
The Synthesis and Utility of Metal-Nitrosophenolato Compounds—Highlighting the Baudisch Reaction



Abstract
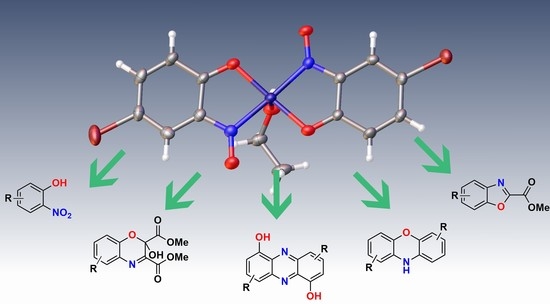
:
1. Introduction to Metal-Nitrosophenolato Complexes
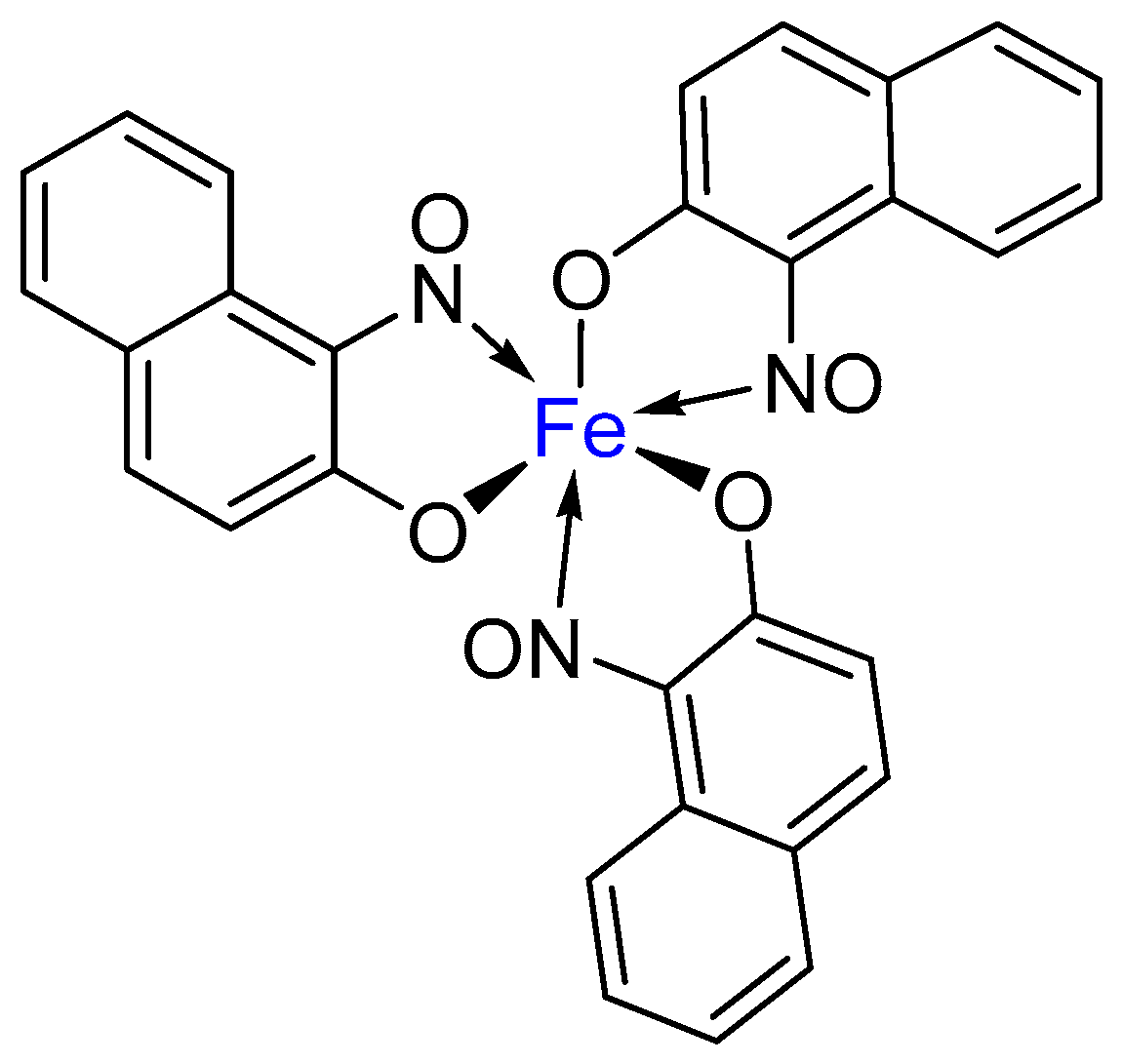
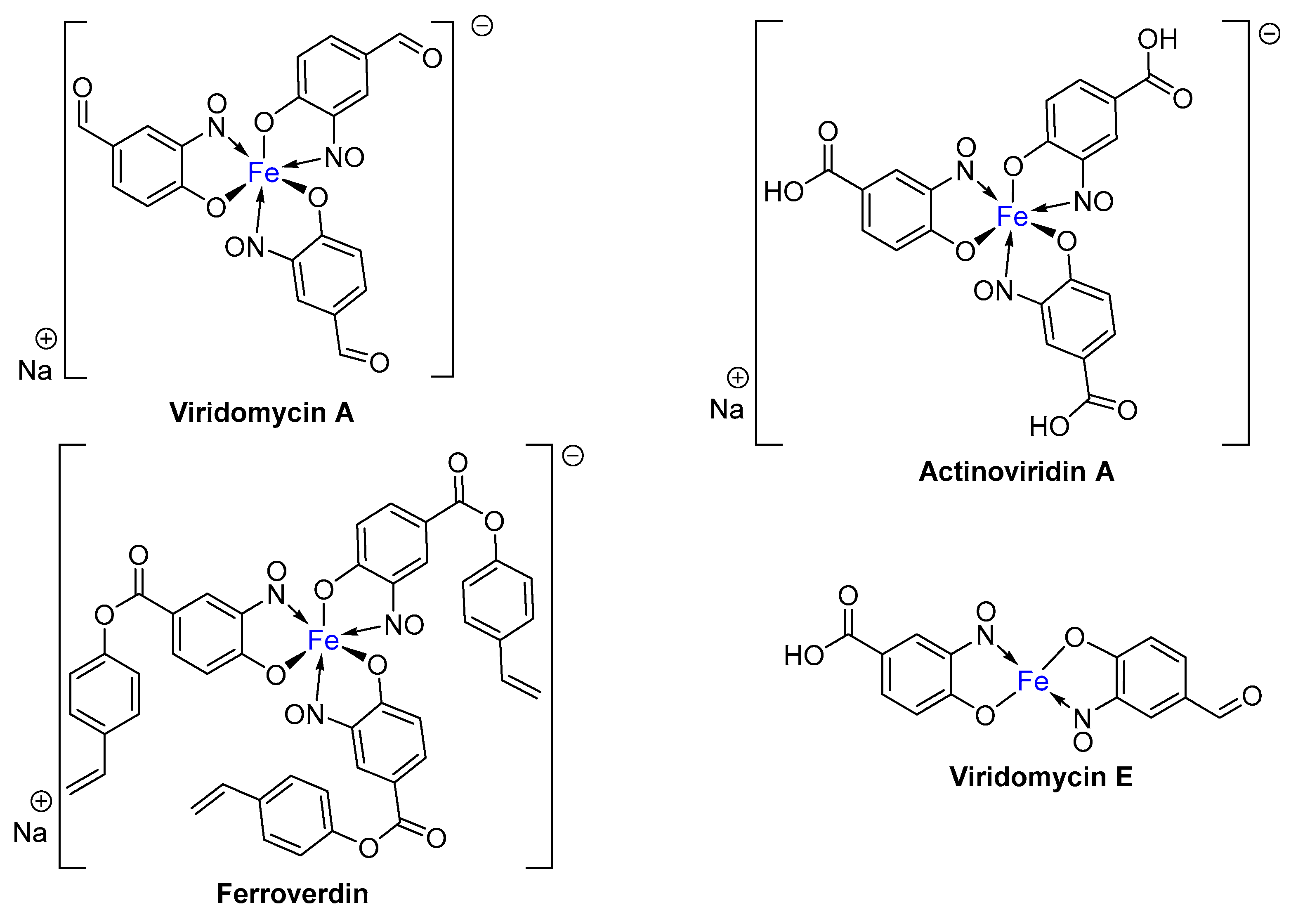
1.1. Metal-Nitrosophenolato Complexes in Nature
1.2. A Brief History of Metal-Nitrosophenolato Synthesis
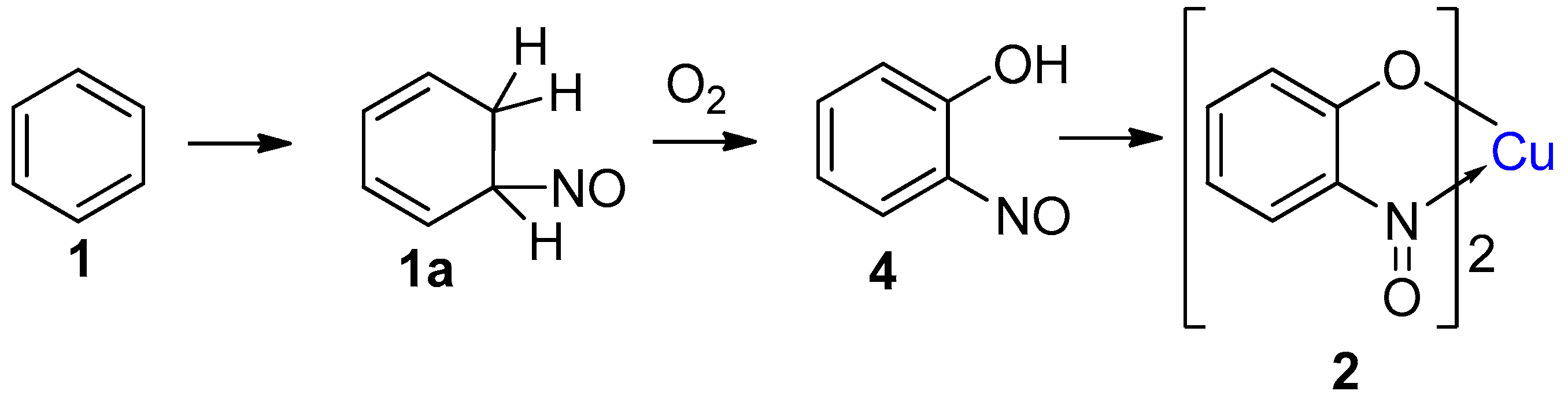
1.3. Introduction to the Baudisch Reaction
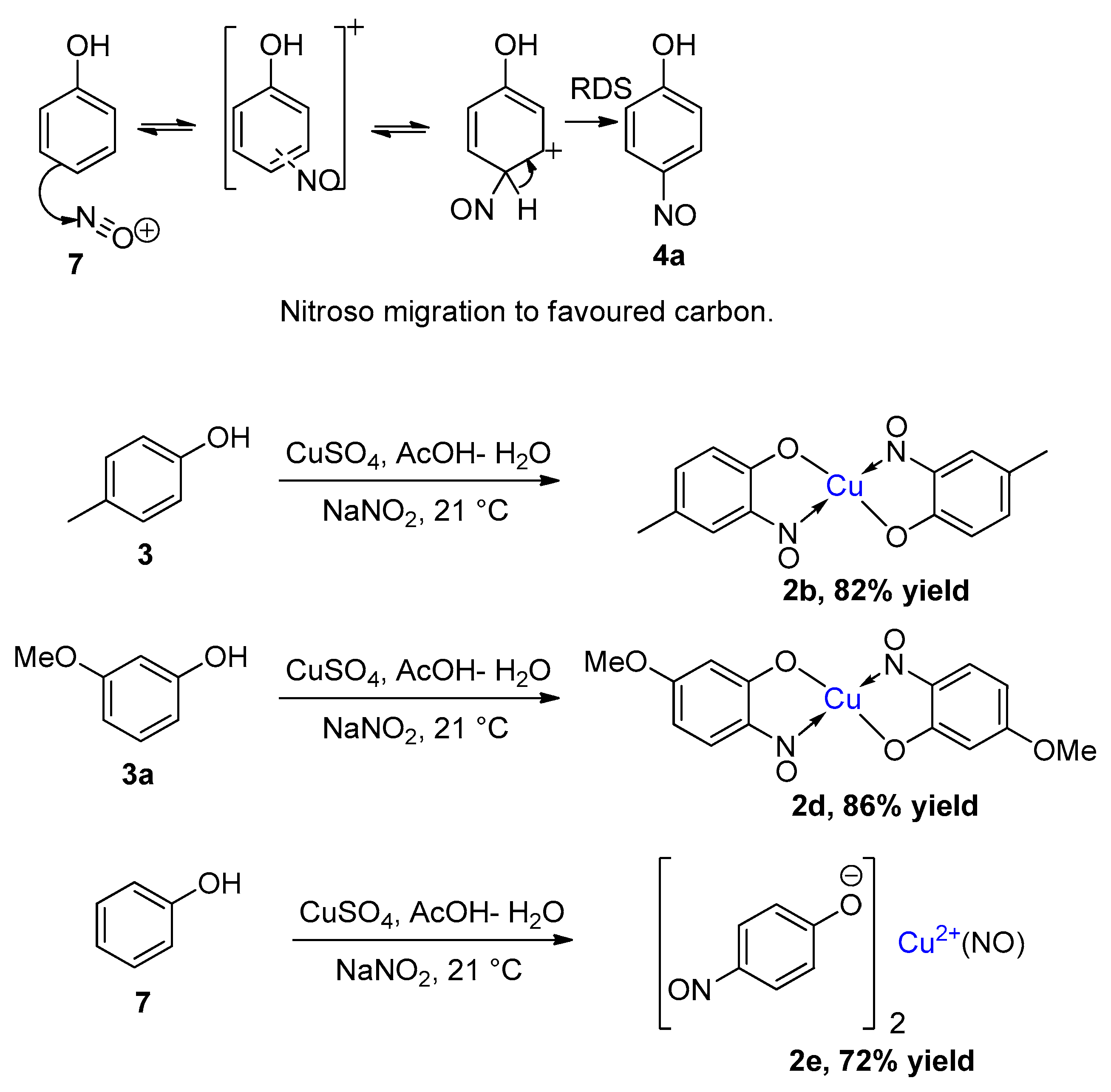
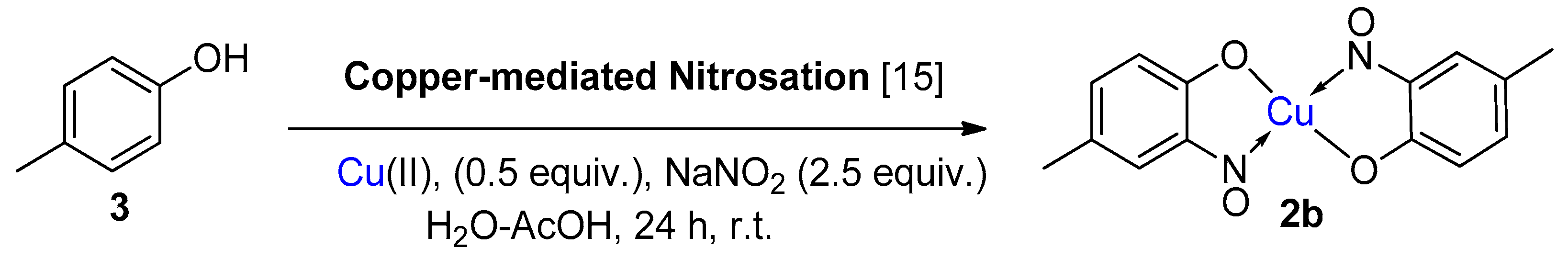
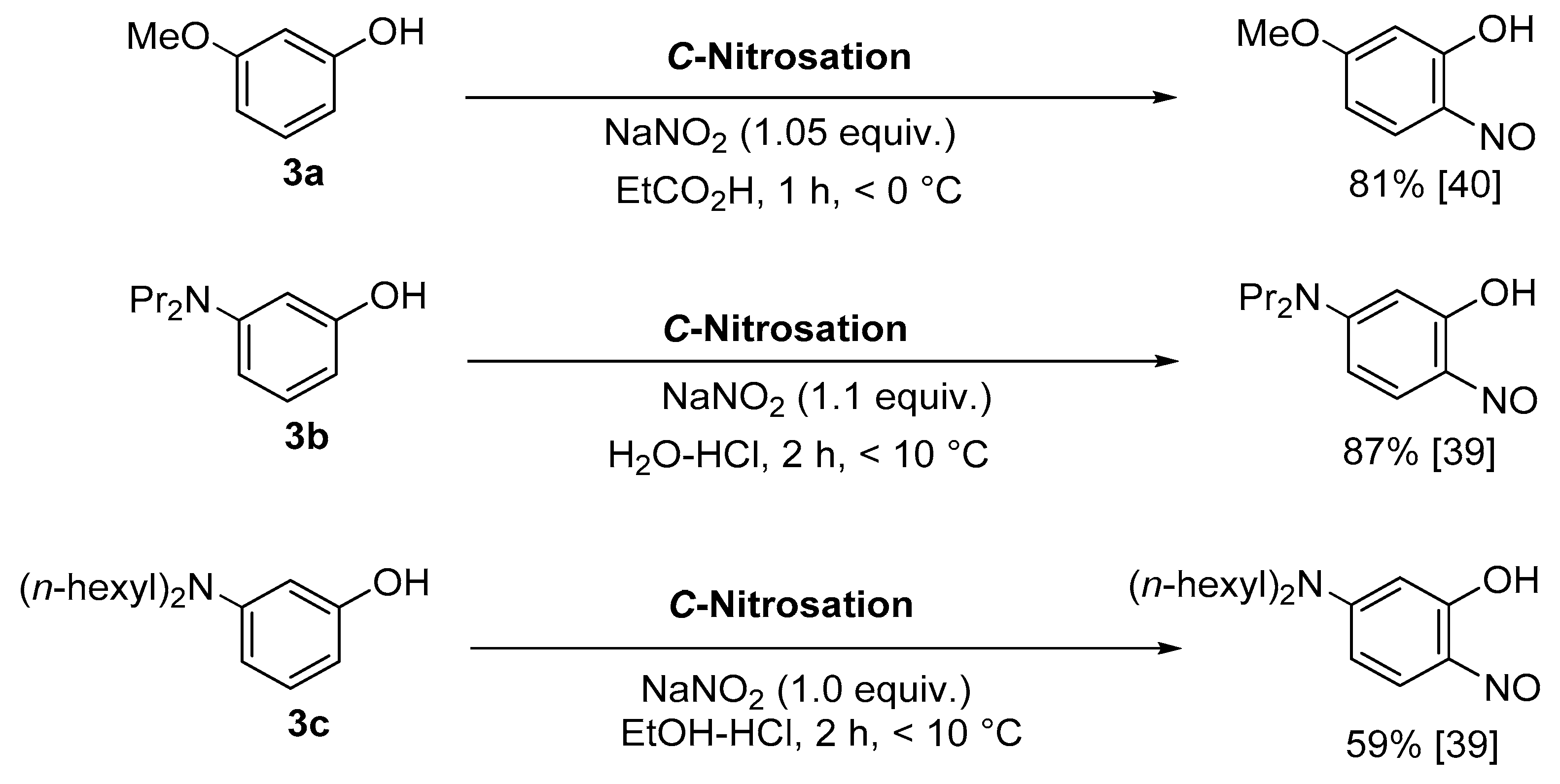
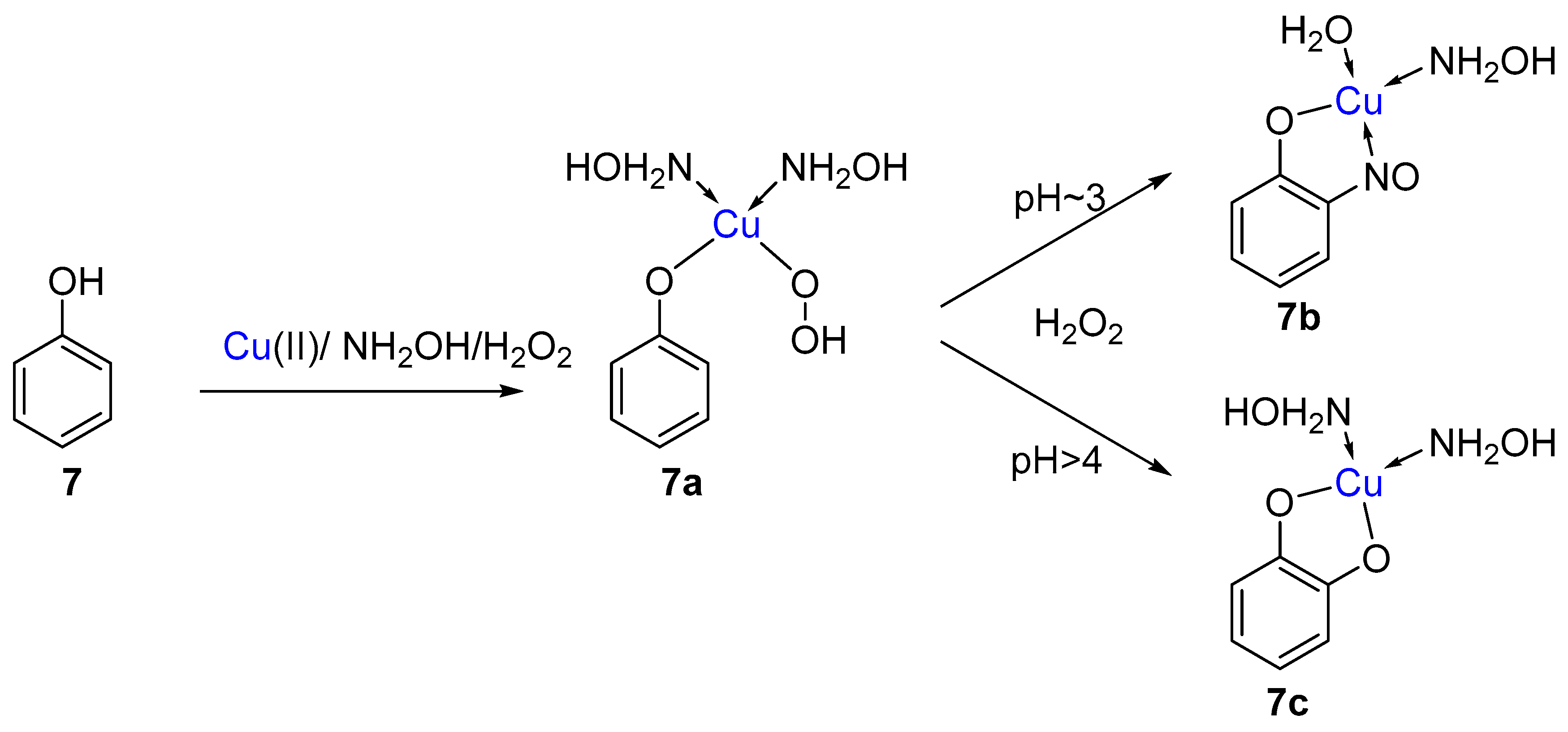
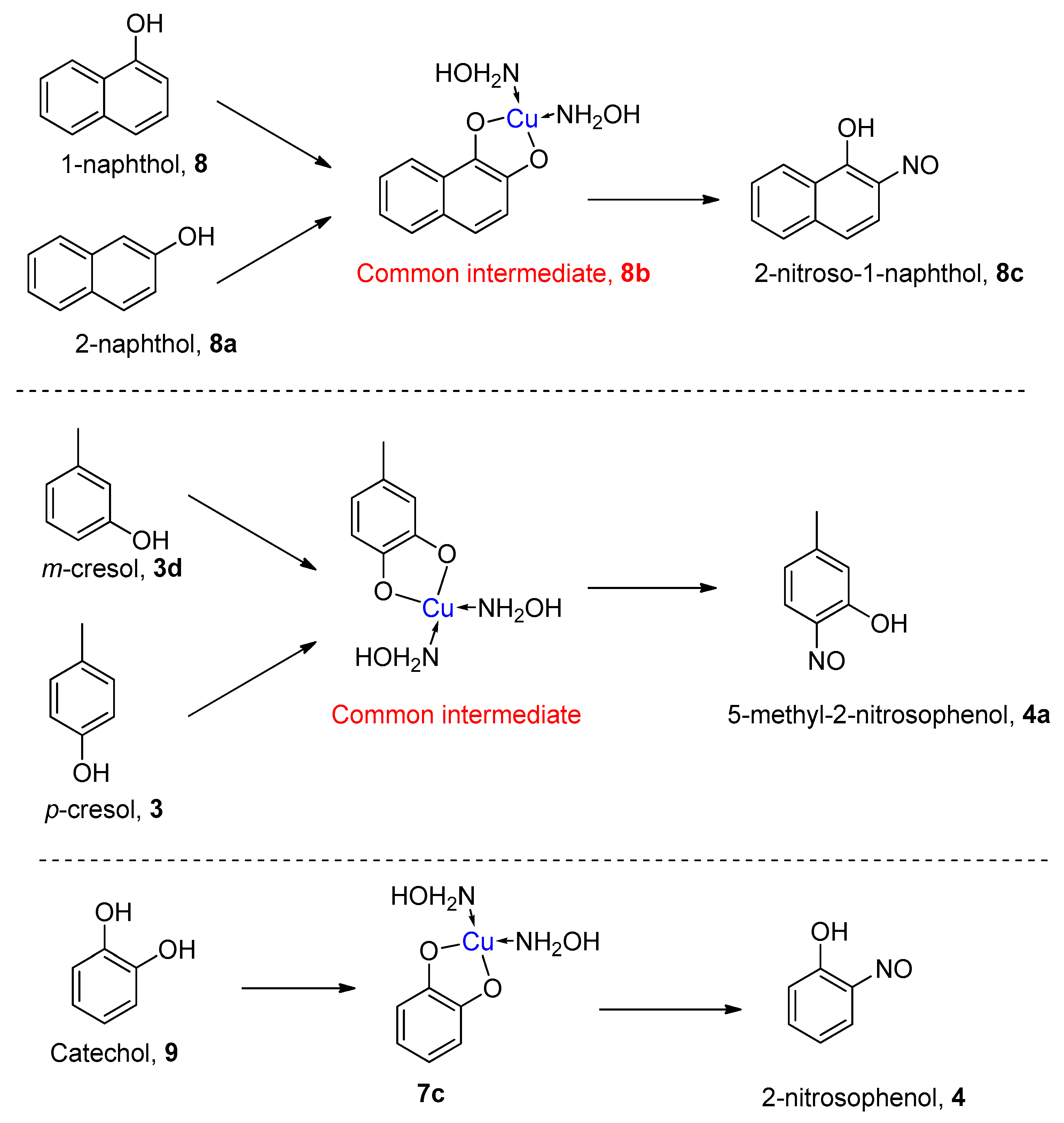
1.4. Copper-Mediated Aromatic Nitrosation
1.5. Additional Syntheses of Metal-Nitrosophenolato Complexes
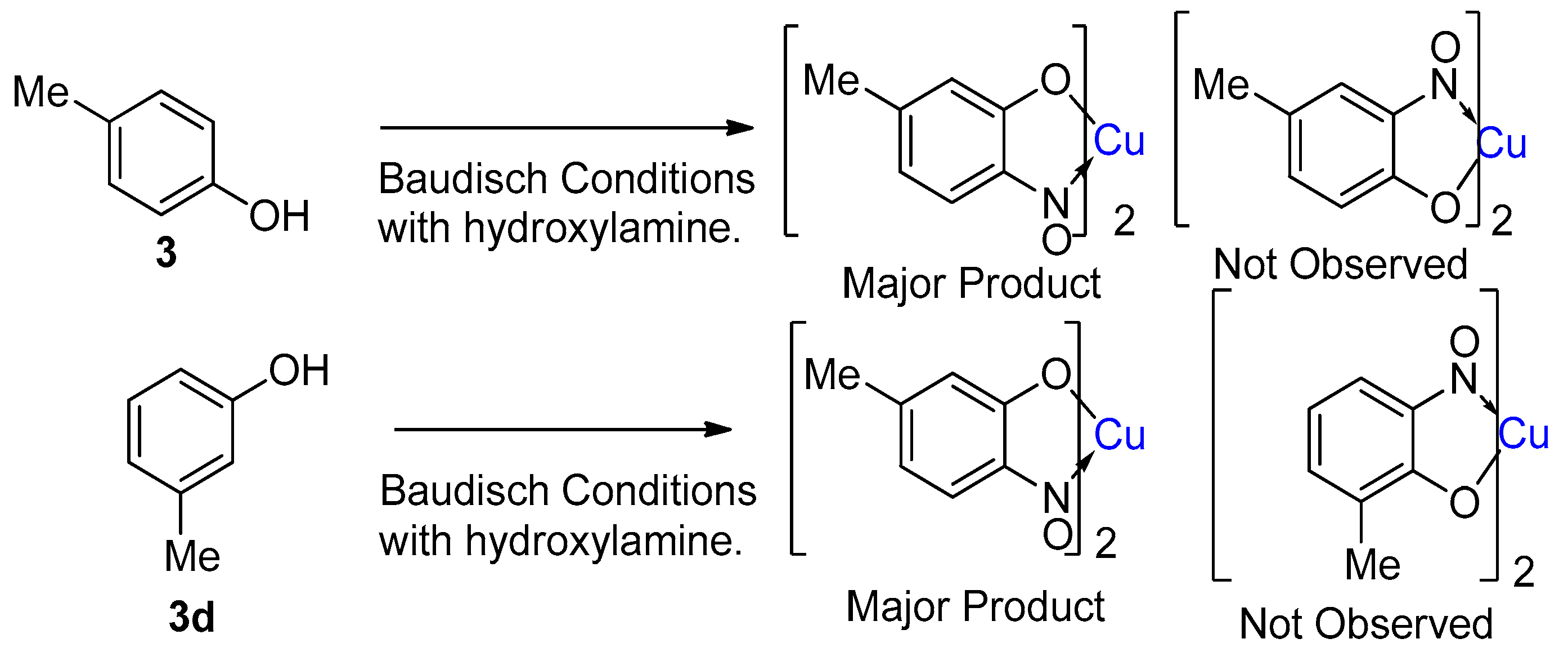
1.6. Scope of Metal-Nitrosophenolato Synthesis
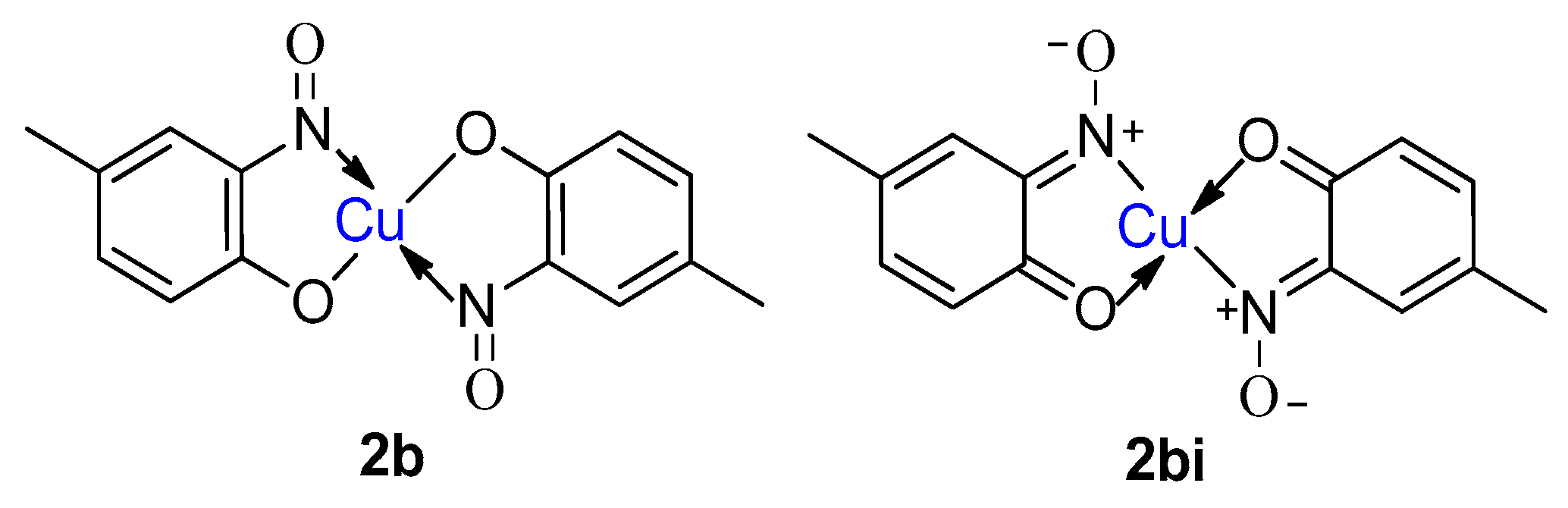
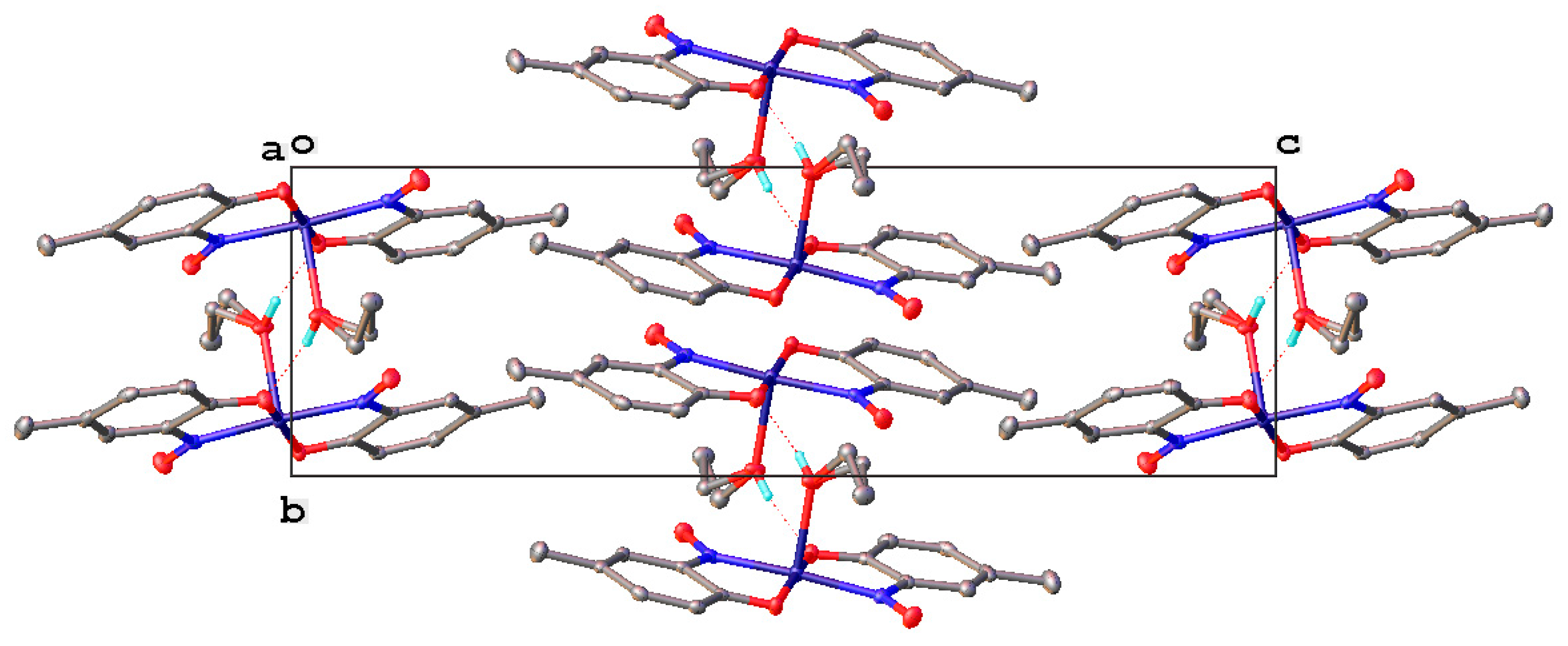
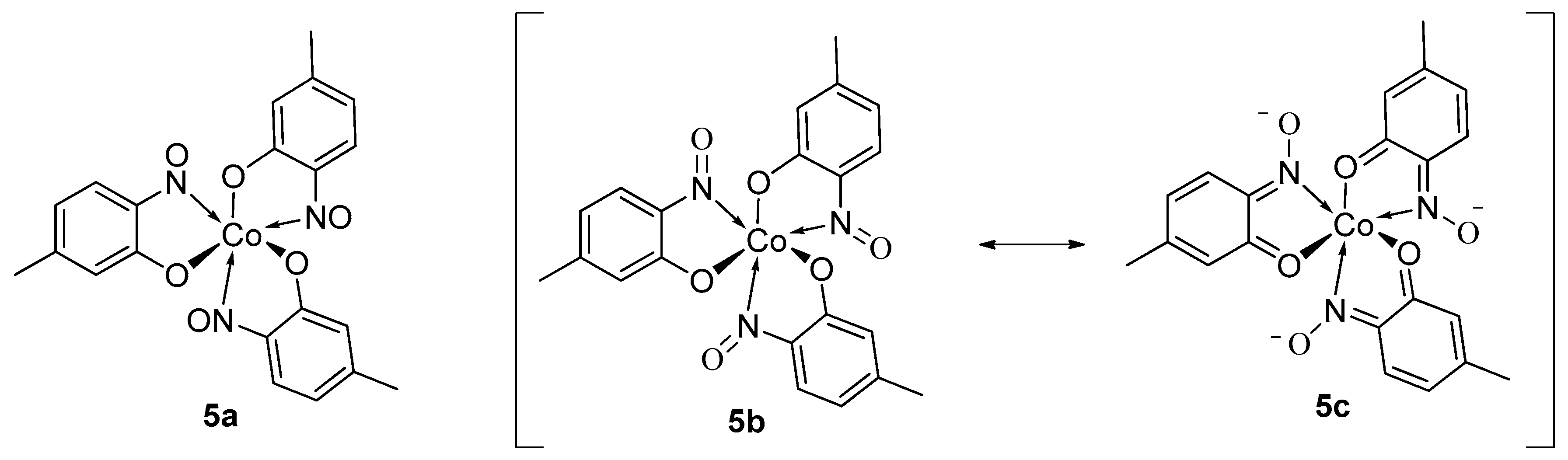
1.7. Properties of Metal-2-Nitrosophenolato Complexes
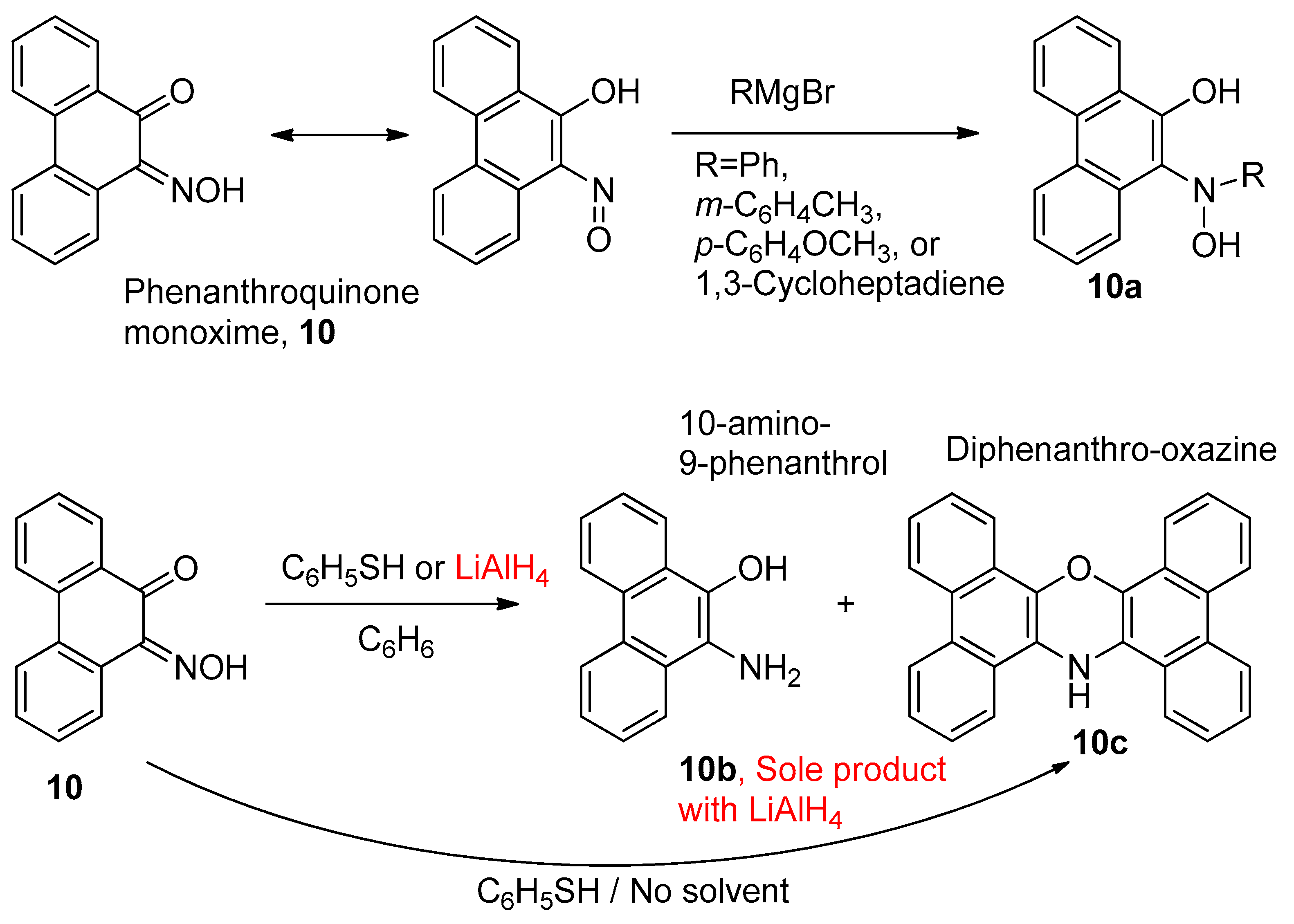
1.8. Derivatisation of Complexes
2. History and Development of the Complex Formation
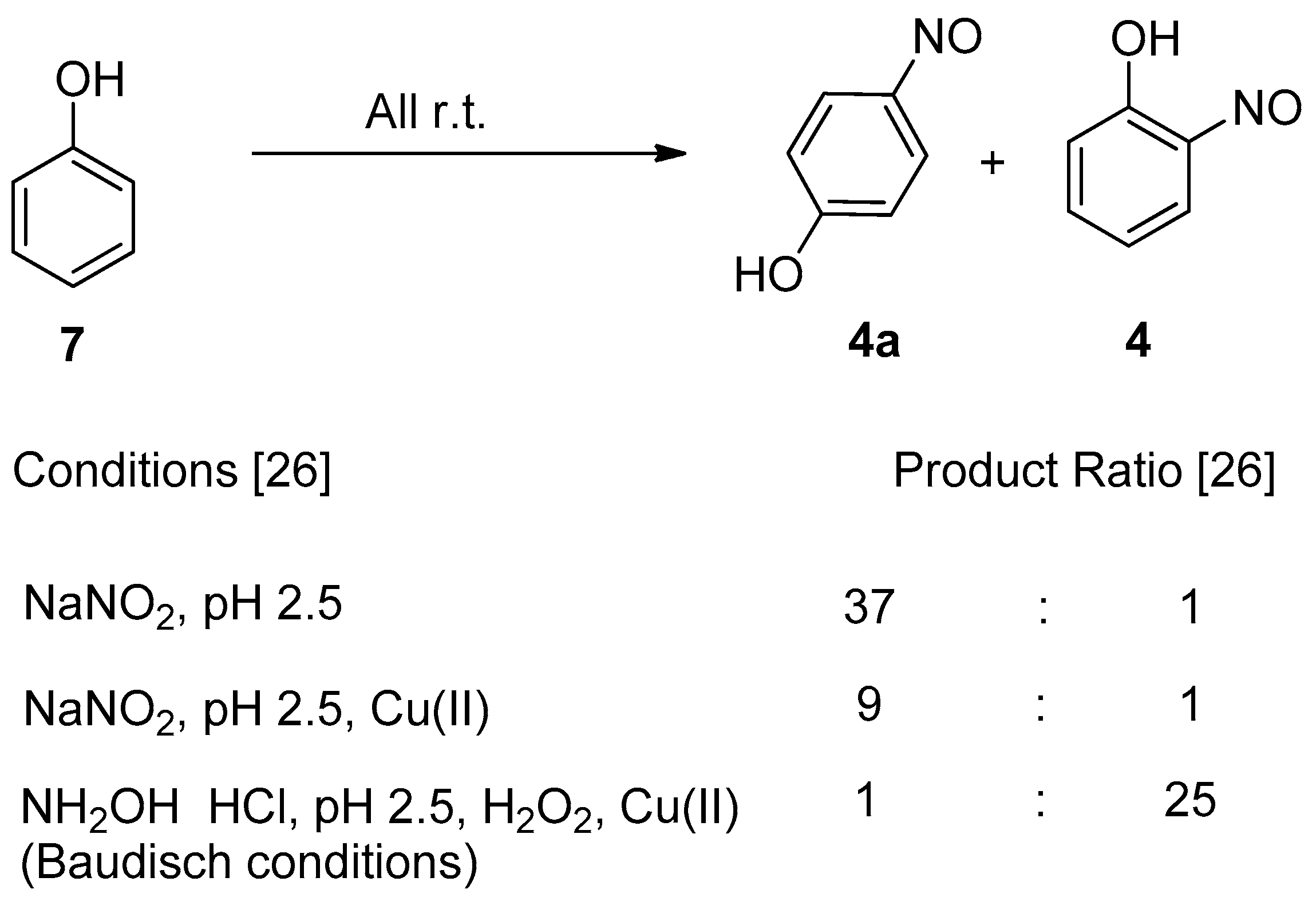
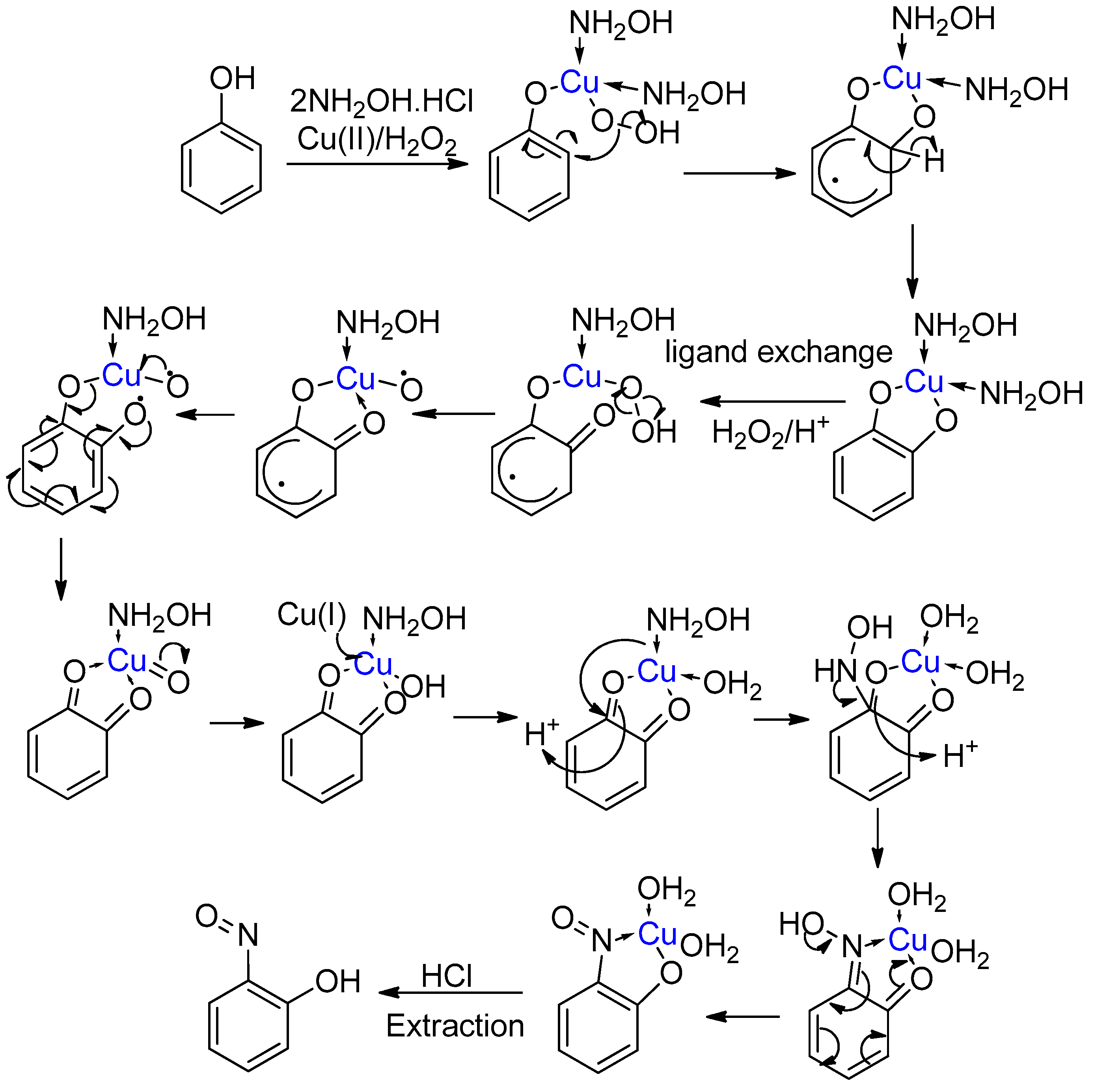
2.1. Development of the Baudisch Reaction Mechanism
2.2. The Accepted Mechanism
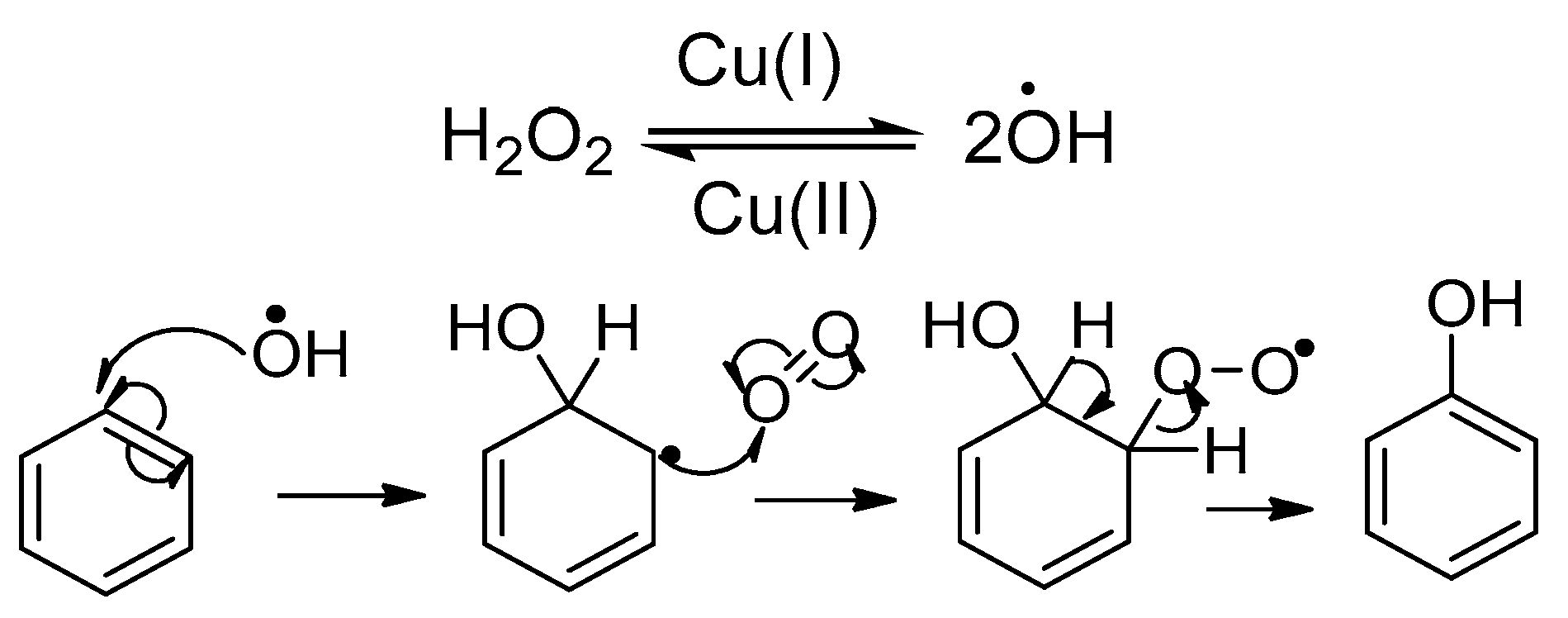
2.3. Mechanism of the Alternative Copper-Mediated Nitrosation
3. Applications of Copper-Nitrosophenolato Complexes
3.1. Colourimetry
3.2. Nucleophilic Addition of Grignard Reagents
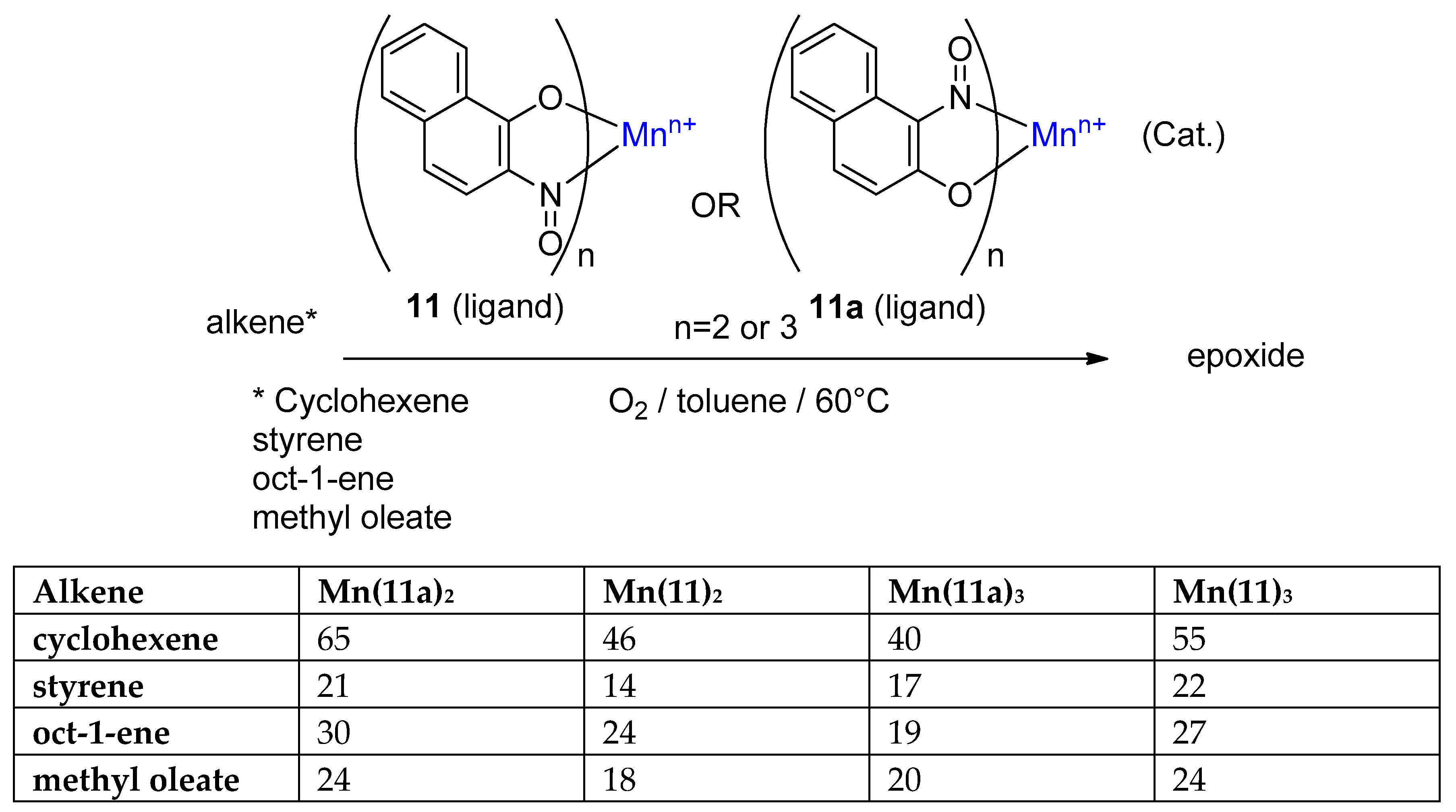
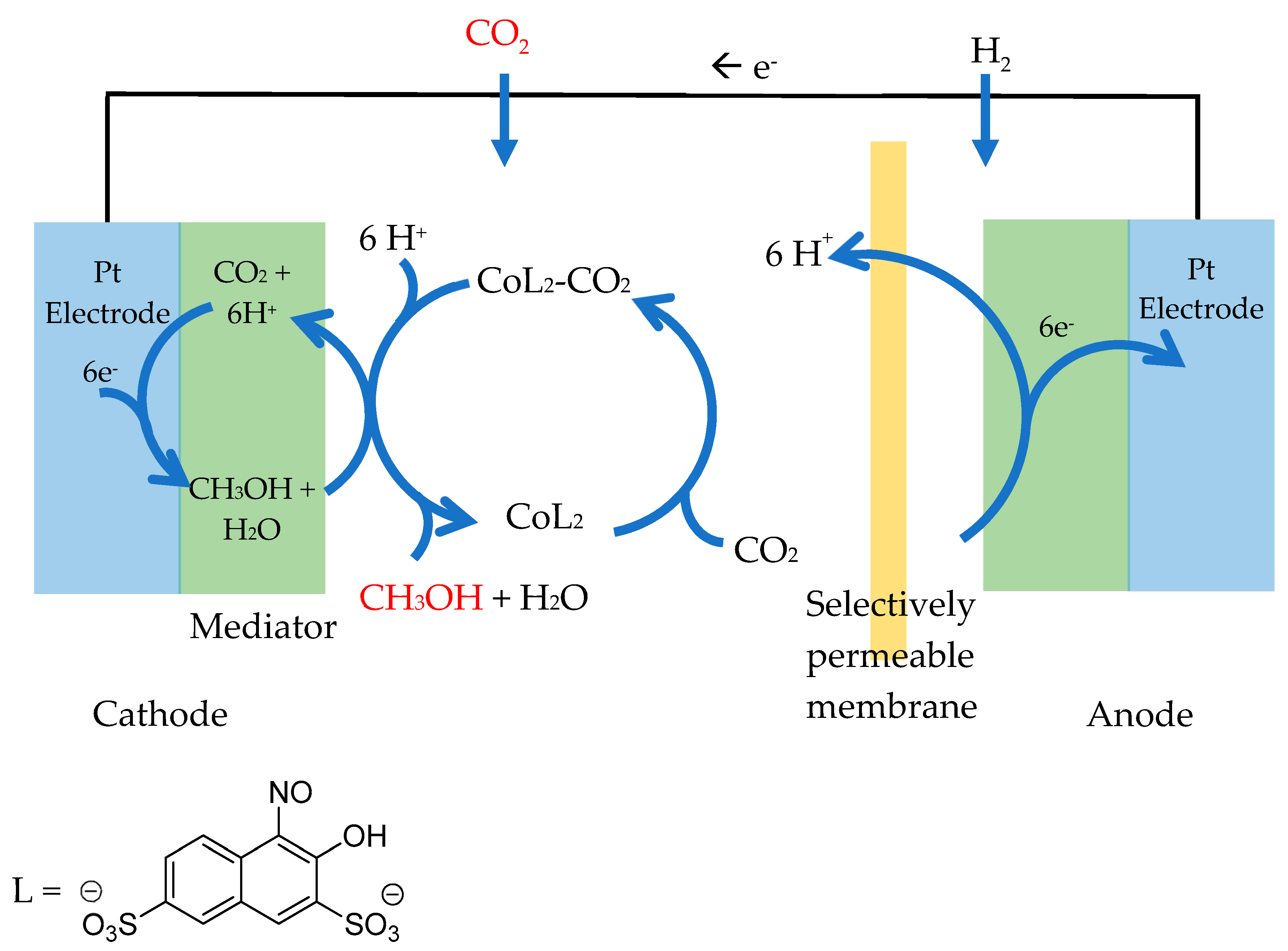
3.3. The Use of Complexes as a Redox Catalyst
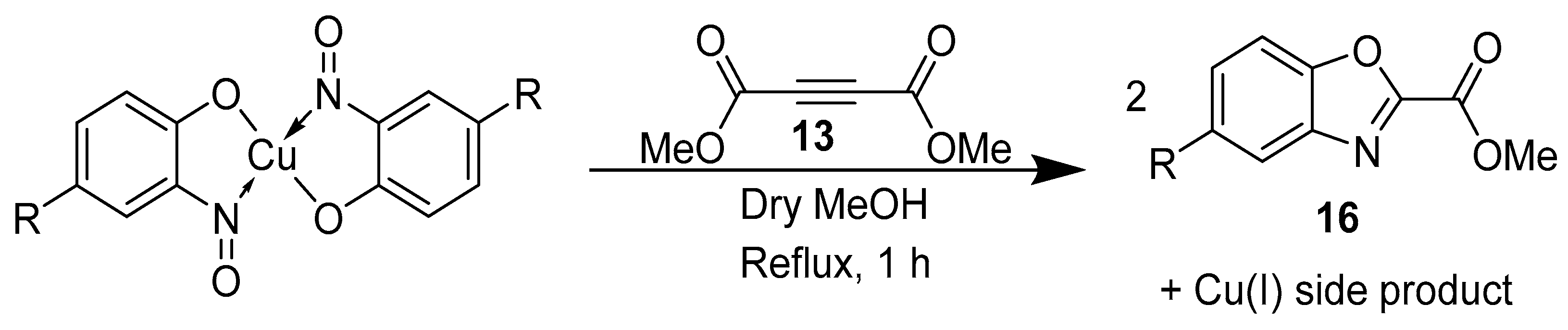
3.4. Use in [4 + 2] Cycloadditions
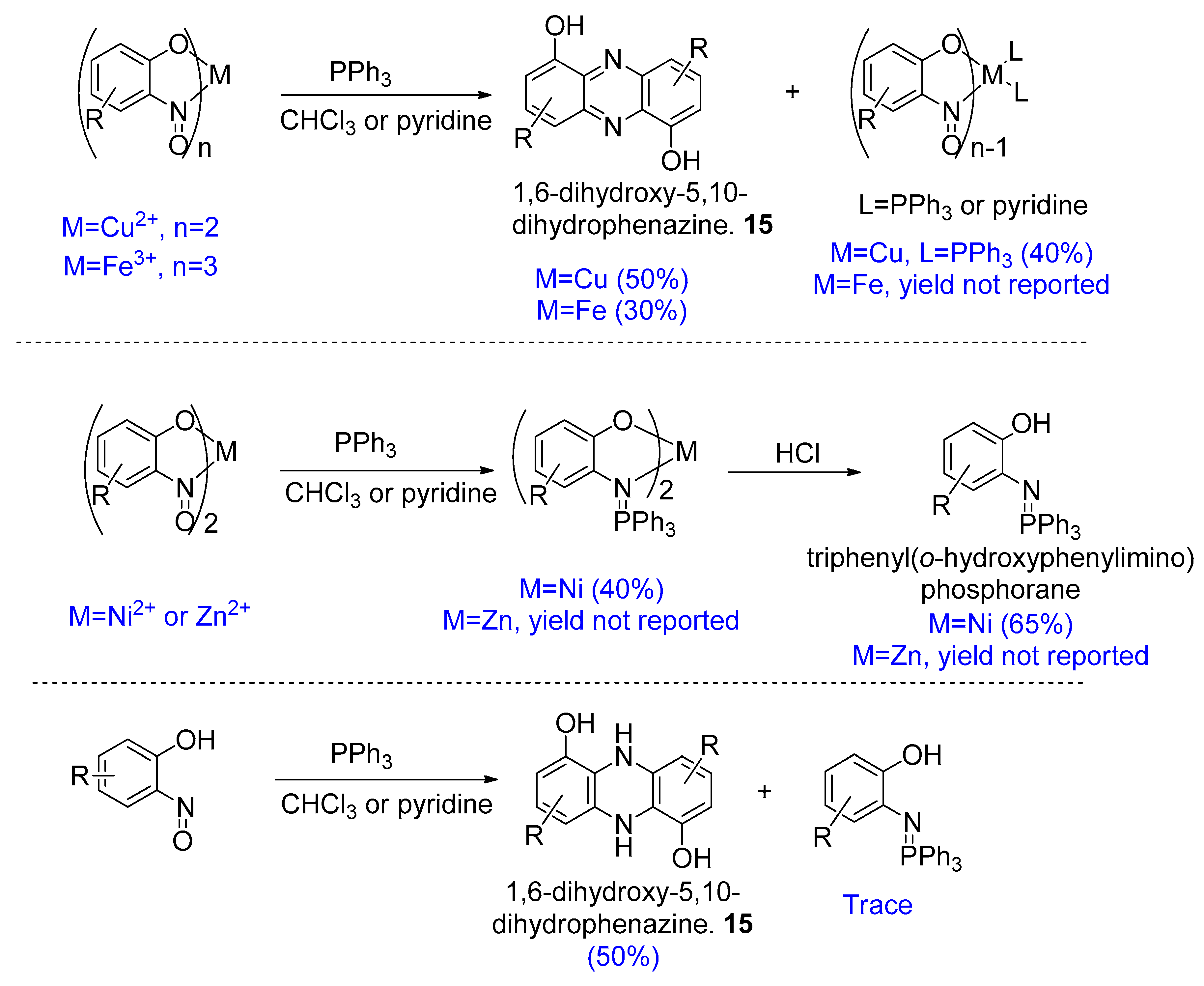
3.5. Synthesis of Phenazines
3.6. Synthesis of Oxazoles
4. Future Applications of Copper-Nitrosophenolato Complexes
4.1. Functionalisation of Natural Products
4.2. Lignin Functionalisation
4.3. Addition of Nucleophilic Amines to Aromatic Rings
4.4. Summary of Uses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Charalambous, J.; Frazer, M.J.; Taylor, F.B. Complexes of copper(II) with o-nitrosophenols (monoximes of ortho-benzoquinones). J. Chem. Soc. A Inorg. Phys. Theor. 1969, 2787. [Google Scholar] [CrossRef]
- Al-Obaidi, U.; Moodie, R.B. The nitrous acid-catalysed nitration of phenol. J. Chem. Soc. Perkin Trans. 2 1985, 467. [Google Scholar] [CrossRef]
- Hayton, T.W.; Legzdins, P.; Sharp, W.B. Coordination and Organometallic Chemistry of Metal − NO Complexes. Chem. Rev. 2002, 102, 935–991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M. Production Device of Rubber Colorant Pigment Green B 2017. Available online: https://patents.google.com/patent/CN108239422A/en?oq=CN108239422A (accessed on 28 July 2019).
- Wegener, J.W.; Klamer, J.C.; Govers, H.; Brinkman, U.A.T. Determination of organic colorants in cosmetic products by high-performance liquid chromatography. Chromatographia 1987, 24, 865–875. [Google Scholar] [CrossRef]
- American Association of Textile Chemists and Colorists (AATCC). Available online: https://www.standardsportal.org/usa_en/trade_associations/aatcc.aspx (accessed on 5 November 2019).
- Shi, H.; Zhang, T.; An, T.; Li, B.; Wang, X. Photocatalytic Hydroxylation of Phenol to Catechol and Hydroquinone by Using Organic Pigment as Selective Photocatalyst. Curr. Org. Chem. 2012, 16, 3002–3007. [Google Scholar] [CrossRef]
- Chain, E.B.; Tonolo, A.; Carilli, A. Ferroverdin, a Green Pigment containing Iron produced by a Streptomycete. Nature 1955, 176, 645. [Google Scholar] [CrossRef]
- Candeloro, S.; Grdenic, D.; Taylor, N.; Thompson, B.; Viswamitra, M.; Hodgkin, D.C. Structure of Ferroverdin. Nature 1969, 224, 589–591. [Google Scholar] [CrossRef]
- Maciejewska, M.; Pessi, I.S.; Arguelles-Arias, A.; Noirfalise, P.; Luis, G.; Ongena, M.; Barton, H.; Carnol, M.; Rigali, S. Streptomyces lunaelactis sp. nov., a novel ferroverdin A-producing Streptomyces species isolated from a moonmilk speleothem. Antonie Van Leeuwenhoek 2015, 107, 519–531. [Google Scholar] [CrossRef]
- Tomoda, H.; Tabata, N.; Shinose, M.; Takahashi, Y.; Woodruff, H.B.; Omura, S. Ferroverdins, Inhibitors of Cholesteryl Ester Transfer Protein Produccd by Streptomyces sp. WK-5344. I. Production, Isolation and Biological Properties. J. Antibiot. (Tokyo) 1999, 52, 1101–1107. [Google Scholar] [CrossRef]
- KUROBANE, I.; DALE, P.L.; VINING, L.C. Characterization of new viridomycins and requirements for production in cultures of Streptomyces griseus. J. Antibiot. (Tokyo) 1987, 40, 1131–1139. [Google Scholar] [CrossRef]
- Yang, C.C.; Leong, J. Mode of antibiotic action of 4-hydroxy-3-nitrosobenzaldehyde from Streptomyces viridans. Antimicrob. Agents Chemother. 1981, 20, 558–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, P.; Pal, S.; Chakravorty, A. Redox in a ferroverdin analogue: Recognition of isomeric co-ordination spheres by Fe II and Fe III. J. Chem. Soc. Chem. Commun. 1989, 977. [Google Scholar] [CrossRef]
- Cronheim, G. O-Nitrosophenols. J. Org. Chem. 1947, 12, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Millon, E. Sur un Réacrif Propre aux composés proteiques. Compt. Rend. Acad. 1849, 132, 156. [Google Scholar]
- Vaubel, W. Zur Kenntniss der Millon’schen Reaction. Zeitschrift für Angew. Chemie 1900, 13, 1125–1130. [Google Scholar] [CrossRef]
- Kido, Y.; Tamura, Z. An improved method for the millon reaction. Chem. Pharm. Bull. (Tokyo) 1981, 29, 2296–2302. [Google Scholar] [CrossRef]
- Gibbs, H.D. PHENOL TESTS.* II. Nitrous Acid Tests. The Millon and Similar Spectrophotometric Iinvestigations. J. Biol. Chem. 1926, 71, 445–459. [Google Scholar]
- Baudisch, O. A New Chemical Reaction With the Nitrosyl Radical Noh. Science 1940, 92, 336–337. [Google Scholar] [CrossRef]
- Baudisch, O.; Smith, S. Ftir die kurzen Originalmitteilungen ist ausschlieBlich der Verfasser verantwortlich. Kurze Orig. 1939, 17, 768–769. [Google Scholar]
- Deka, H.; Ghosh, S.; Gogoi, K.; Saha, S.; Mondal, B. Nitric Oxide Reactivity of a Cu(II) Complex of an Imidazole-Based Ligand: Aromatic C-Nitrosation Followed by the Formation of N-Nitrosohydroxylaminato Complex. Inorg. Chem. 2017, 56, 5034–5040. [Google Scholar] [CrossRef]
- Kundu, S.; Kim, W.Y.; Bertke, J.A.; Warren, T.H. Copper(II) activation of nitrite: Nitrosation of nucleophiles and generation of NO by thiols. J. Am. Chem. Soc. 2017, 139, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Cone, M.C.; Melville, C.R.; Carney, J.R.; Gore, M.P.; Gould, S.J. 4-Hydroxy-3-nitrosobenzamide and its ferrous chelate from Streptomyces murayamaensis. Tetrahedron 1995, 51, 3095–3102. [Google Scholar] [CrossRef]
- Dessouky, Y.M.; Gal El Rub, L.N. Colorimetric determination of procaine hydrochloride in pharmaceutical preparations. Analyst 1976, 101, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Goto, R.; Tanimoto, I. Studies on the Baudisch Reaction. I. The Synthesis of O-Nitrosophenols. J. Org. Chem. 1967, 32, 2516–2520. [Google Scholar] [CrossRef]
- Masoud, M.S.; Hindawey, A.M.; Mostafa, M.A.; Ramadan, A.M. Synthesis and structural chemistry of 2,4-dinitrosoresorcionl, 1-nitroso-2-naphthol and 4-carboxy-2-nitrosophenol. Spectrosc. Lett. 1997, 30, 1227–1247. [Google Scholar] [CrossRef]
- Porta, F.; Prati, L. Catalytic synthesis of C-nitroso compounds by cis-Mo(O)2(acac)2. J. Mol. Catal. A Chem. 2000, 157, 123–129. [Google Scholar] [CrossRef]
- Williams, D.L.H. Nitrosation Reactions and the Chemistry of Nitric Oxide, 1st ed.; Elsevier B.V: Bodmin, UK, 2004. [Google Scholar]
- Baluch, D.; Charalambous, J.; Haines, L.I.B. Manganese Complexes of 1,2-Naphthoquinone Mono-oximes (2-Nitrosophenols) as Catalysts for Alkene Epoxidation Dosten. J. Chem. Soc. Chem. Commun. 1988, 1178–1179. [Google Scholar] [CrossRef]
- Aljaar, N.; Malakar, C.C.; Conrad, J.; Beifuss, U. Base-Promoted Domino Reaction of 5-Substituted 2-Nitrosophenols with Bromomethyl Aryl Ketones: A Transition-Metal-Free Approach to 2-Aroylbenzoxazoles. J. Org. Chem. 2015, 80, 10829–10837. [Google Scholar] [CrossRef]
- Li, H.; Pang, M.; Wu, B.; Meng, J. Synthesis, crystal structure and photochromism of a novel spiro[indoline-naphthaline]oxazine derivative. J. Mol. Struct. 2015, 1087, 23–29. [Google Scholar] [CrossRef]
- Atherton, J.H.; Moodie, R.B.; Noble, D.R. Kinetics and mechanism of nitrosation of toluene, o-xylene, and m-xylene in trifluoroacetic acid, or in acetic–sulfuric acid mixtures, under nitric oxide. J. Chem. Soc. Perkin Trans. 2 1999, 699–706. [Google Scholar] [CrossRef]
- Leis, J.R.; Ríos, A.; Rodríguez-Sánchez, L. Reactivity of phenolic nucleophiles towards nitroso compounds. Part 2.1 Reaction with alkyl nitrites (O-nitroso compounds). J. Chem. Soc. Perkin Trans. 2 1998, 2729–2734. [Google Scholar] [CrossRef]
- Charalambous, J.; Kensett, M.J.; Jenkins, J.M. Deoxygenation of 2-nitrosophenols and of their metal complexes with triphenylphosphine. Synthesis of phenazines, dihydrophenazines, triphenyl(o-hydroxyphenylimino)phosphoranes and their metal complexes. J. Chem. Soc. Chem. Commun. 1977, 400–401. [Google Scholar] [CrossRef]
- Atherton, J.H.; Moodie, R.B.; Noble, D.R.; O’Sullivan, B. Nitrosation of m-xylene, anisole, 4-nitrophenyl phenyl ether and toluene in trifluoroacetic acid or in acetic–sulfuric acid mixtures under nitric oxide. J. Chem. Soc. Perkin Trans. 2 1997, 2, 663–664. [Google Scholar] [CrossRef]
- Ramozzi, R.; Chéron, N.; El Kaïm, L.; Grimaud, L.; Fleurat-Lessard, P. Predicting new ugi-smiles couplings: A combined experimental and theoretical study. Chem.-A Eur. J. 2014, 20, 9094–9099. [Google Scholar] [CrossRef] [PubMed]
- Bosch, E.; Kochi, J.K. Direct Nitrosation of Aromatic Hydrocarbons and Ethers with the Electrophilic Nitrosonium Cation. J. Org. Chem. 1994, 59, 5573–5586. [Google Scholar] [CrossRef]
- Crossley, M.L.; Dreisbach, P.F.; Hofmann, C.M.; Parker, R.P. Chemotherapeutic Dyes. I. 5-Aralkylamino-9-alkylaminobenzo [a]phenoxazines 1. J. Am. Chem. Soc. 1952, 74, 573–578. [Google Scholar] [CrossRef]
- Maleski, R.J.; Kluge, M.; Sicker, D. A facile access to substituted 2-nitrosophenols and 2-nitrophenols via regioselective nitrosation of resorcinol monoethers. Synth. Commun. 1995, 25, 2327–2335. [Google Scholar] [CrossRef]
- Nicholls, A.J.; Batsanov, A.S.; Baxendale, I.R. Copper-Mediated Nitrosation: 2-Nitrosophenolato Complexes and their use in the Synthesis of Heterocycles. Molecules 2019. under review. [Google Scholar]
- Maruyama, K.; Tanimoto, I. Studies on the Baudisch Reaction. IV. The Reaction Mechanism. Bull. Chem. Soc. Jpn. 1971, 44, 3120–3123. [Google Scholar] [CrossRef] [Green Version]
- Konecny, J.O. Hydroxylation of Benzene in Aqueous Solution in the Presence of Hydroxylamine Hydrochloride. J. Am. Chem. Soc. 1955, 77, 5748–5750. [Google Scholar] [CrossRef]
- 1-Nitroso-2-Naphthol (2019). Available online: https://www.sigmaaldrich.com/catalog/product/aldrich/114693?lang=en®ion=GB (accessed on 1 August 2019).
- Patil, S.V.; Mohite, B.S. Palladium (II) Chelates of some o-Nitrosophenols. J. Mol. Catal. 1977, 46, 638. [Google Scholar]
- Ka-Hong Lee, K.; Wong, W.-T. Syntheses and molecular structures of ruthenium carbonyl complexes containing 1,2-naphthoquinone-1-oximate ligands. J. Chem. Soc. Dalt. Trans. 1997, 2987–2996. [Google Scholar] [CrossRef]
- Lyubchenko, S.N.; Ionov, A.M.; Shcherbakov, I.N.; Aleksandrov, G.G.; Kogan, V.A.; Tsivadze, A.Y. Coordination compounds based on 4,6-di-tert-butyl-2-nitrosophenol. Russ. J. Coord. Chem. 2006, 32, 539–544. [Google Scholar] [CrossRef]
- Rout, K.C.; Mondal, B. Aromatic C-nitrosation by a copper(ii)–nitrosyl complex. Dalt. Trans. 2015, 44, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Baluch, D. Studies of 1,2-Quinone Mono-Oximato Complexes and Their Redox Reactions; The Polytechnic of North London: London, UK, 1987. Available online: https://core.ac.uk/display/161337781 (accessed on 5 November 2019).
- Castellani, C.B.; Carugo, O. Studies on copper(II) complexes of o-quinone monooximes. 8. Penta and hexacoordinated adducts of bis(4-chloro-1,2-benzaquinone 2-oximato)copper(II), [Cu(Clqo)2], with imidazole and N-methylimidazole. Inorg. Chim. Acta 1988, 150, 119–123. [Google Scholar] [CrossRef]
- The Cambridge Crystallographic Data Centre Home—The Cambridge Crystallographic Data Centre (CCDC). Available online: https://www.ccdc.cam.ac.uk (accessed on 5 November 2019).
- Basu, P. Cobalt analogue of ferroverdin: Synthesis and geometric structure. Polyhedron 1992, 11, 3037–3040. [Google Scholar] [CrossRef]
- Castellani, C.B.; Buttafava, A.; Carugo, O.; Poggi, A. Studies on metal complexes of ortho-quinone mono-oximes. Part 6. Redox behaviour of bis(4-chloro-1,2-benzoquinone 2-oximato)-copper(II) and -nickel(II); synthesis and characterization of reduced species containing a paramagnetic ligand. J. Chem. Soc. Dalt. Trans. 1988, 1497. [Google Scholar] [CrossRef]
- Masoud, M.S.; Haggag, S.S.; Ramadan, A.M.; Mahmoud, S. a Nitrosophenol complexes of transition metal salts. Transit. Met. Chem. 1998, 23, 343–347. [Google Scholar] [CrossRef]
- Castellani, C.B.; Gatti, G.; Millini, R. Studies on copper(II) Complexes of o-quinone monooximes. adducts of bis(4-chioro-1 -benzoquinone 2-oximato) copper(II) with Some heterocyclic bases. structure of the 2,2’-bipyridine adduct. Inorg. Chem. 1984, 23, 4004–4008. [Google Scholar] [CrossRef]
- Castellani, C.B.; Carugo, O.; Coda, A. Studies on copper(II) complexes of o-quinone monooximes. 3. Reactivity of bis(4-chloro-1-benzoquinone 2-oximato)copper(II) with halides. Crystal structure of potassium bis(.mu.-iodo)tris[bis(4-chloro-1-benzoquinone 2-oximato)cuprate(II)]. Inorg. Chem. 1987, 26, 671–675. [Google Scholar] [CrossRef]
- Adatia, T.; Chakrabarti, J.; Charalambous, J.; Carugo, O.; Castellani, C.B. Synthesis and structural characterization of lithium and sodium complexes of 5-ethylamino-4-methyl-1,2-benzoquinone 2-oxime. Polyhedron 1996, 15, 1331–1338. [Google Scholar] [CrossRef]
- Li, F.; Liu, Q.; Hu, J.; Feng, Y.; He, P.; Ma, J. Recent advances in cathode materials for rechargeable lithium–sulfur batteries. Nanoscale 2019, 11, 15418–15439. [Google Scholar] [CrossRef] [PubMed]
- Kasumov, V.T.; Kartal, I.; Koksal, F. Synthesis and ESR studies of redox reactivity of bis (3,5-di-tert-butyl-1,2-benzoquinone-2-monooximato)Cu(II). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2000, 56, 841–850. [Google Scholar] [CrossRef]
- Clayden, J.; Greeves, N.; Warren, S.; Wothers, P. Organic Chemistry, 1st ed.; Oxford University Press: Oxford, OH, USA, 2001. [Google Scholar]
- Shchavlev, A.E.; Pankratov, A.N.; Enchev, V. Intramolecular Hydrogen-Bonding Interactions in 2-Nitrosophenol and Nitrosonaphthols: Ab Initio, Density Functional, and Nuclear Magnetic Resonance Theoretical Study Intramolecular Hydrogen-Bonding Interactions in 2-Nitrosophenol and Nitrosonaphthols. J. Phys. Chem. A 2007, 111, 30. [Google Scholar] [CrossRef] [PubMed]
- Radner, F.; Wall, A.; Loncar, M. Nitrosation of Anisole and Related Compounds. Direct synthesis of 4-nitrosoanisole. Acta Chem. Scand. 1990, 44, 152–157. [Google Scholar] [CrossRef]
- Lindeman, S.V.; Bosch, E.; Kochi, J.K. Electrophilic aromatic nitrosation. Isolation and X-ray crystallography of the metastable NO+ complex with nitrosoarene. J. Chem. Soc. Perkin Trans. 2 2000, 4, 1919–1923. [Google Scholar] [CrossRef]
- Carla Bisi Castellani—Researchgate (2019). Available online: https://www.researchgate.net/profile/Carla_Castellani (accessed on 30 July 2019).
- John Charalambous—LinkedIn (2019). Available online: https://www.linkedin.com/in/profjohncharalambous/?originalSubdomain=uk (accessed on 30 July 2019).
- Röder, L.; Nicholls, A.J.; Baxendale, I.R. Flow hydrodediazoniation of aromatic heterocycles. Molecules 2019, 24, 1996. [Google Scholar] [CrossRef]
- Sandmeyer, T. Ueber die Ersetzung der Amidgruppe durch Chlor in den aromatischen Substanzen. Berichte der Dtsch. Chem. Gesellschaft 1884, 17, 1633–1635. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, A.; Kamel, M. Reactions of o-Quinone Monoximes, Monoimines and Triketoindane-2-oxime. J. Am. Chem. Soc. 1954, 76, 124–127. [Google Scholar] [CrossRef]
- Noble, D.R.; Williams, D.L.H. Nitrosation products from S-nitrosothiols via preliminary nitric oxide formation. J. Chem. Soc. Perkin Trans. 2 2002, 1834–1838. [Google Scholar] [CrossRef]
- Nishino, H.; Satoh, H.; Yamashita, M.; Kurosawa, K. (Nitrosonaphtholato)metal complex-catalyzed oxidation of phenols and alkenes. J. Chem. Soc. Perkin Trans. 2 1999, 1919–1924. [Google Scholar] [CrossRef]
- Ogura, K.; Yoshida, I. Electrocatalytic reduction of CO2 to methanol. Part 9: Mediation with metal porphyrins. J. Mol. Catal. 1988, 47, 51–57. [Google Scholar] [CrossRef]
- Ogura, K.; Migita, C.T.; Nagaoka, T. Electrochemical reduction of carbon dioxide with an electrode mediator and homogeneous catalysts. J. Mol. Catal. 1989, 56, 276–283. [Google Scholar] [CrossRef]
- Ogura, K.; Migita, C.T.; Uchida, H. Exclusion of CO and CO2 from ammonia synthesis gas by an electrocatalytic process. J. Appl. Electrochem. 1990, 20, 240–244. [Google Scholar] [CrossRef]
- Ogura, K.; Mine, K.; Yano, J.; Sugihara, H. Electrocatalytic Reduction of Carbon Dioxide at a Composite Film Electrode with a Surface-Confined Metal Complex. Denki Kagaku Kyōkai 1993, 61, 810–811. [Google Scholar]
- Castellani, C.B.; Millini, R. Notes: Studies on copper(II) complexes of ortho-quinone mono-oximes: Reaction of bis(4-chloro-1,2-benzoquinone 2-oximato)copper(II) with dimethyl acetylenedicarboxylate under various experimental conditions. J. Chem. Soc. Dalt. Trans. 1984, 1461–1462. [Google Scholar] [CrossRef]
- McKillop, A.; Sayer, T.S.B. Metal complexes in organic synthesis. I. Cycloaddition of dimethyl acetylenedicarboxylate with the bis copper(II) complexes formed from o-nitrosophenols. Synthesis of 2,3-dicarbomethoxy-4-hydroxy-1,4-benzoxazines. J. Org. Chem. 1976, 41, 1079–1080. [Google Scholar] [CrossRef]
- Buckley, R.G.; Charalambous, J.; Kensett, M.J.; McPartlin, M.; Mukerjee, D.; Brain, E.G.; Jenkins, J.M. Reaction of triphenylphosphine with copper complexes derived from 2-nitrosophenols (1,2-quinone mono-oximes). J. Chem. Soc. Perkin Trans. 1 1983, 693. [Google Scholar] [CrossRef]
- Buckley, R.G.; Charalambous, J.; Brain, E.G. Interaction of copper complexes derived from 1,2-quinone monooximes (2-nitroso-phenols) with amines. J. Chem. Soc. Perkin Trans. 1 1982, 1075. [Google Scholar] [CrossRef]
- Li, S.; Lo, C.-Y.; Pan, M.-H.; Lai, C.-S.; Ho, C.-T. Black tea: Chemical analysis and stability. Food Funct. 2013, 4, 10–18. [Google Scholar] [CrossRef]
- Lee, S.H.; Doherty, T.V.; Linhardt, R.J.; Dordick, J.S. Ionic liquid-mediated selective extraction of lignin from wood leading to enhanced enzymatic cellulose hydrolysis. Biotechnol. Bioeng. 2009, 102, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, T.V.; Phillips, C.B. Renewable fuels via catalytic hydrodeoxygenation. Appl. Catal. A Gen. 2011, 397, 1–12. [Google Scholar] [CrossRef]
- Rossberg, C.; Bremer, M.; Machill, S.; Koenig, S.; Kerns, G.; Boeriu, C.; Windeisen, E.; Fischer, S. Separation and characterisation of sulphur-free lignin from different agricultural residues. Ind. Crops Prod. 2015, 73, 81–89. [Google Scholar] [CrossRef]
- Toledano, A.; Serrano, L.; Pineda, A.; Romero, A.A.; Luque, R.; Labidi, J. Microwave-assisted depolymerisation of organosolv lignin via mild hydrogen-free hydrogenolysis: Catalyst screening. Appl. Catal. B Environ. 2014, 145, 43–55. [Google Scholar] [CrossRef]
- Snelders, J.; Dornez, E.; Benjelloun-Mlayah, B.; Huijgen, W.J.J.; de Wild, P.J.; Gosselink, R.J.A.; Gerritsma, J.; Courtin, C.M. Biorefining of wheat straw using an acetic and formic acid based organosolv fractionation process. Bioresour. Technol. 2014, 156, 275–282. [Google Scholar] [CrossRef]
- Karagöz, S.; Bhaskar, T.; Muto, A.; Sakata, Y. Effect of Rb and Cs carbonates for production of phenols from liquefaction of wood biomass. Fuel 2004, 83, 2293–2299. [Google Scholar] [CrossRef]
- Molinari, V.; Clavel, G.; Graglia, M.; Antonietti, M.; Esposito, D. Mild Continuous Hydrogenolysis of Kraft Lignin over Titanium Nitride-Nickel Catalyst. ACS Catal. 2016, 6, 1663–1670. [Google Scholar] [CrossRef]
- Kwart, H.; Evans, E.R. The Vapor Phase Rearrangement of Thioncarbonates and Thioncarbamates. J. Org. Chem. 1966, 31, 410–413. [Google Scholar] [CrossRef]
- Bay, E.; Bak, D.A.; Leone-Bay, A.; Timony, P.E. Preparation of aryl chlorides from phenols. J. Org. Chem. 1990, 55, 3415–3417. [Google Scholar] [CrossRef]
- Newman, M.S.; Karnes, H.A. The Conversion of Phenols to Thiophenols via Dialkylthiocarbamates. J. Org. Chem. 1966, 31, 3980–3984. [Google Scholar] [CrossRef]
- Bean, F.R.; Donovan, T.S.; Kodak, E. Amination of Dihydricphenols. U.S. Patent No. 2,376,112 1945, 15 May 1945. [Google Scholar]
- Joncour, R.; Duguet, N.; Métay, E.; Ferreira, A.; Lemaire, M. Green Chemistry. Green Chem. 2014, 16, 2997–3002. [Google Scholar] [CrossRef]
- Bamberger, E. Weiteres uber die Oxydation des Orthoaminobenzaldehyds und iiber seine Beziehungen aum Benzoxazol. Berichte der Dtsch. Chem. Gesellschaft 1903, 36, 2042–2055. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
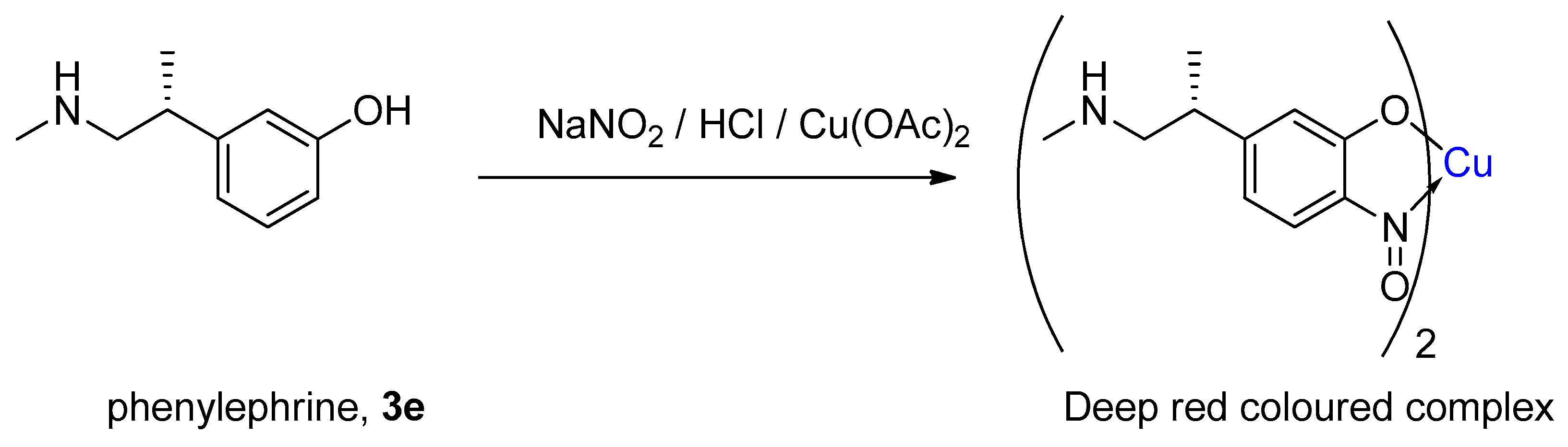{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
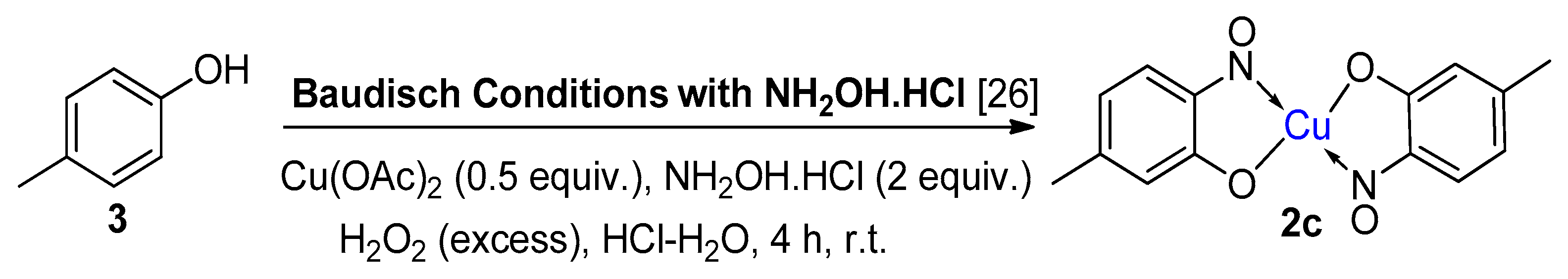{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Accepted Starting Materials |
|---|---|
| Baudisch conditions with hydroxylamine | A range of aromatics including benzene, phenols, catechols, naphthalenes and phenylsulfinic acids [20,42,43]. |
 | |
| Baudisch conditions with nitrous acid | Shown to accept benzene. Similar aromatics expected to work, though scope not investigated [20]. |
 | |
| Copper-mediated aromatic nitrosation | Phenols with sufficiently electron-rich aromatic ring and at least one non-functionalised carbon atom ortho to the phenol [15,41]. |
 | |
| Association of free 2-nitrosophenols with copper | STable 2-nitrosophenol compounds [1,15,41]. |
 |
 |
 |
| Starting Phenol | Expected Major Product | Reported Major Product (with Yield) | Reference |
|---|---|---|---|
 |  |  (not reported) (not reported) | [43] |
 |  |  (70%) (70%) | [1] |
 |  |  (71%) a (71%) a | [49] |
 |  |  (50%) b (50%) b | [1] |
 |  |  (not reported) (not reported) | [25] |
 |
 |
 |
 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicholls, A.J.; Barber, T.; Baxendale, I.R. The Synthesis and Utility of Metal-Nitrosophenolato Compounds—Highlighting the Baudisch Reaction. Molecules 2019, 24, 4018. https://doi.org/10.3390/molecules24224018
Nicholls AJ, Barber T, Baxendale IR. The Synthesis and Utility of Metal-Nitrosophenolato Compounds—Highlighting the Baudisch Reaction. Molecules. 2019; 24(22):4018. https://doi.org/10.3390/molecules24224018
Chicago/Turabian StyleNicholls, Alexander J., Thomas Barber, and Ian R. Baxendale. 2019. "The Synthesis and Utility of Metal-Nitrosophenolato Compounds—Highlighting the Baudisch Reaction" Molecules 24, no. 22: 4018. https://doi.org/10.3390/molecules24224018
APA StyleNicholls, A. J., Barber, T., & Baxendale, I. R. (2019). The Synthesis and Utility of Metal-Nitrosophenolato Compounds—Highlighting the Baudisch Reaction. Molecules, 24(22), 4018. https://doi.org/10.3390/molecules24224018