
A Study of the Influence of the HCl Concentration on the Composition and Structure of (Hydroxy)Arylsiloxanes from the Hydrolysis–Condensation Reaction of Aryltrichlorosilanes

 ,
,
Abstract
:
1. Introduction
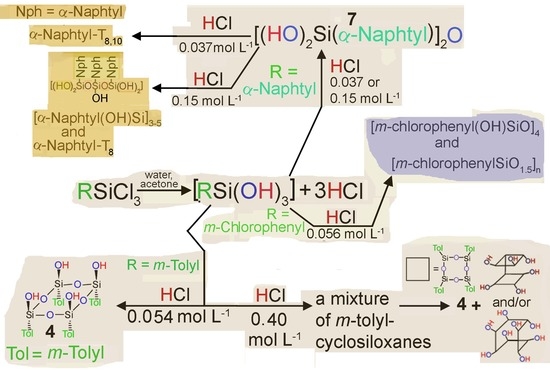
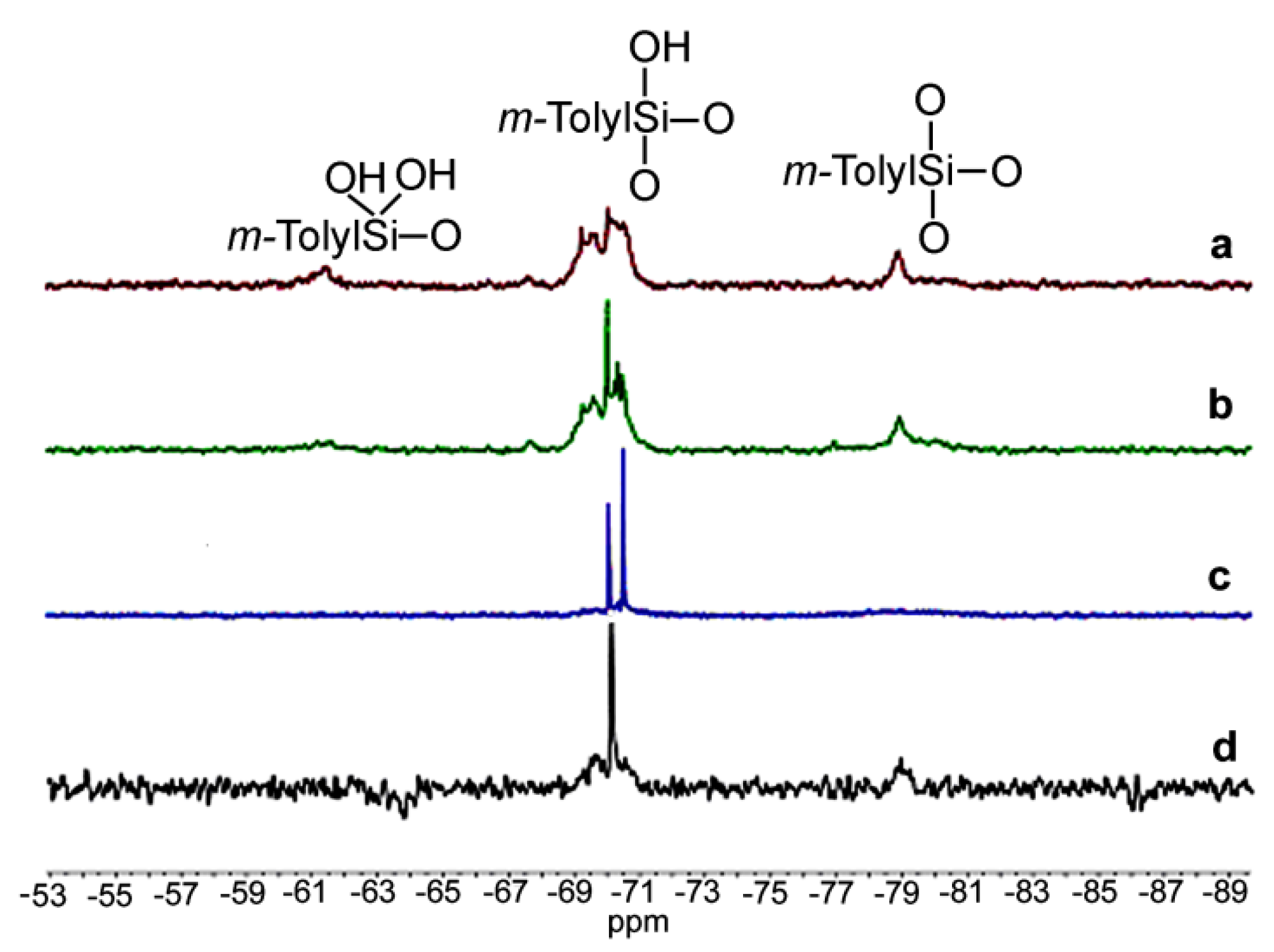
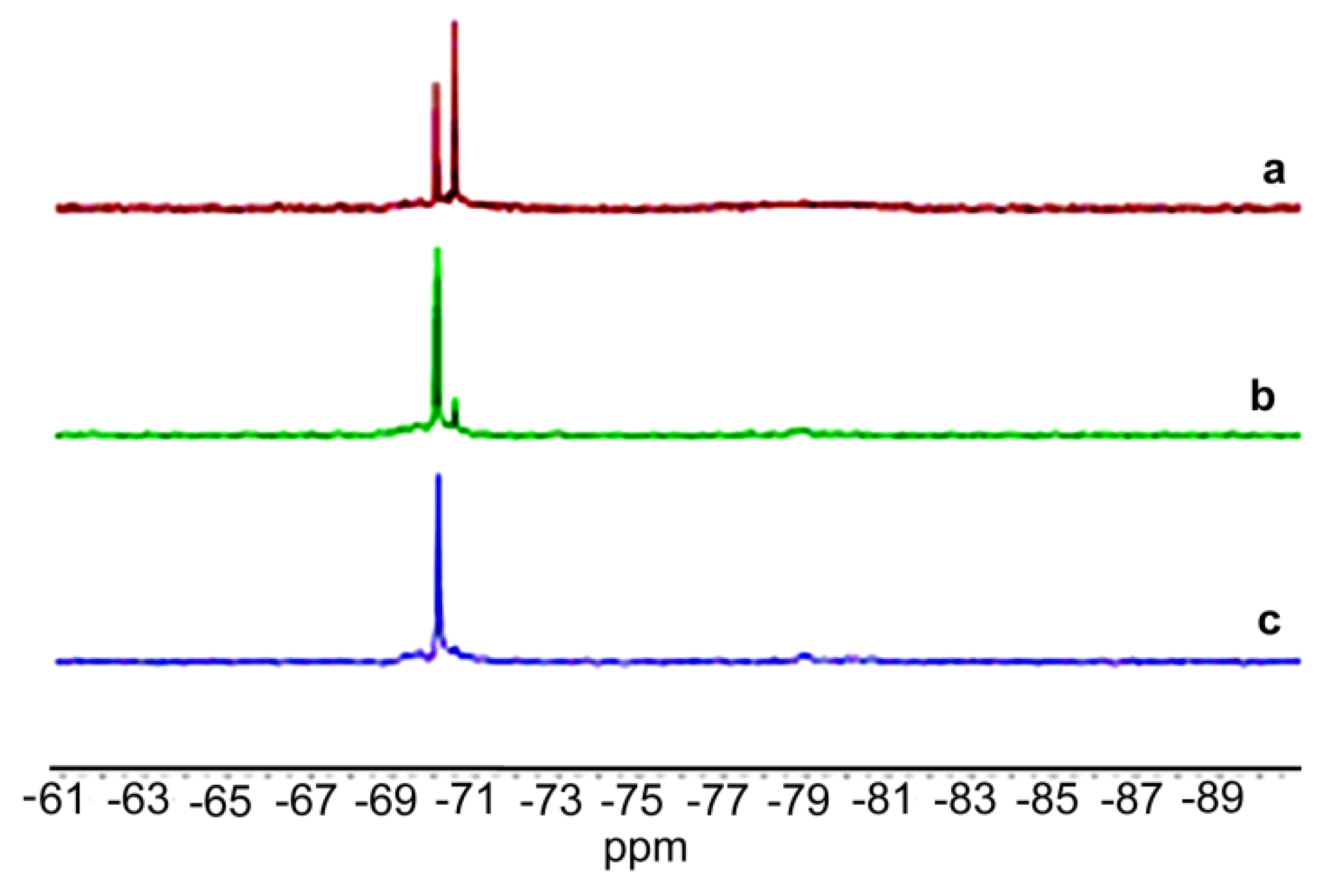
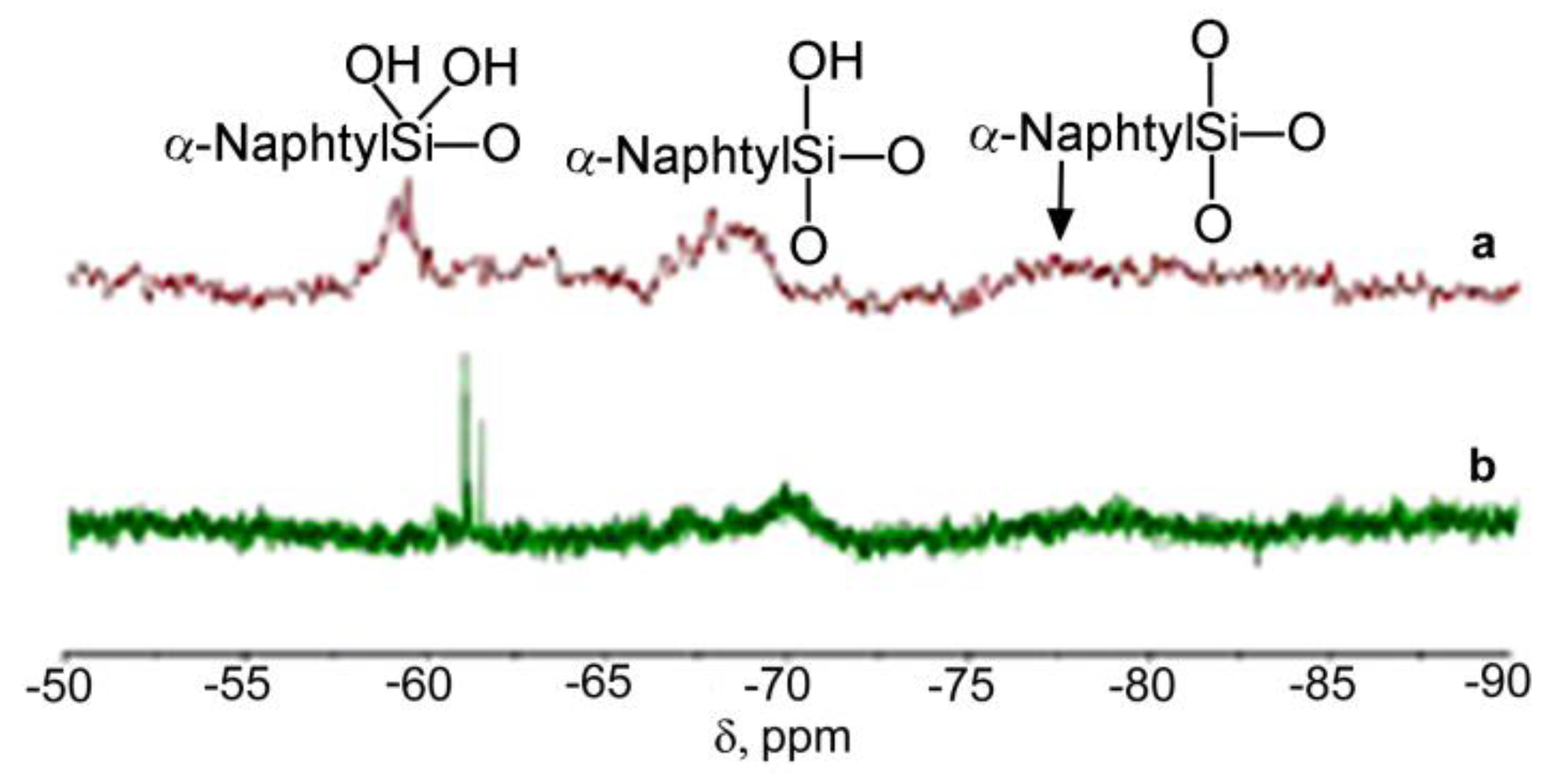
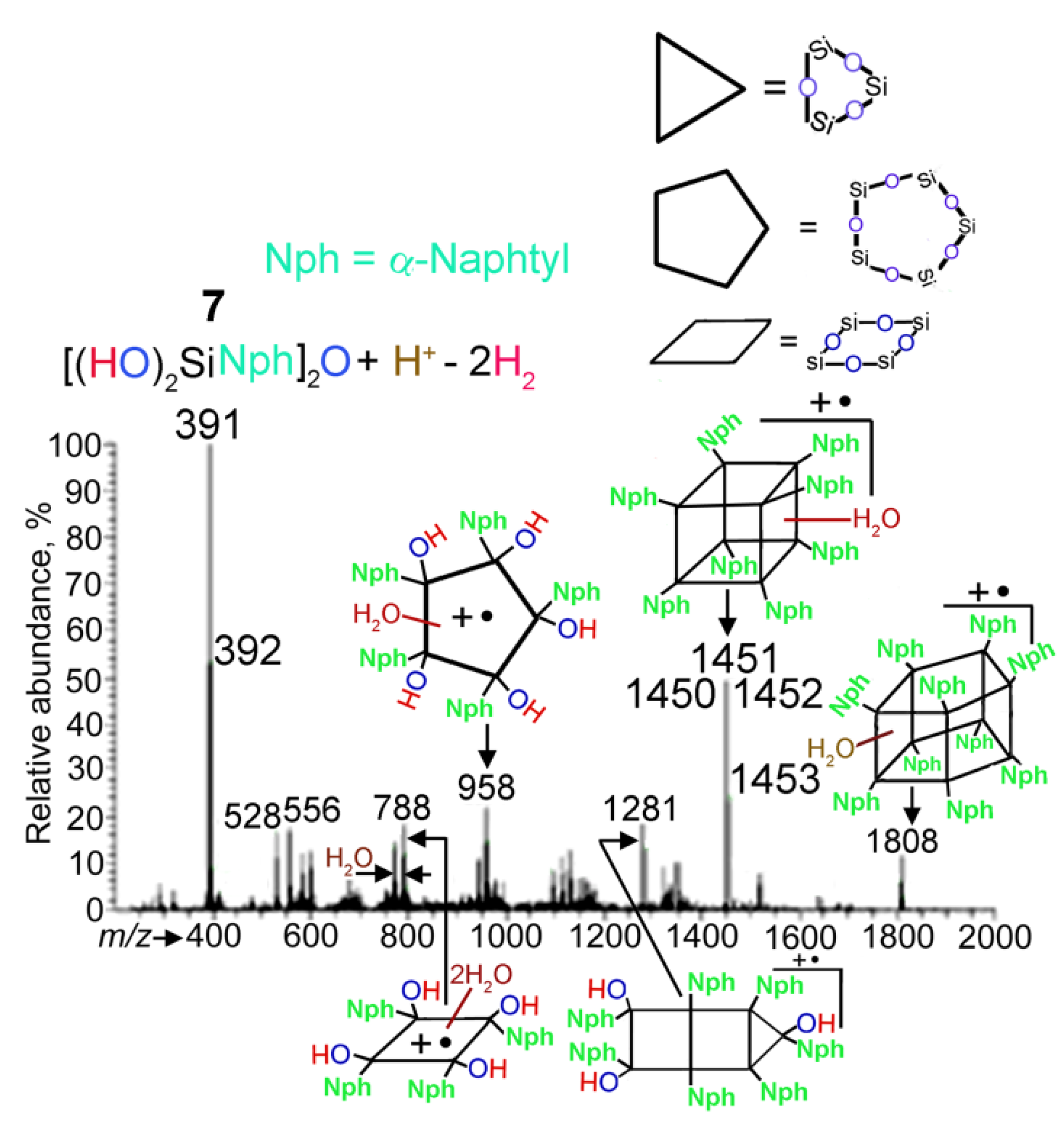
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, G.; Wang, L.; Ni, H.; Pittman, C.U. Polyhedral oligomeric silsesquioxane (POSS) polymers and copolymers: A Review. J. Inorg. Organometal. Polym. 2001, 11, 123–154. [Google Scholar] [CrossRef]
- Li, Z.; Kong, J.; Wang, F.; He, C. Polyhedral oligomeric silsesquioxanes (POSSs): An important building block for organic optoelectronic materials. J. Mater. Chem. C 2017, 5, 5283–5298. [Google Scholar] [CrossRef]
- Dong, F.; Lu, L.; Ha, C.-S. Silsesquioxane-containing hybrid nanomaterials: Fascinating platforms for advanced applications. Macromol. Chem. Phys. 2019, 220, 1800324. [Google Scholar] [CrossRef]
- Ayandele, E.; Sarkar, B.; Alexandridis, P. Polyhedral oligomeric silsesquioxane (POSS)—containing polymer nanocomposites. Nanomaterials 2012, 2, 445–475. [Google Scholar] [CrossRef]
- Baney, R.H.; Itoh, M.; Sakakibara, A.; Suzuki, T. Silsesquioxanes. Chem. Rev. 1995, 95, 1409–1430. [Google Scholar] [CrossRef]
- Brown, J.F.; Vogt, L.H.; Katchman, A.; Eustance, J.W.; Kister, K.M.; Krantz, K.W. Double chain polymers of phenylsilsesquioxane. J. Am. Chem. Soc. 1960, 82, 6194–6195. [Google Scholar] [CrossRef]
- Brown, J.F.; Vogt, L.H.; Prescott, P.I. Preparation and characterization of the lower equilibrated phenylsilsesquioxane. J. Am. Chem. Soc. 1964, 86, 1120–1125. [Google Scholar] [CrossRef]
- Brown, J.F.; Vogt, L.H. The Polycondensation of cyclohexylsilanetriol. J. Am. Chem. Soc. 1965, 87, 4313–4317. [Google Scholar] [CrossRef]
- Brown, J.F. The Polycondensation of phenylsilanetriol. J. Am. Chem. Soc. 1965, 87, 4317–4324. [Google Scholar] [CrossRef]
- Hambley, T.W.; Maschmeyer, T.; Masters, A.F. The synthesis and characterization of norbornylsilsesquioxanes. Appl. Organomet. Chem. 1992, 6, 253–260. [Google Scholar] [CrossRef]
- Feher, F.J.; Newnan, D.A.; Walzer, J.F. Silsesquioxanes as models for silica surfaces. J. Am. Chem. Soc. 1989, 111, 1741–1748. [Google Scholar] [CrossRef]
- Feher, F.J.; Budzichowski, T.A.; Blanski, R.L.; Weller, K.J.; Ziller, J.W. Facile syntheses of new incompletely condensed polyhedral organosilsesquioxanes: [(c-C5H9)7Si7O9(OH)3], [(c-C7H13)7Si7O9(OH)3], and [(c-C7H13)7Si6O7(OH)4]. Organometallics 1991, 10, 2526–2528. [Google Scholar] [CrossRef]
- Feher, F.J.; Newnan, D.A. Enhanced silylation reactivity of a model for silica surfaces. J. Am. Chem. Soc. 1990, 112, 1931–1936. [Google Scholar] [CrossRef]
- Birchall, J.D.; Carey, J.G.; Howard, A.J. Silane triols of high stability in aqueous solution. Nature 1977, 266, 154–156. [Google Scholar] [CrossRef]
- Smith, K.A. Polycondensation of methylmethoxysilane. Macromolecules 1987, 20, 2514–2520. [Google Scholar] [CrossRef]
- Yamamoto, S.; Yasuda, N.; Ueyama, A.; Adachi, H.; Ishikawa, M. Mechanism for the formation of poly(phenylsilsesquioxane). Macromolecules 2004, 37, 2775–2778. [Google Scholar] [CrossRef]
- Lee, A.S.; Choi, S.-S.; Lee, H.S.; Baek, K.-Y.; Hwang, S.S. A new, higher yielding synthetic route towards dodecaphenyl cage silsesquioxanes synthesis and mechanistic insights. Dalton Trans. 2012, 41, 10585–10588. [Google Scholar] [CrossRef]
- Choi, S.-S.; Lee, A.S.; Hwang, S.S.; Baek, K.-Y. Structural control of fully condensed polysilsesquioxanes: Ladder like vs. cage structured polyphenylsilsesquioxanes. Macromolecules 2015, 48, 6063–6070. [Google Scholar] [CrossRef]
- Zhang, X.; Xie, P.; Shen, Z.; Jiang, J.; Zhu, C.; Li, H.; Zhang, T.; Han, C.C.; Wan, L.; Yan, S.; et al. Confined synthesis of a cis-isotactic ladder polysilsesquioxane by using a π-stacking and H-bonding superstructure. Angew. Chem. Int. Ed. 2006, 45, 3112–3116. [Google Scholar] [CrossRef]
- Zhang, Z.-X.; Hao, J.; Xie, P.; Zhang, X.; Han, C.C.; Zhang, R. A well-defined ladder polyphenylsilsesquioxane (Ph-LPSQ) synthesized via a new three-step approach: Monomer self-organization–lyophilization-surface-confined polycondensation. Chem. Mater. 2008, 20, 1322–1330. [Google Scholar] [CrossRef]
- Yagihashi, F.; Igarashi, M.; Nakajima, Y.; Ando, W.; Sato, K.; Yumoto, Y.; Matsui, C.; Shimada, S. Acid-catalyzed condensation reaction of phenylsilanetriole: Unexpected formation of cis,trans-1,3,5-trihydroxy-1,3,5-triphenylcyclotrisiloxane as the main product and its isolation. Organometallics 2014, 33, 6278–6281. [Google Scholar] [CrossRef]
- Ito, R.; Kakihana, Y.; Kawakami, Y. Cyclic tetrasiloxanetetraols: Formation, isolation, and characterization. Chem. Lett. 2009, 38, 364–365. [Google Scholar] [CrossRef]
- Fujita, M.; Oguro, D.; Mlyazawa, M.; Oka, H.; Yamaguchi, K.; Ogura, K. Self-assembly of ten molecules into nanometre-sized organic host frameworks. Nature 1995, 378, 469–471. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Kusukawa, T.; Fujita, M.; Sakamoto, S.; Yamaguchi, K. Ship-in-a-bottle synthesis of otherwise labile cyclic trimers of siloxanes in a self-assembled coordination cage. J. Am. Chem. Soc. 2000, 122, 6311–6312. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Kusukawa, T.; Fujita, M.; Yamaguchi, K. Cavity-directed synthesis of labile silanol oligomers within self-assembled coordinatiom cages. J. Am. Chem. Soc. 2001, 123, 10454–10459. [Google Scholar] [CrossRef] [PubMed]
- Kusukawa, T.; Yoshizawa, M.; Fujita, M. Probing quest geometry and dynamics through host-quest interactions. Angew. Chem. Inter. Ed. 2001, 40, 1879–1884. [Google Scholar] [CrossRef]
- Shchegolikhina, O.I.; Pozdnyakova, Y.A.; Molodtsova, Y.A.; Korkin, S.D.; Bukalov, S.S.; Leites, L.A.; Lyssenko, K.A.; Peregudov, A.S.; Auner, N.; Katsoulis, D.E. Synthesis and properties of stereoregular cyclic polysilanols: Cis-[PhSi(O)OH]4, cis-[PhSi(O)OH]6 and tris -cis–tris-trans-[PhSi(O)OH]12. Inorg. Chem. 2002, 41, 6892–6904. [Google Scholar] [CrossRef]
- Pozdnyakova, Y.A.; Korlyukov, A.A.; Kononova, E.G.; Lyssenko, K.A.; Peregudov, A.S.; Shchegolikhina, O.I. Cyclotetrasiloxanetetrols with methyl groups at silicon: Isomers all-cis and cis-trans-cis-[MeSi(O)OH]4. Inorg. Chem. 2010, 49, 572–577. [Google Scholar]
- Molodtsova, Y.A.; Pozdnyakova, Y.A.; Blagodatskikh, I.V.; Peregudov, A.S.; Shchegolikhina, O.I. Hydrolytic condensation of trialkoxysilanes in the presence of alkali metal and copper(II) ions. Influence of the reaction conditions on the structure of Cu/M organosiloxanes. Russ. Chem. Bull. 2003, 52, 2722–2731. [Google Scholar] [CrossRef]
- Bilyachenko, A.N.; Sergeenko, N.V.; Korlyukov, A.A.; Antipin, M.Y.; Zavin, B.G.; Levitskii, M.M. New heterometallic organosiloxanes. Russ. Chem. Bull. 2006, 55, 943–945. [Google Scholar] [CrossRef]
- Bilyachenko, A.N.; Korlyukov, A.A.; Levitskii, M.M.; Antipin, M.Y.; Zavin, B.G. New cage-like metallasiloxane containing FeIII ions in different coordination spheres. Russ. Chem. Bull. 2007, 56, 543–545. [Google Scholar] [CrossRef]
- Bilyachenko, A.N.; Korlyukov, A.A.; Levitsky, M.M.; Zavin, B.G.; Antipin, M.Y.; Filippov, O.A.; Tsupreva, V.I. Dinuclear cage-like metalloorganosiloxane containing CrIII ions. Russ. Chem. Bull. 2008, 57, 2204. [Google Scholar] [CrossRef]
- Bilyachenko, A.N.; Dronova, M.S.; Korlyukov, A.A.; Levitskii, M.M.; Antipin, M.Y.; Zavin, B.G. Cage-like manganesephenylsiloxane with an unusual structure. Russ. Chem. Bull. 2011, 60, 1762–1765. [Google Scholar] [CrossRef]
- Sergienko, N.V.; Cherkun, N.V.; Korlyukov, A.A.; Sochikhin, A.A.; Myakushev, V.D.; Strelkova, T.V.; Zavin, B.G. Synthesis and study of the structure of cage-like metallosiloxanes with pair combinations of 3d transition and alkaline earth metals. Russ. Chem. Bull. 2013, 62, 1999–2006. [Google Scholar] [CrossRef]
- Petrova, I.M.; Buryak, A.K.; Peregudov, A.S.; Strelkova, T.V.; Kononova, E.G.; Bushmarinov, I.S.; Makarova, N.N. Effect of stereoisomerism of tetrakis(hydroxy)(phenyl)-cyclotetrasiloxane on the siloxane framework in polyphenylsilsesquioxanes obtained by polycondensation in the presence of layered-architecture compounds. Mendeleev Commun. 2015, 25, 229–231. [Google Scholar] [CrossRef]
- Makarova, N.N.; Petrova, I.M.; Petrovskii, P.V.; Kaznacheev, A.V.; Volkova, L.M.; Shcherbina, M.A.; Bessonova, N.P.; Chvalun, S.N.; Godovskii, Y.K. Synthesis of new stereoregular 2,4,6,8-tetraphenylcyclotetrasiloxanes with mesogenic groups and the influence of spatial isomerism on the phase state of individual isomers and their mixtures. Russ. Chem. Bull. 2004, 53, 1983–1995. [Google Scholar] [CrossRef]
- Makarova, N.N.; Lyakhovetsky, Y.I.; Petrova, I.M.; Dolgushin, F.M.; Ikonnikov, N.S.; Peregudov, A.S.; Strelkova, T.V.; Klemenkova, Z.S. A study of the polycondensation of (tetrahydroxy)(tetraaryl)cyclotetrasiloxanes under equilibrium and non-equilibrium conditions in the presence and absence of montmorillonite. Polymers 2018, 10, 422. [Google Scholar] [CrossRef] [Green Version]
- Gabelica, V.; De Pauw, E. Internal energy and fragmentation of ions produced in electrospray sources. Mass Spectrom. Rev. 2005, 24, 566–587. [Google Scholar] [CrossRef]
- Nekrasov, Y.S.; Ikonnikov, N.S.; Birisov, Y.A.; Kiselev, S.S.; Kornienko, A.G.; Velezheva, V.S.; Lyakhovetsky, Y.I. Reactivity of N-methylidenemalonates of 3-arylaminoindoles and p-dimethylamino-N-phenylaniline in the course of their analysis by electrospray ionization mass spectrometry. Int. J. Anal. Mass Spectrom. Chromatogr. 2017, 5, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Groom, C.R.; Allen, F.H. The Cambridge structural data base in retrospect and prospect. Angew. Chem. Int. Ed. 2014, 53, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Andrianov, K.A.; Makarova, N.N. Polymerization of hydrolysis products of organotrichlorosilanes. Vysokomol. Soed. (Russ.) 1970, 12A, 663–670. [Google Scholar] [CrossRef]
- Petrov, A.D.; Ponomarenko, V.A.; Sokolov, B.A.; Roshal’, V.U. Photochemical chlorination of phenyltrichlorosilane. Zhurnal Obsch. Chim. (Russ.) 1956, 26, 1229–1232. [Google Scholar]
- Andrianov, K.A. Investigations of silico-organic compounds. V. Synthesis of alkyl and arylhalogenomonosilanes. Zhurnal Obsch. Khim. (Russ.) 1946, 16, 487–492. [Google Scholar]
- Zhukov, S.G.; Chernyshev, V.V.; Babaev, E.V.; Sonneveld, E.J.; Schenk, H.Z. Application of simulated annealing approach for structure solution of molecular crystals from X-ray laboratory powder data. Z. Kristallogr. 2001, 216, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Zlokazov, V.B.; Chernyshev, V.V. MRIA-a program for a full profile analysis of powder multiphase neutron-diffraction time-of-flight (direct and Fourier) spectra. J. Appl. Cryst. 1992, 25, 447–451. [Google Scholar] [CrossRef] [Green Version]
- Logacheva, N.M.; Baulin, V.E.; Tsivadze, A.Y.; Pyatova, E.N.; Ivanova, I.S.; Velikodny, Y.A.; Chernyshev, V.V. Ni(II), Co(II), Cu(II), Zn(II) and Na(I) complexes of a hybrid ligand 4′-(4′′-benzo-15-crown-5)-methyloxy-2,2′,6′,2′-terpyridine. Dalton Trans. 2009, 2482–2489. [Google Scholar] [CrossRef]
- Veselovsky, V.V.; Lozanova, A.V.; Isaeva, V.I.; Lobova, A.A.; Fitch, A.N.; Chernyshev, V.V. Optically active derivatives of terephthalic acid: Four crystal structures from two powder patterns. Acta Crystallogr. 2018, C74, 248–255. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | C20H18O5Si2 |
|---|---|
| Mr | 394.52 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Wavelength, Å | 1.54059 |
| Unit cell dimensions | |
| a, Å | 13.5727(12) |
| b, Å | 8.4084(7) |
| c, Å | 8.2589(6) |
| β,o | 96.880(11) |
| Volume, Å3 | 935.76(13) |
| Z | 2 |
| Dx (Mg m−3) | 1.400 |
| μ, mm−1 | 1. 981 |
| 2θmin–2θmax, ∆2θ (o) | 3.00–90.00, 0.01 |
| No. parameters/restraints | 327/402 |
| Rp, Rwp, Rexp | 0.0331, 0.0420, 0.0193 |
| D−H…A | D−H | H…A | D…A | D−H…A |
|---|---|---|---|---|
| O2−H2…O3i | 0.82 | 1.89 | 2.674(5) | 160 |
| O3−H3…O2ii | 0.82 | 2.24 | 2.759(5) | 122 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrova, I.M.; Lyakhovetsky, Y.I.; Chernyshev, V.V.; Ikonnikov, N.S.; Makarova, N.N. A Study of the Influence of the HCl Concentration on the Composition and Structure of (Hydroxy)Arylsiloxanes from the Hydrolysis–Condensation Reaction of Aryltrichlorosilanes. Molecules 2019, 24, 4195. https://doi.org/10.3390/molecules24224195
Petrova IM, Lyakhovetsky YI, Chernyshev VV, Ikonnikov NS, Makarova NN. A Study of the Influence of the HCl Concentration on the Composition and Structure of (Hydroxy)Arylsiloxanes from the Hydrolysis–Condensation Reaction of Aryltrichlorosilanes. Molecules. 2019; 24(22):4195. https://doi.org/10.3390/molecules24224195
Chicago/Turabian StylePetrova, Irina M., Yury I. Lyakhovetsky, Vladimir V. Chernyshev, Nikolai S. Ikonnikov, and Nataliya N. Makarova. 2019. "A Study of the Influence of the HCl Concentration on the Composition and Structure of (Hydroxy)Arylsiloxanes from the Hydrolysis–Condensation Reaction of Aryltrichlorosilanes" Molecules 24, no. 22: 4195. https://doi.org/10.3390/molecules24224195
APA StylePetrova, I. M., Lyakhovetsky, Y. I., Chernyshev, V. V., Ikonnikov, N. S., & Makarova, N. N. (2019). A Study of the Influence of the HCl Concentration on the Composition and Structure of (Hydroxy)Arylsiloxanes from the Hydrolysis–Condensation Reaction of Aryltrichlorosilanes. Molecules, 24(22), 4195. https://doi.org/10.3390/molecules24224195