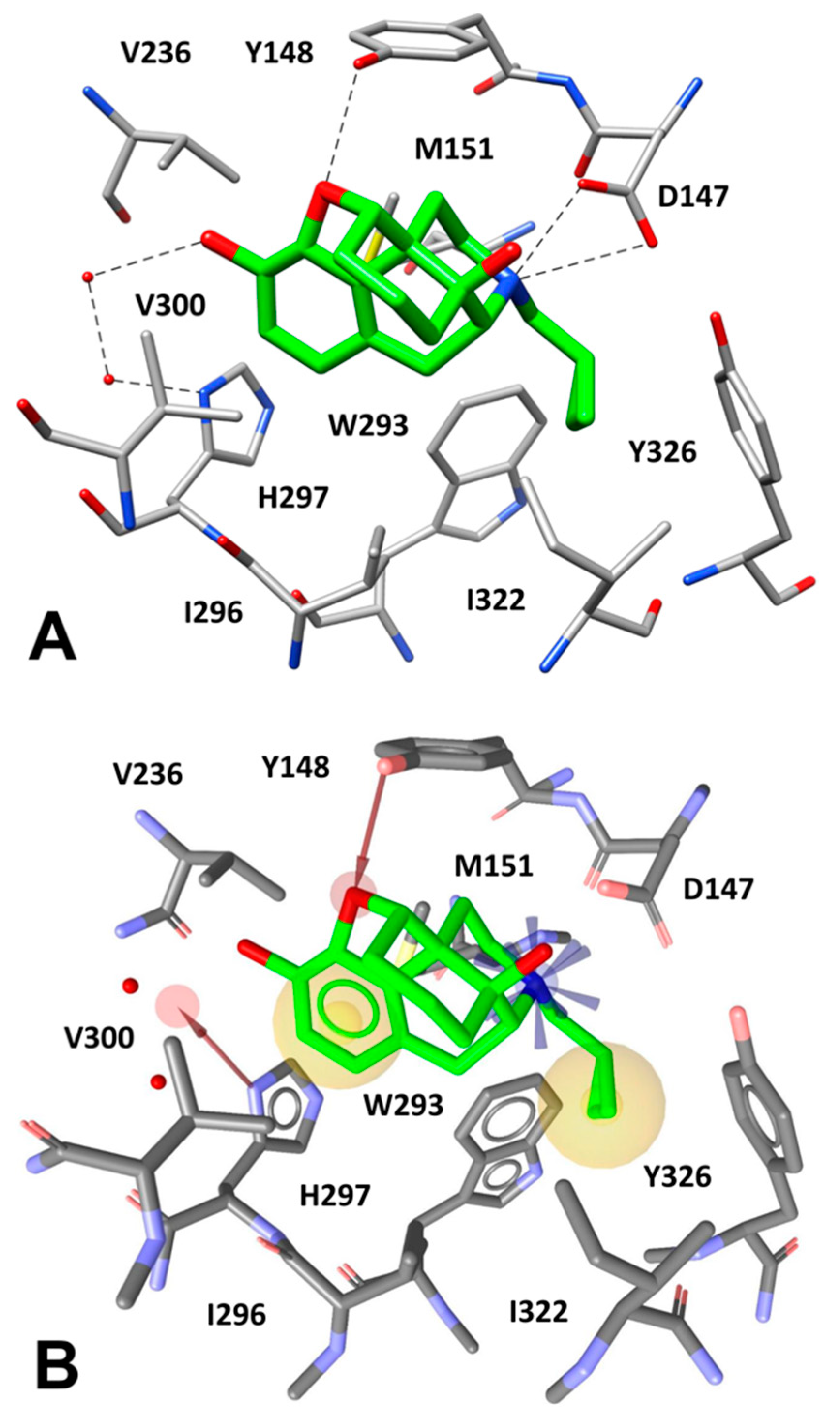
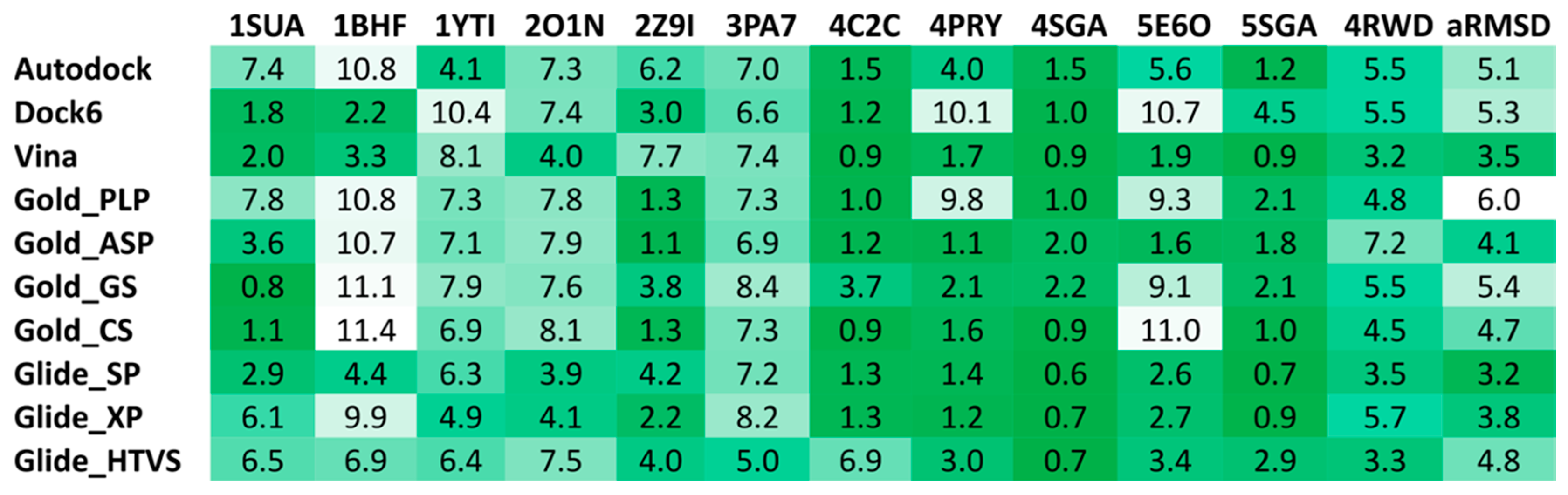
2.4. Docking Reliability Evaluation
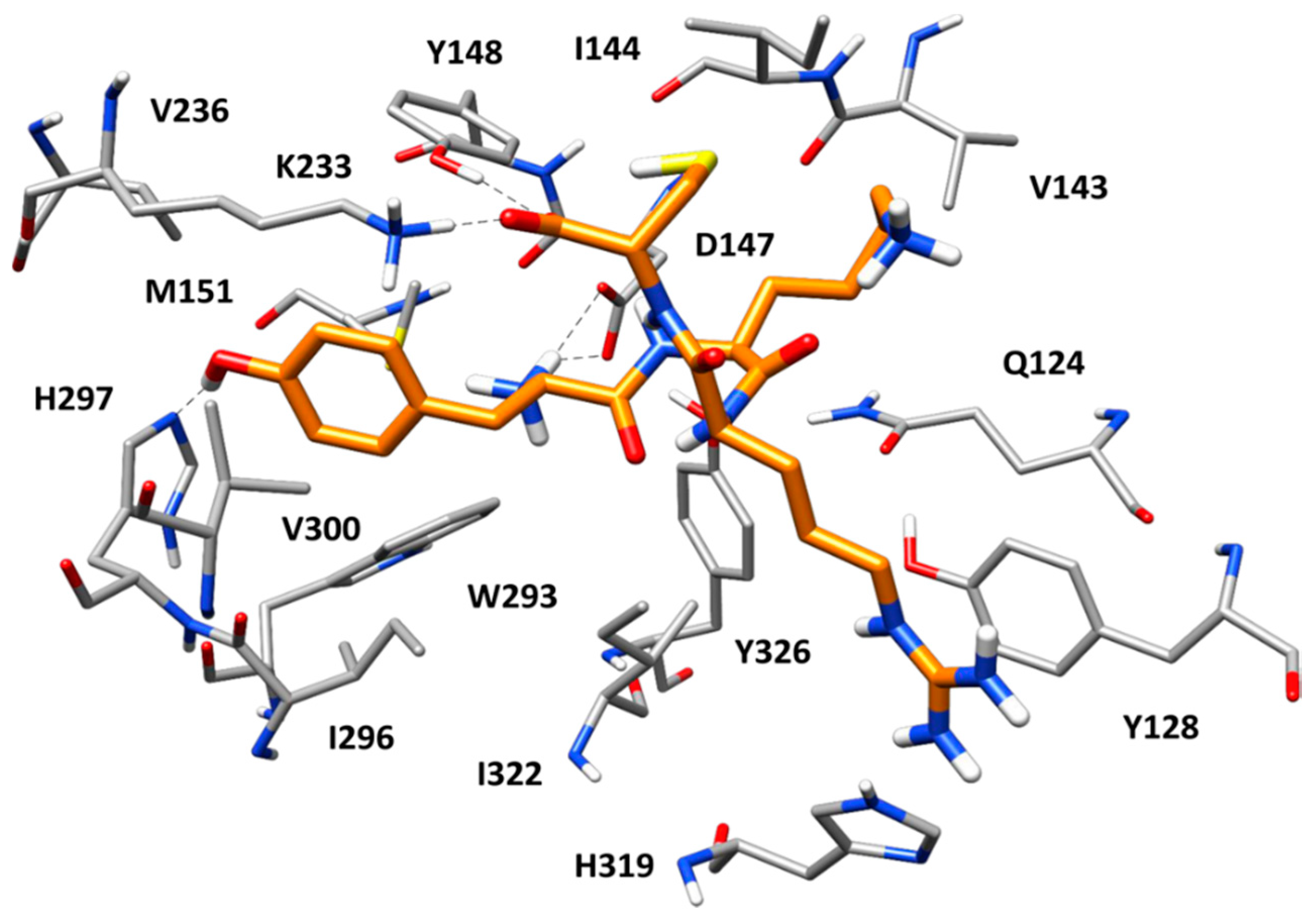
2.5. Docking of Screening Compounds and Pose Filtering
2.6. Molecular Dynamics
3. Chemistry
3.1. Materials and Methods
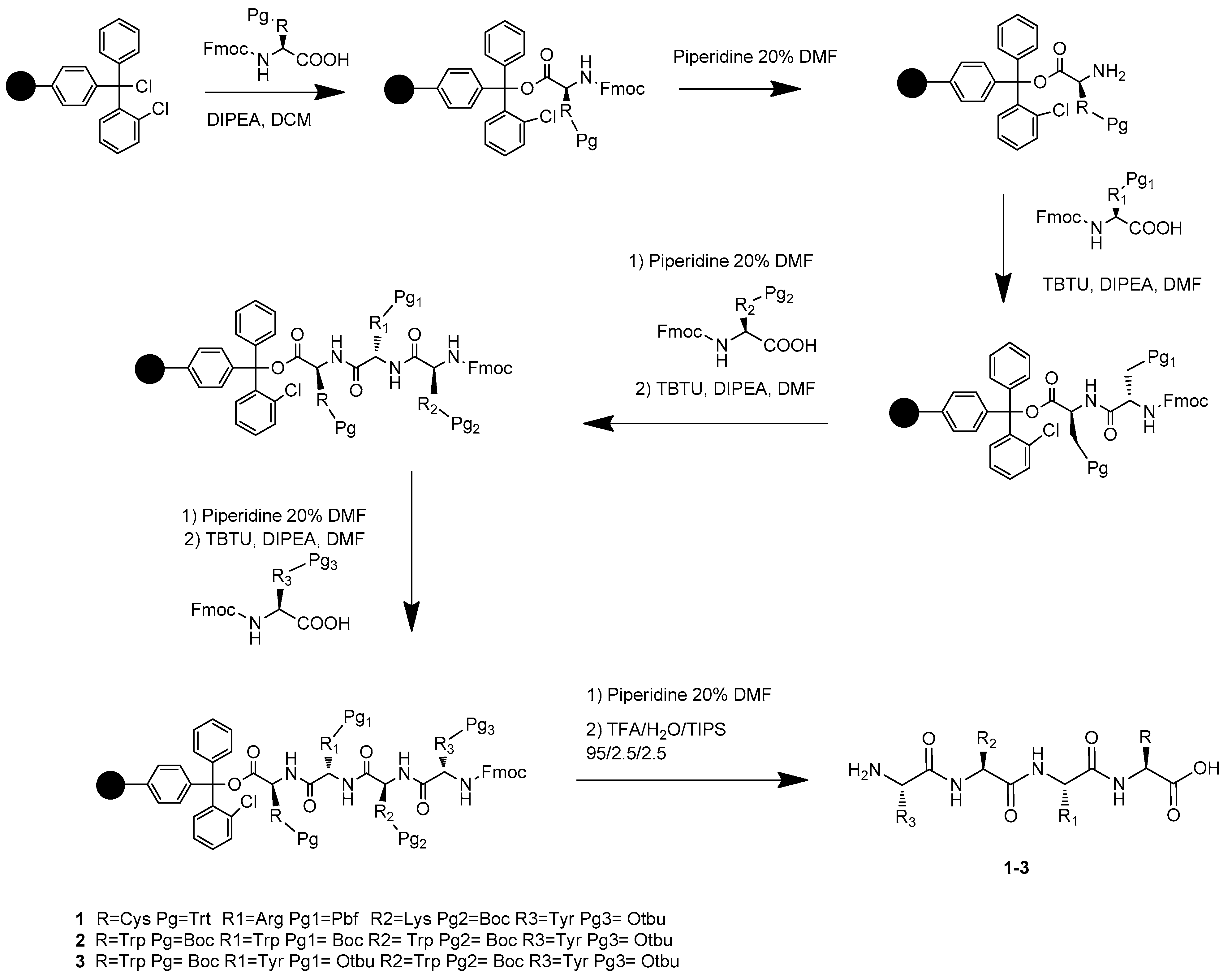
3.2. Synthesis of Compounds
3.3. Resin Loading
3.4. Cleavage and Purification
4. Biological Assays
4.1. Chemicals
4.2. Animals
4.3. Rat Membrane Preparations
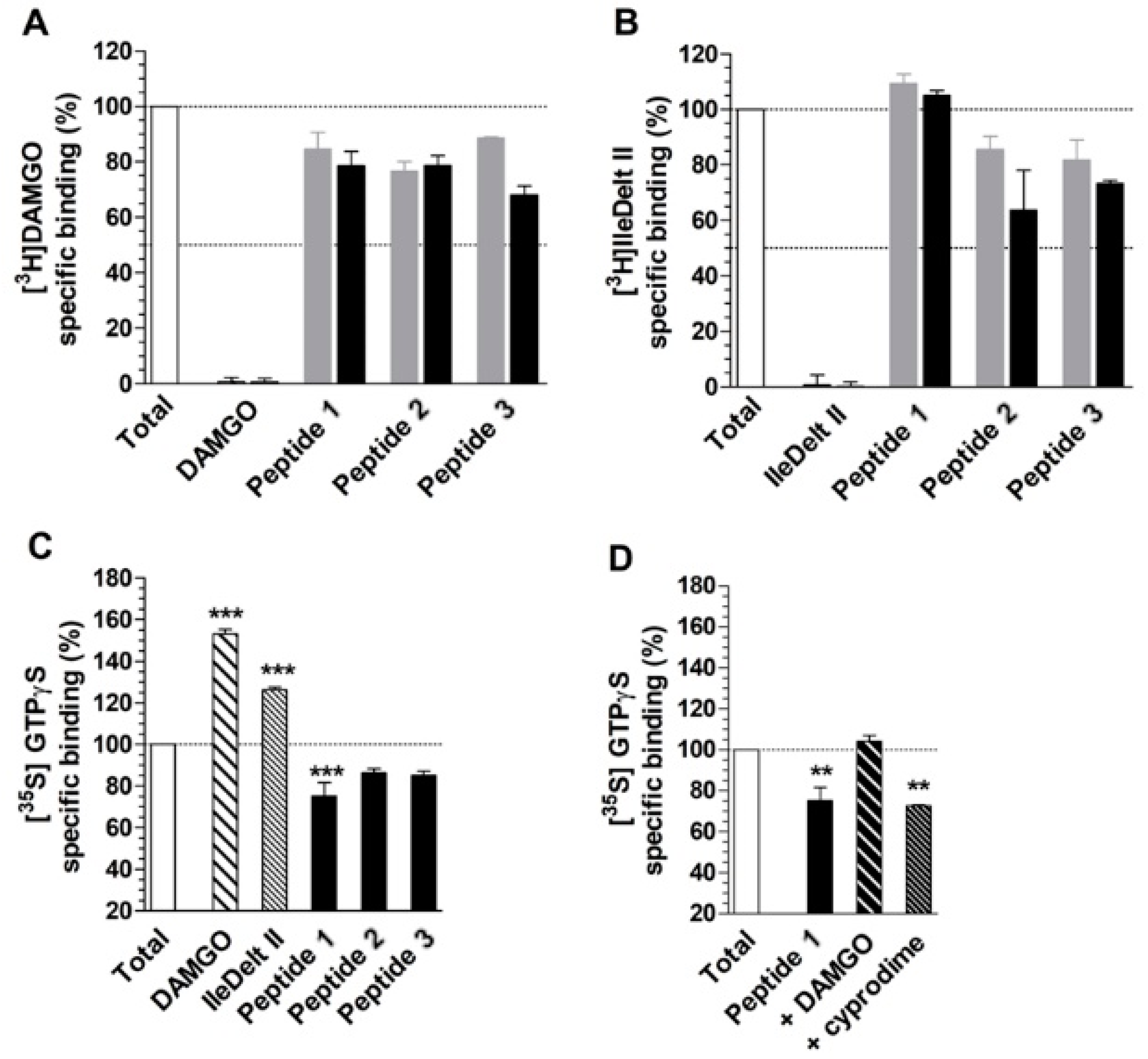
4.4. Radioligand Competition Binding Experiments
4.5. Functional [35S]GTPγS Binding Experiments
4.6. Data Analysis
5. Results and Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Anand, J.P.; Montgomery, D. Multifunctional Opioid Ligands. Handb. Exp. Pharmacol. 2018, 247, 21–51. [Google Scholar] [PubMed]
- Machelska, H.; Celik, M.O. Advances in Achieving Opioid Analgesia Without Side Effects. Front. Pharmacol. 2018, 9, 1388. [Google Scholar] [CrossRef] [PubMed]
- Noori, S.A.; Aiyer, R.; Yu, J.; White, R.S.; Mehta, N.; Gulati, A. Nonopioid versus opioid agents for chronic neuropathic pain, rheumatoid arthritis pain, cancer pain and low back pain. Pain Manag. 2019, 9, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Navarro, M.; Maldonado, R.; Banos, J.E. Why mu-opioid agonists have less analgesic efficacy in neuropathic pain? Eur. J. Pain (London, England) 2019, 23, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Nourbakhsh, F.; Atabaki, R.; Roohbakhsh, A. The role of orphan G protein-coupled receptors in the modulation of pain: A review. Life Sci. 2018, 212, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.; Herz, A. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 7321–7325. [Google Scholar] [CrossRef]
- Maack, C.; Cremers, B.; Flesch, M.; Höper, A.; Südkamp, M.; Böhm, M. Different intrinsic activities of bucindolol, carvedilol and metoprolol in human failing myocardium. Br. J. Pharmacol. 2000, 130, 1131–1139. [Google Scholar] [CrossRef]
- Morisset, S.; Rouleau, A.; Ligneau, X.; Gbahou, F.; Tardivel-Lacombe, J.; Stark, H.; Schunack, W.; Ganellin, C.R.; Arrang, J.-M. High constitutive activity of native H 3 receptors regulates histamine neurons in brain. Nature 2000, 408, 860. [Google Scholar] [CrossRef]
- Martin, D.J.; FitzMorris, P.E.; Li, B.; Ayestas, M.; Sally, E.J.; Dersch, C.M.; Rothman, R.B.; Deveau, A.M. An efficient synthesis of 3-OBn-6β, 14-epoxy-bridged opiates from naltrexone and identification of a related dual MOR inverse agonist/KOR agonist. Bioorganic Med. Chem. Lett. 2012, 22, 6801–6805. [Google Scholar] [CrossRef]
- Zhu, L.; Cui, Z.; Zhu, Q.; Zha, X.; Xu, Y. Novel opioid receptor agonists with reduced morphine-like side effects. Mini Rev. Med. Chem. 2018, 18, 1603–1610. [Google Scholar] [CrossRef]
- Perlikowska, R.; Janecka, A. Rubiscolins-highly potent peptides derived from plant proteins. Mini Rev. Med. Chem. 2018, 18, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Luger, D.; Poli, G.; Wieder, M.; Stadler, M.; Ke, S.; Ernst, M.; Hohaus, A.; Linder, T.; Seidel, T.; Langer, T.; et al. Identification of the putative binding pocket of valerenic acid on GABAA receptors using docking studies and site-directed mutagenesis. Br. J. Pharmacol. 2015, 172, 5403–5413. [Google Scholar] [CrossRef] [PubMed]
- Daga, P.R.; Polgar, W.E.; Zaveri, N.T. Structure-based virtual screening of the nociceptin receptor: Hybrid docking and shape-based approaches for improved hit identification. J. Chem. Inf. Modeling 2014, 54, 2732–2743. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Zengin, G.; Durdagi, S.; Ekhteiari Salmas, R.; Macedonio, G.; Stefanucci, A.; Dimmito, M.P.; Novellino, E. Combinatorial peptide library screening for discovery of diverse α-glucosidase inhibitors using molecular dynamics simulations and binary QSAR models. J. Biomol. Struct. Dyn. 2019, 37, 726–740. [Google Scholar] [CrossRef]
- Maestro, version 10.7. Portland (OR): Schrödinger Inc. 2016. Available online: https://www.schrodinger.com/ (accessed on 25 January 2019).
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321. [Google Scholar] [CrossRef]
- Case David, A. Amber 2015 Reference Manual; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Modeling 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Poli, G.; Seidel, T.; Langer, T. Conformational Sampling of Small Molecules With iCon: Performance Assessment in Comparison With OMEGA. Front. Chem. 2018, 6, 229. [Google Scholar] [CrossRef]
- Yang, S.; Yunden, J.; Sonoda, S.; Doyama, N.; Lipkowski, A.W.; Kawamura, Y.; Yoshikawa, M. Rubiscolin, a delta selective opioid peptide derived from plant Rubisco. Febs Lett. 2001, 509, 213–217. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Poli, G.; Martinelli, A.; Tuccinardi, T. Reliability analysis and optimization of the consensus docking approach for the development of virtual screening studies. J. Enzym. Inhib. Med. Chem. 2016, 31, 167–173. [Google Scholar] [CrossRef]
- Tuccinardi, T.; Poli, G.; Romboli, V.; Giordano, A.; Martinelli, A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J. Chem. Inf. Modeling 2014, 54, 2980–2986. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.W. Improved protein–ligand docking using GOLD. Proteins: Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glide, version 5.0; Schrodinger Inc.: Portland, OR, USA, 2009.
- Poli, G.; Gelain, A.; Porta, F.; Asai, A.; Martinelli, A.; Tuccinardi, T. Identification of a new STAT3 dimerization inhibitor through a pharmacophore-based virtual screening approach. J. Enzym. Inhib Med Chem. 2016, 31, 1011–1017. [Google Scholar] [CrossRef]
- Tuccinardi, T.; Poli, G.; Corchia, I.; Granchi, C.; Lapillo, M.; Macchia, M.; Minutolo, F.; Ortore, G.; Martinelli, A. A virtual screening study for lactate dehydrogenase 5 inhibitors by using a pharmacophore-based approach. Mol. Inform. 2016, 35, 434–439. [Google Scholar] [CrossRef]
- Poli, G.; Scarpino, A.; Aissaoui, M.; Granchi, C.; Minutolo, F.; Martinelli, A.; Tuccinardi, T. Identification of lactate dehydrogenase 5 inhibitors using pharmacophore-driven consensus docking. Curr. Bioact. Compd. 2018, 14, 197–204. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef]
- Mollica, A.; Costante, R.; Novellino, E.; Stefanucci, A.; Pieretti, S.; Zador, F.; Samavati, R.; Borsodi, A.; Benyhe, S.; Vetter, I. Design, synthesis and biological evaluation of two opioid agonist and Cav2. 2 blocker multitarget ligands. Chem. Biol. Drug Des. 2015, 86, 156–162. [Google Scholar] [CrossRef]
- Mollica, A.; Pinnen, F.; Azzurra, S.; Costante, R. The evolution of peptide synthesis: From early days to small molecular machines. Curr. Bioact. Compd. 2013, 9, 184–202. [Google Scholar] [CrossRef]
- Stefanucci, A.; Novellino, E.; Macedonio, G.; Dimmito, M.P.; Mirzaie, S.; Cardoso, F.C.; Lewis, R.; Zádor, F.; Erdei, A.I.; Dvorácskó, S. Design, synthesis and biological profile of mixed opioid agonist/N-VGCC blocker peptides. New J. Chem. 2018, 42, 5656–5659. [Google Scholar] [CrossRef] [Green Version]
- Erdei, A.I.; Borbély, A.; Magyar, A.; Szűcs, E.; Ötvös, F.; Gombos, D.; Al-Khrasani, M.; Stefanucci, A.; Dimmito, M.P.; Luisi, G. Biochemical and pharmacological investigation of novel nociceptin/OFQ analogues and N/OFQ-RYYRIK hybrid peptides. Peptides 2019, 112, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Stefanucci, A.; Dimmito, M.P.; Zengin, G.; Luisi, G.; Mirzaie, S.; Novellino, E.; Mollica, A. Discovery of novel amide tripeptides as pancreatic lipase inhibitors by virtual screening. New J. Chem. 2019, 43, 3208–3217. [Google Scholar] [CrossRef]
- Stefanucci, A.; Luisi, G.; Zengin, G.; Macedonio, G.; Dimmito, M.P.; Novellino, E.; Mollica, A. Discovery of arginine-containing tripeptides as a new class of pancreatic lipase inhibitors. Future Med. Chem. 2019, 11, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Costante, R.; Stefanucci, A.; Pinnen, F.; Lucente, G.; Fidanza, S.; Pieretti, S. Antinociceptive profile of potent opioid peptide AM94, a fluorinated analogue of biphalin with non-hydrazine linker. J. Pept. Sci. 2013, 19, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Ioja, E.; Tourwé, D.; Kertész, I.; Tóth, G.; Borsodi, A.; Benyhe, S. Novel diastereomeric opioid tetrapeptides exhibit differing pharmacological activity profiles. Brain Res. Bull. 2007, 74, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Benyhe, S.; Farkas, J.; Tóth, G.; Wollemann, M. Met5-enkephalin-Arg 6-Phe7, an endogenous neuropeptide, binds to multiple opioid and nonopioid sites in rat brain. J. Neurosci. Res. 1997, 48, 249–258. [Google Scholar] [CrossRef]
- Zádor, F.; Kocsis, D.; Borsodi, A.; Benyhe, S. Micromolar concentrations of rimonabant directly inhibits delta opioid receptor specific ligand binding and agonist-induced G-protein activity. Neurochem. Int. 2014, 67, 14–22. [Google Scholar] [CrossRef]
- Traynor, J.R.; Nahorski, S.R. Modulation by mu-opioid agonists of guanosine-5′-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995, 47, 848–854. [Google Scholar]
- Burford, N.T.; Danxin, W.; Sadèe, W. G-protein coupling of μ-opioid receptors (OP3): Elevated basal signalling activity. Biochem. J. 2000, 348, 531–537. [Google Scholar] [CrossRef]
- Lapillo, M.; Tuccinardi, T.; Martinelli, A.; Macchia, M.; Giordano, A.; Poli, G. Extensive reliability evaluation of docking-based target-fishing strategies. Int. J. Mol. Sci. 2019, 20, 1023. [Google Scholar] [CrossRef]
- Tuccinardi, T.; Poli, G.; Dell’Agnello, M.; Granchi, C.; Minutolo, F.; Martinelli, A. Receptor-based virtual screening evaluation for the identification of estrogen receptor β ligands. J. Enzym. Inhib. Med. Chem. 2015, 30, 662–670. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1,2,3 are available from the authors. |







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poli, G.; Dimmito, M.P.; Mollica, A.; Zengin, G.; Benyhe, S.; Zador, F.; Stefanucci, A. Discovery of Novel µ-Opioid Receptor Inverse Agonist from a Combinatorial Library of Tetrapeptides through Structure-Based Virtual Screening. Molecules 2019, 24, 3872. https://doi.org/10.3390/molecules24213872
Poli G, Dimmito MP, Mollica A, Zengin G, Benyhe S, Zador F, Stefanucci A. Discovery of Novel µ-Opioid Receptor Inverse Agonist from a Combinatorial Library of Tetrapeptides through Structure-Based Virtual Screening. Molecules. 2019; 24(21):3872. https://doi.org/10.3390/molecules24213872
Chicago/Turabian StylePoli, Giulio, Marilisa Pia Dimmito, Adriano Mollica, Gokhan Zengin, Sandor Benyhe, Ferenc Zador, and Azzurra Stefanucci. 2019. "Discovery of Novel µ-Opioid Receptor Inverse Agonist from a Combinatorial Library of Tetrapeptides through Structure-Based Virtual Screening" Molecules 24, no. 21: 3872. https://doi.org/10.3390/molecules24213872
APA StylePoli, G., Dimmito, M. P., Mollica, A., Zengin, G., Benyhe, S., Zador, F., & Stefanucci, A. (2019). Discovery of Novel µ-Opioid Receptor Inverse Agonist from a Combinatorial Library of Tetrapeptides through Structure-Based Virtual Screening. Molecules, 24(21), 3872. https://doi.org/10.3390/molecules24213872