Nasal Drug Delivery of Anticancer Drugs for the Treatment of Glioblastoma: Preclinical and Clinical Trials

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
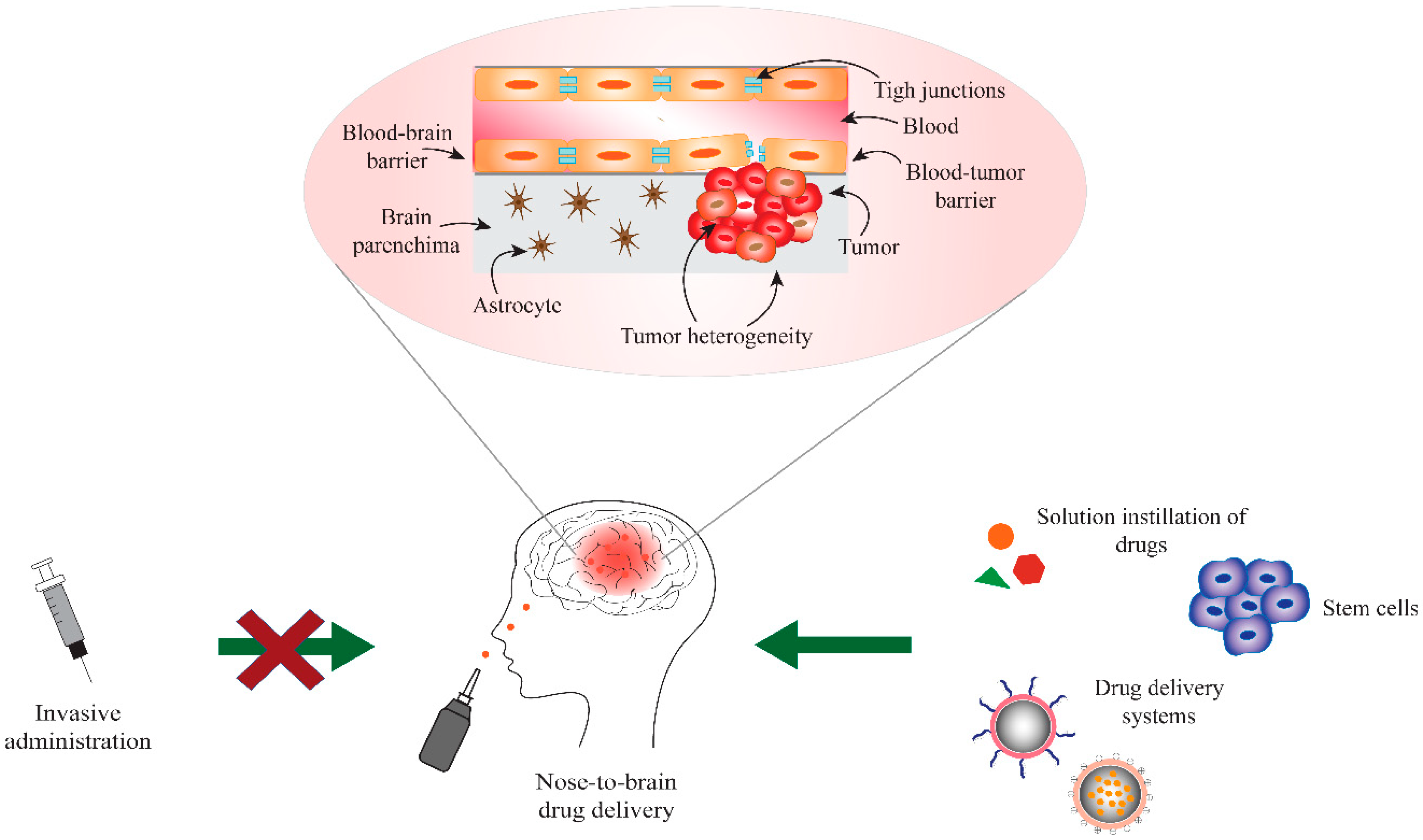
2. Blood-Brain and Blood-Tumor Barriers (BBB/BTB)
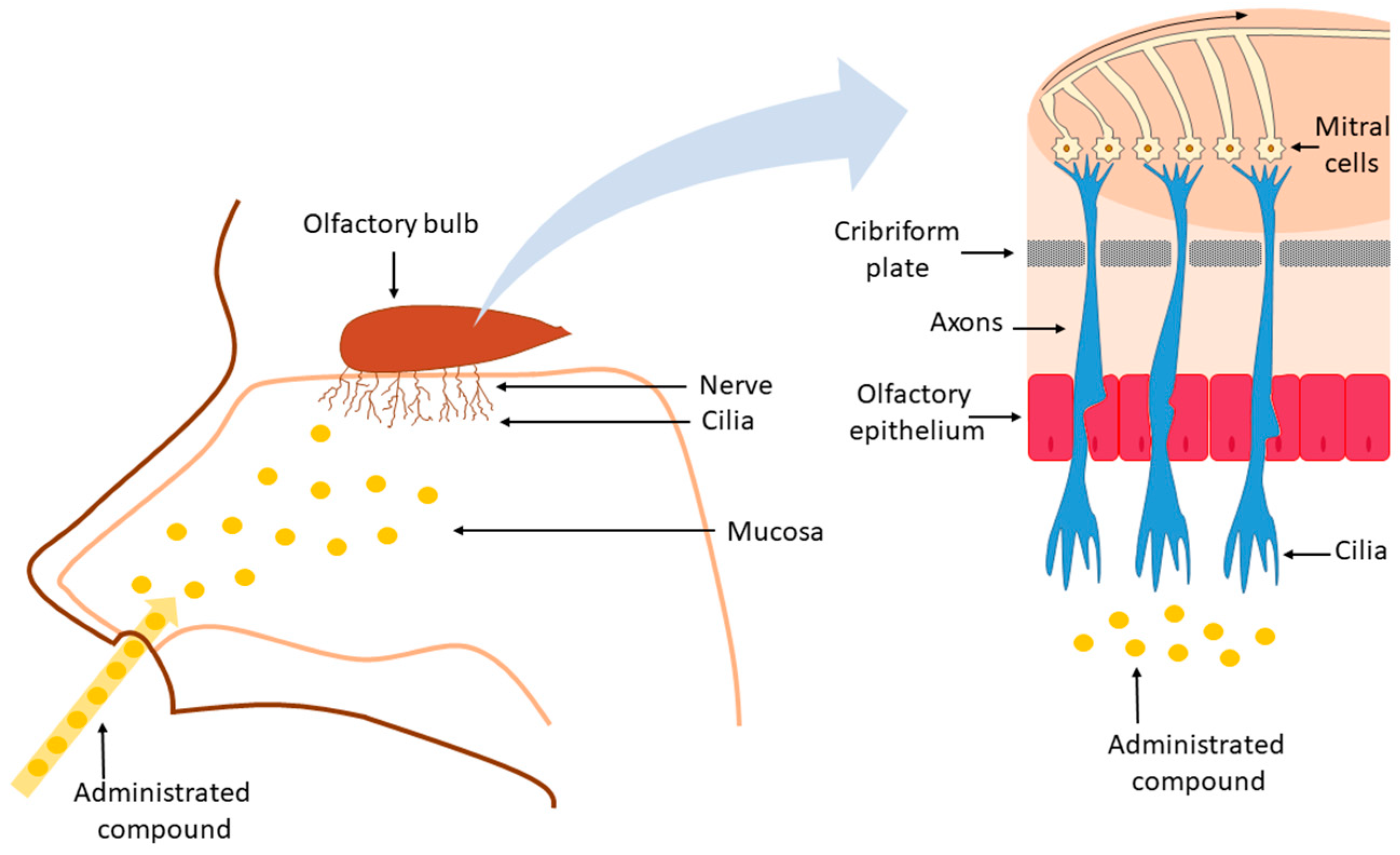
3. Nose-to-Brain Drug Delivery
4. Drugs for GBM Treatment Administered Intranasally
Clinical Trials on the Use of Intranasal Perillyl Alcohol for Glioblastoma Treatment
5. Drug Delivery Systems for Nose-to-Brain Delivery in Glioblastoma Therapy
5.1. Polymer-based NP
5.2. Lipid-Based NP
5.3. Polysaccharide NP
5.4. Targeted NP
6. Stem Cells for Treatment of GBM
7. Conclusions
Funding
Conflicts of Interest
References
- Leece, R.; Xu, J.; Ostrom, Q.T.; Chen, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. Global incidence of malignant brain and other central nervous system tumors by histology, 2003–2007. Neuro Oncol. 2017, 19, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Izycka-Swieszewska, E.; Bien, E.; Stefanowicz, J.; Szurowska, E.; Szutowicz-Zielinska, E.; Koczkowska, M.; Sigorski, D.; Kloc, W.; Rogowski, W.; Adamkiewicz-Drozynska, E. Malignant gliomas as second neoplasms in pediatric cancer survivors: Neuropathological study. BioMed Res. Int. 2018, 2018, 4596812. [Google Scholar] [CrossRef] [PubMed]
- Morsy, A.A.; Ng, W.H. Re-do craniotomy for recurrent glioblastoma. CNS Oncol. 2015, 4, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.A.; Karajannis, M.A.; Harter, D.H. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef]
- Ozdemir-Kaynak, E.; Qutub, A.A.; Yesil-Celiktas, O. Advances in glioblastoma multiforme treatment: New models for nanoparticle therapy. Front. Physiol. 2018, 19, 170. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Prev. Biomark. 2014, 23, 1985–1996. [Google Scholar] [CrossRef]
- Song, W.; Ruder, A.M.; Hu, L.; Li, Y.; Ni, R.; Shao, W.; Kaslow, R.A.; Butler, M.; Tang, J. Genetic epidemiology of glioblastoma multiforme: Confirmatory and new findings from analyses of human leukocyte antigen alleles and motifs. PLoS ONE 2009, 4, e7157. [Google Scholar] [CrossRef]
- Han, S.J.; Englot, D.J.; Birk, H.; Molinaro, A.M.; Chang, S.M.; Clarke, J.L.; Prados, M.D.; Taylor, J.W.; Berger, M.S.; Butowski, N.A. Incidence rates of glioblastoma increase, with rates highest in individuals age 75 to 84 years. Neurosurgery 2015, 62, 160–165. [Google Scholar] [CrossRef]
- Wang, H.; Cai, S.; Ernstberger, A.; Bailey, B.J.; Wang, M.Z.; Cai, W.; Goebel, W.S.; Czader, M.B.; Crean, C.; Suvannasankha, A.; et al. Temozolomide-mediated DNA methylation in human myeloid precursor cells: Differential involvement of intrinsic and extrinsic apoptotic pathways. Clin. Cancer Res. 2013, 19, 2699–2709. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, T.; Gao, L.; Zheng, J.; Shao, A.; Zhang, J. Efficacy and safety of long-term therapy for high-grade glioma with temozolomide: A meta-analysis. Oncotarget 2017, 8, 51758–51765. [Google Scholar] [CrossRef] [PubMed]
- Tiek, D.M.; Rone, J.D.; Graham, G.T.; Pannkuk, E.L.; Haddad, B.R.; Riggins, R.B. Alterations in cell motility, proliferation, and metabolism in novel models of acquired temozolomide resistant glioblastoma. Sci. Rep. 2018, 8, 7222. [Google Scholar] [CrossRef] [PubMed]
- Syro, L.V.; Rotondo, F.; Camargo, M.; Ortiz, L.D.; Serna, C.A.; Kovacs, K. Temozolomide and pituitary tumors: Current understanding, unresolved issues, and future directions. Front. Endocrinol. 2018, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Suki, D.; Hess, K.; Sawaya, R. The influence of maximum safe resection of glioblastoma on survival in 1229 patients: Can we do better than gross-total resection? J. Neurosurg. 2016, 124, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Pirzkall, A.; McGue, C.; Saraswathy, S.; Cha, S.; Liu, R.; Vandenberg, S.; Lamborn, K.R.; Berger, M.S.; Chang, S.M.; Nelson, S.J. Tumor regrowth between surgery and initiation of adjuvant therapy in patients with newly diagnosed glioblastoma. Neuro Oncol. 2009, 11, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of disease and treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in blood-brain barrier penetrance and drug repurposing for improved treatment of glioblastoma. Front. Oncol. 2018, 8, 462. [Google Scholar] [CrossRef]
- Paolillo, M.; Boselli, C.; Schinelli, S. Glioblastoma under siege: An overview of current therapeutic strategies. Brain Sci. 2018, 8, 15. [Google Scholar] [CrossRef]
- Guo, J.; Schlich, M.; Cryan, J.F.; O’Driscoll, C.M. Targeted drug delivery via folate receptors for the treatment of brain cancer: Can the promise deliver? J. Pharm. Sci. 2017, 106, 3413–3420. [Google Scholar] [CrossRef]
- Greene, C.; Campbell, M. Tight junction modulation of the blood brain barrier: CNS delivery of small molecules. Tissue Barriers 2016, 4, e1138017. [Google Scholar] [CrossRef] [PubMed]
- Burgo, L.S.; Hernández, R.M.; Orive, G.; Pedraz, J.L. Nanotherapeutic approaches for brain cancer management. Nanomedicine 2014, 10, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Illum, L. Transport of drugs from the nasal cavity to the central nervous system. Eur. J. Pharm. Sci. 2000, 11, 1–18. [Google Scholar] [CrossRef]
- Mc Carthy, D.J.; Malhotra, M.; O’Mahony, A.M.; Cryan, J.F.; O’Driscoll, C.M. Nanoparticles and the blood-brain barrier: Advancing from in-vitro models towards therapeutic significance. Pharm. Res. 2015, 32, 1161–1185. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Warren, K.E. Beyond the blood:brain barrier: The importance of central nervous system (CNS) pharmacokinetics for the treatment of CNS tumors, including diffuse intrinsic pontine glioma. Front. Oncol. 2018, 8, 239. [Google Scholar] [CrossRef]
- Van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Updates 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Li, M.; Deng, H.; Peng, H.; Wang, Q. Functional nanoparticles in targeting glioma diagnosis and therapies. J. Nanosci. Nanotechnol. 2014, 14, 415–432. [Google Scholar] [CrossRef]
- Chacko, A.M.; Li, C.; Pryma, D.A.; Brem, S.; Coukos, G.; Muzykantov, V. Targeted delivery of antibody-based therapeutic and imaging agents to CNS tumors: Crossing the blood-brain barrier divide. Expert Opin. Drug Deliv. 2013, 10, 907–926. [Google Scholar] [CrossRef]
- Meng, J.; Agrahari, V.; Youm, I. Advances in targeted drug delivery approaches for the central nervous system tumors: The inspiration of nanobiotechnology. J. Neuroimmune Pharmacol. 2017, 12, 84–98. [Google Scholar] [CrossRef]
- Ganipineni, L.P.; Danhier, F.; Préat, V. Drug delivery challenges and future of chemotherapeutic nanomedicine for glioblastoma treatment. J. Control. Release 2018, 281, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Luo, Z.; Xia, Z.; Shen, X.; Cai, K. Time-sequenced drug delivery approaches towards effective chemotherapeutic treatment of glioma. Mater. Horiz. 2017, 4, 977. [Google Scholar] [CrossRef]
- Lyday, R.W.; Etters, A.M.; Kim, C.; Magana, F.; Pontipiedra, G.M.; Singh, N.K.M.; Kadavakollu, S. PDE5 inhibitors offer novel mechanisms in combination and solo cancer therapy. Curr. Cancer Ther. Rev. 2017, 13, 107–119. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, W. Recent advances in brain tumor-targeted nano-drug delivery systems. Expert Opin. Drug Deliv. 2012, 9, 671–686. [Google Scholar] [CrossRef]
- Larjavaara, S.; Mäntylä, R.; Salminen, T.; Haapasalo, H.; Raitanen, J.; Jääskeläinen, J.; Auvinen, A. Incidence of gliomas by anatomic location. Neuro Oncol. 2007, 9, 319–325. [Google Scholar] [CrossRef]
- Hadziahmetovic, M.; Shirai, K.; Chakravarti, A. Recent advancements in multimodality treatment of gliomas. Future Oncol. 2011, 7, 1169–1183. [Google Scholar] [CrossRef] [Green Version]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus procarbazine, CCNU, and vincristine in low-grade glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef]
- Cairncross, G.; Wang, M.; Shaw, E.; Jenkins, R.; Brachman, D.; Buckner, J.; Fink, K.; Souhami, L.; Laperriere, N.; Curran, W.; et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: Long-term results of RTOG 9402. J. Clin. Oncol. 2013, 31, 337–343. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Hopkins, K.; Tonn, J.C.; Stupp, R.; Falini, A.; Cohen-Jonathan-Moyal, E.; Frappaz, D.; Henriksson, R.; Balana, C.; et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014, 15, e395–e403. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.C.; Pentheroudakis, G.; ESMO Guidelines Working Group. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. 3), 93–101. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, T.S.; Shade, M.Y.; Breton, G.; Gilbert, M.R.; Mahajan, A.; Scheurer, M.E.; Vera, E.; Berger, A.M. Sleep-wake disturbance in patients with brain tumors. Neuro Oncol. 2017, 19, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Taphoorn, M.J.; Heimans, J.J.; van der Veen, E.A.; Karim, A.B. Endocrine functions in long-term survivors of low-grade supratentorial glioma treated with radiation therapy. J. Neurooncol. 1995, 2, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Gregor, A.; Cull, A.; Traynor, E.; Stewart, M.; Lander, F.; Love, S. Neuropsychometric evaluation of long-term survivors of adult brain tumours: Relationship with tumour and treatment parameters. Radiother. Oncol. 1996, 41, 55–59. [Google Scholar] [CrossRef]
- Surma-aho, O.; Niemela, M.; Vilkki, J.; Kouri, M.; Brander, A.; Salonen, O.; Paetau, A.; Kallio, M.; Pyykkönen, J.; Jääskeläinen, J. Adverse long-term effects of brain radiotherapy in adult low-grade glioma patients. Neurology 2001, 56, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef] [Green Version]
- Grossman, S.A.; Trump, D.L.; Chen, D.C.; Thompson, G.; Camargo, E.E. Cerebrospinal fluid flow abnormalities in patients with neoplastic meningi tis. An evaluation using 111indium-DTPA ventriculography. Am. J. Med. 1982, 73, 641–647. [Google Scholar] [CrossRef]
- Blakeley, J. Drug delivery to brain tumors. Curr. Neurol. Neurosci. Rep. 2008, 8, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef]
- Chapman, C.D.; Frey, W.H.; Craft, S.; Danielyan, L.; Hallschmid, M.; Schioth, H.B.; Benedict, C. Intranasal treatment of central nervous system dysfunction in humans. Pharm. Res. 2013, 30, 2475–2484. [Google Scholar] [CrossRef]
- Chauhan, M.B.; Chauhan, N.B. Brain Uptake of Neurotherapeutics after Intranasal versus Intraperitoneal Delivery in Mice. J. Neurol. Neurosurg. 2015, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H., II. Intranasal Delivery to the Central Nervous System: Mechanisms and Experimental Considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef] [PubMed]
- Lledo, P.M.; Gheusi, G.; Vincent, J.D. Information processing in the mammalian olfactory system. Physiol. Rev. 2005, 85, 281–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, R.; Mygind, N. Anatomy, physiology and function of the nasal cavities in health and disease. Adv. Drug Deliv. Rev. 1998, 29, 3–12. [Google Scholar]
- Geurkink, N. Nasal anatomy, physiology and function. J. Allergy Clin. Immunol. 1983, 72, 123–128. [Google Scholar] [CrossRef]
- Watelet, J.B.; Van Cauwenberge, P. Applied anatomy and physiology of the nose and paranasal sinuses. Allergy 1999, 57, 14–25. [Google Scholar] [CrossRef]
- Jafek, B.W. Ultrastructure of human nasal mucosa. Laryngoscope 1983, 93, 1576–1599. [Google Scholar] [CrossRef]
- Junqueira, L.C.; Carneiro, J. Biologia Celular e Molecular, 9th ed.; Guanabara Koogan: Rio de Janeiro, Brazil, 2012; pp. 337–338. [Google Scholar]
- Leopold, D.A. The relationship between nasal anatomy and human olfaction. Laryngoscope 1988, 98, 1232–1238. [Google Scholar] [CrossRef]
- Gizurarson, S. Anatomical and histological factors affecting intranasal drug and vaccine delivery. Curr. Drug Deliv. 2012, 9, 566–582. [Google Scholar] [CrossRef] [Green Version]
- Mistry, A.; Stolnik, S.; Illum, L. Nanoparticles for direct nose-to-brain delivery of drugs. Int. J. Pharm. 2009, 379, 146–157. [Google Scholar] [CrossRef]
- Caggiano, M.; Kauer, J.S.; Hunter, D.D. Globose basal cells are neuronal progenitors in the olfactory epithelium: A lineage analysis using a replication-incompetent retrovirus. Neuron 1994, 13, 339–352. [Google Scholar] [CrossRef]
- Arora, P.; Sharma, S.; Garg, S. Permeability issues in nasal drug delivery. Drug Discov. Today 2002, 7, 967–975. [Google Scholar] [CrossRef]
- Pires, A.; Fortuna, A.; Alves, G.; Falcão, A. Intranasal Drug Delivery: How, Why and What for? J. Pharm. Sci. 2009, 12, 288–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, T.K.; Babbar, A.K.; Sharma, R.K.; Singh, S.; Misra, A. Intranasal mucoadhesive microemulsions of clonazepam: Preliminary studies on brain targeting. J. Pharm. Sci. 2006, 95, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Martinez, J.A.; Hanson, L.R.; Frey, W.H., II; Toth, C.C. Intranasal treatment of neurodegenerative diseases and stroke. Front. Biosci. 2012, 4, 74–89. [Google Scholar] [CrossRef]
- Einer-Jensen, N.; Hunter, R. Counter-current transfer in reproductive biology. Reproduction 2005, 129, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Charlton, S.; Jones, N.S.; Davis, S.S.; Illum, L. Distribution and clearance of bioadhesive formulations from the olfactory region in man: Effect of polymer type and nasal delivery device. Eur. J. Pharm. Sci. 2007, 30, 295–302. [Google Scholar] [CrossRef]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef]
- Illum, L. Is nose-to-brain transport of drugs in man a reality? J. Pharm. Pharmacol. 2004, 56, 3–17. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Jansson, B.; Björk, E. Visualization of In Vivo Olfactory Uptake and Transfer Using Fluorescein Dextran. J. Drug Target. 2002, 10, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.L.; Böttger, B.; Silver, W.L.; Finger, T.E. Trigeminal collaterals in the nasal epithelium and olfactory bulb: A potential route for direct modulation of olfactory information by trigeminal stimuli. J. Comp. Neurol. 2002, 444, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Wolak, D.J.; Pizzo, M.E.; Thorne, R.G. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J. Cereb. Blood Flow Metab. 2015, 35, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Groothuis, D.R.; Vavra, M.W.; Schlageter, K.E.; Kang, E.W.; Itskovich, A.C.; Hertzler, S.; Allen, C.V.; Lipton, H.L. Efflux of drugs and solutes from brain: The interactive roles of diffusional transcapillary transport, bulk flow and capillary transportes. J. Cereb. Blood Flow Metab. 2007, 27, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Grevers, G.; Herrmann, U. Fenestrated endothelia in vessels of the nasal mucosa. An electron-microscopic study in the rabbit. Arch. Otorhinolaryngol. 1987, 244, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.H. Enzymatic barriers to peptide and protein absorption. Crit. Rev. Ther. Drug Carr. Syst. 1988, 5, 69–97. [Google Scholar] [CrossRef]
- Quintana, D.S.; Westlye, L.T.; Rustan, Ø.G.; Tesli, N.; Poppy, C.L.; Smevik, H.; Tesli, M.; Røine, M.; Mahmoud, R.A.; Smerud, K.T.; et al. Low-dose oxytocin delivered intranasally with Breath Powered device affects social-cognitive behavior: A randomized four-way crossover trial with nasal cavity dimension assessment. Transl. Psychiatry 2015, 5, e602. [Google Scholar] [CrossRef]
- Wang, X.; Deng, J.; Yuan, J.; Tang, X.; Wang, Y.; Chen, H.; Liu, Y.; Zhou, L. Curcumin exerts its tumor suppressive function via inhibition of NEDD4 oncoprotein in glioma cancer cells. Int. J. Oncol. 2017, 51, 467–477. [Google Scholar] [CrossRef]
- Nasr, M. Development of an optimized hyaluronic acid-based lipidic nanoemulsion co-encapsulating two polyphenols for nose to brain delivery. Drug Deliv. 2016, 23, 1444–1452. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Baidoo, J.; Fried, A.; Atwi, D.; Dolai, S.; Boockvar, J.; Symons, M.; Ruggieri, R.; Raja, K.; Banerjee, P. Curcumin changes the polarity of tumor-associated microglia and eliminates glioblastoma. Int. J. Cancer 2016, 139, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.X.; Xia, W.; Yue, W.; Peng, C.; Rahman, K.; Zhang, H. Rhein: A Review of Pharmacological Activities. Evid. Based Complement. Altern. Med. 2015, 2015, 578107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Luo, G.; Chen, D.; Xiang, Z.A. Comprehensive and System Review for the Pharmacological Mechanism of Action of Rhein, an Active Anthraquinone Ingredient. Front. Pharmacol. 2016, 7, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, X.D.; Ding, M.J.; Dai, D.Z.; Wu, Y.; Zhang, Y.; Dai, Y. ER stress, P66shc, and P-Akt/Akt mediate adjuvant-induced inflammation, which is blunted by argirein, a supermolecule and rhein in rats. Inflammation 2012, 35, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Blacher, E.; Baruch, B.B.; Levy, A.; Geva, N.; Green, K.D.; Garneau-Tsodikova, S.; Fridman, M.; Stein, R. Inhibition of glioma progression by a newly discovered CD38 inhibitor. Int. J. Cancer 2015, 136, 1422–1433. [Google Scholar] [CrossRef] [PubMed]
- Shingaki, T.; Hidalgo, I.J.; Furubayashi, T.; Katsumi, H.; Sakane, T.; Yamamoto, A.; Yamashita, S. The transnasal delivery of 5-fluorouracil to the rat brain is enhanced by acetazolamide (the inhibitor of the secretion of cerebrospinal fluid). Int. J. Pharm. 2009, 377, 85–91. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.P. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur. J. Cancer 2004, 40, 939–950. [Google Scholar] [CrossRef]
- Diasio, R.B.; Harris, B.E. Clinical pharmacology of 5-fluorouracil. Clin. Pharmacokinet. 1989, 16, 215–237. [Google Scholar] [CrossRef]
- Silva, P. Farmacologia, 8th ed.; Guanabara Koogan: Rio de Janeiro, Brazil, 2010; pp. 1064–1071. [Google Scholar]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Flurouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Uldall, M.; Botfield, H.; Jansen-Olesen, I.; Sinclair, A.; Jensen, R. Acetazolamide lowers intracranial pressure and modulates the cerebrospinal fluid secretion pathway in healthy rats. Neurosci. Lett. 2017, 645, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Shingaki, T.; Inoue, D.; Furubayashi, T.; Sakane, T.; Katsumi, H.; Yamamoto, A.; Yamashita, S. Transnasal Delivery of Methotrexate to Brain Tumors in Rats: A New Strategy for Brain Tumor Chemotherapy. Mol. Pharm. 2010, 7, 1561–1568. [Google Scholar] [CrossRef]
- Hagner, N.; Joerger, M. Cancer chemotherapy: Targeting folic acid synthesis. Cancer Manag. Res. 2010, 2, 293–301. [Google Scholar] [PubMed]
- Abolmaali, S.S.; Tamaddon, A.M.; Dinarvand, R. A review of therapeutic challenges and achievements of methotrexate delivery systems for treatment of cancer and rheumatoid arthritis. Cancer Chemother. Pharmacol. 2013, 71, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shi, K.; Wan, F.; Cui, F. Methotrexate-loaded microspheres for nose to brain delivery: In vitro/in vivo evaluation. J. Drug Deliv. Sci. Technol. 2012, 22, 167–174. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Liu, G.; Zhou, X.; Wang, Y.; Wang, Y.; Ma, L. Intranasal administration of temozolomide for brain-targeting delivery: Therapeutic effect on glioma in rats. Nan Fang Yi Ke Da Xue Xue Bao 2014, 34, 631–635. [Google Scholar]
- Baker, S.D.; Wirth, M.; Statkevich, P. Absorption, metabolism and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin. Cancer Res. 1999, 5, 309–317. [Google Scholar]
- Mrugala, M.M.; Chamberlain, M.C. Mechanisms of Disease: Temozolomide and glioblastoma-look to the future. Nat. Clin. Pract. Oncol. 2008, 5, 476–486. [Google Scholar] [CrossRef]
- Pineda, J.R.; Jeitany, M.; Andrieux, A.; Junier, M.P.; Chneiweiss, H.; Boussin, F.D. Intranasal Administration of Temozolomide Delayed the Development of Brain Tumors Initiated by Human Glioma Stem-Like Cell in Nude Mice. Cancer Sci. Ther. 2017, 9, 374–378. [Google Scholar]
- Chen, T.C.; Fonseca, C.O.; Schönthal, A.H. Preclinical development and clinical use of perillyl alcohol for chemoprevention and cancer therapy. Am. J. Cancer Res. 2015, 5, 1580–1593. [Google Scholar]
- da Fonseca, C.O.; Khandelia, H.; Salazar, M.D.A.; Schönthal, A.H.; Meireles, O.C.; Quirico-Santos, T. Perillyl alcohol: Dynamic interactions with the lipid bilayer and implications for long-term inhalational chemotherapy for gliomas. Surg. Neurol. Int. 2016, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.C.; da Fonseca, C.O.; Schönthal, A.H. Intranasal Perillyl Alcohol for Glioma Therapy: Molecular Mechanisms and Clinical Development. Int. J. Mol. Sci. 2018, 19, 3905. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02704858 (accessed on 22 September 2019).
- da Fonseca, C.O.; Simão, M.; Lins, I.R.; Caetano, R.O.; Futuro, D.; Quirico-Santos, T. Efficacy of monoterpene perillyl alcohol upon survival rate of patients with recurrent glioblastoma. J. Cancer. Res. Clin. Oncol. 2011, 137, 287–293. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, C.O.; Teixeira, R.M.; Silva, J.C.; De Saldanha, D.A.; Gama Fischer, J.; Meirelles, O.C.; Landeiro, J.A.; Quirico-Santos, T. Long-term outcome in patients with recurrent malignant glioma treated with Perillyl alcohol inhalation. Anticancer Res. 2013, 33, 5625–5631. [Google Scholar] [PubMed]
- da Fonseca, C.O.; Schwartsmann, G.; Fischer, J.; Nagel, J.; Futuro, D.; Quirico-Santos, T.; Gattass, C.R. Preliminary results from a phase I/II study of perillyl alcohol intranasal administration in adults with recurrent malignant gliomas. Surg. Neurol. 2008, 70, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.G.; Da Cruz, W.M.S.; Schönthal, A.H.; Salazar, M.D.; Fontes, C.A.P.; Quirico-Santos, T.; Da Fonseca, C.O. Efficacy of a ketogenic diet with concomitant intranasal perillyl alcohol as a novel strategy for the therapy of recurrent glioblastoma. Oncol. Lett. 2018, 15, 1263–1270. [Google Scholar] [CrossRef]
- Bernardi, A.; Braganhol, E.; Jäger, E.; Figueiró, F.; Edelweiss, M.I.; Pohlmann, A.R.; Guterres, S.S.; Battastini, A.M. Indomethacin-loaded nanocapsules treatment reduces in vivo glioblastoma growth in a rat glioma model. Cancer Lett. 2009, 281, 53–63. [Google Scholar] [CrossRef]
- Saludas, L.; Pascual-Gil, S.; Roli, F.; Garbayo, E.; Blanco-Prieto, M.J. Heart tissue repair and cardioprotection using drug delivery systems. Maturitas 2018, 110, 1–9. [Google Scholar] [CrossRef]
- Pohlmann, A.R.; Fonseca, F.N.; Paese, K.; Detoni, C.B.; Coradini, K.; Beck, R.C.; Guterres, S.S. Poly(e-caprolactone) microcapsules and nanocapsules in drug delivery. Expert Opin. Drug Deliv. 2013, 10, 623–638. [Google Scholar] [CrossRef]
- Frank, L.A.; Contri, R.V.; Beck, R.C.; Pohlmann, A.R.; Guterres, S.S. Improving drug biological effects by encapsulation into polymeric nanocapsules. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 623–639. [Google Scholar] [CrossRef]
- Comfort, C.; Garrastazu, G.; Pozzoli, M.; Sonvico, F. Opportunities and challenges for the nasal administration of nanoemulsions. Curr. Top. Med. Chem. 2015, 15, 356–368. [Google Scholar] [CrossRef]
- Graff, C.L.; Pollack, G.M. Functional evidence for P-glycoprotein at the nose-brain barrier. Pharm. Res. 2005, 22, 86–93. [Google Scholar] [CrossRef]
- Pires, P.C.; Santos, A.O. Nanosystems in nose-to-brain drug delivery: A review of non-clinical brain targeting studies. J. Control. Release 2018, 270, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Sonvico, F.; Clementino, A.; Buttini, F.; Colombo, G.; Pescina, S.; Guterres, S.S.; Pohlmann, A.R.; Nicoli, S. Surface-Modified Nanocarriers for Nose-to-Brain Delivery: From Bioadhesion to Targeting. Pharmaceutics 2018, 10, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, F.; Dhaliwal, H.K.; Gattacceca, F.; Sarmento, B.; Amiji, M.M. Enhanced anti-angiogenic effects of bevacizumab in glioblastoma treatment upon intranasal administration in polymeric nanoparticles. J. Control. Release 2019, 309, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Azambuja, J.; Schuh, R.S.; Michels, L.R.; Gelsleichter, N.E.; Beckenkamp, L.R.; Iser, I.C.; Lenz, G.S.; de Oliveira, F.H.; Venturin, G.; Greggio, S.; et al. Nasal Administration of Cationic Nanoemulsions as CD73-siRNA Delivery System for Glioblastoma Treatment: A New Therapeutical Approach. Mol. Neurobiol. 2019, 56, 1–15. [Google Scholar] [CrossRef]
- Gadhavea, D.; Gorain, B.; Tagalpallewar, A.; Kokare, C. Intranasal teriflunomide microemulsion: An improved chemotherapeutic approach in glioblastoma. J. Drug Deliv. Sci. Technol. 2019, 51, 276–289. [Google Scholar] [CrossRef]
- de Oliveira Junior, E.R.; Nascimento, T.L.; Salomão, M.A.; da Silva, A.C.G.; Valadares, M.C.; Lima, E.M. Increased Nose-to-Brain Delivery of Melatonin Mediated by Polycaprolactone Nanoparticles for the Treatment of Glioblastoma. Pharm. Res. 2019, 36, 131. [Google Scholar] [CrossRef]
- Chu, L.; Aiping, W.; Ni, L.; Yan, X.; Song, Y.; Zhao, M.; Sun, K.; Mu, H.; Liu, S.; Wu, Z.; et al. Nose-to-brain delivery of temozolomide-loaded PLGA nanoparticles functionalized with anti-EPHA3 for glioblastoma targeting. Drug Deliv. 2018, 25, 1634–1641. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Figueiró, F.; de Fraga Dias, A.; Teixeira, H.F.; Battastini, A.M.O.; Koester, L.S. Kaempferol-loaded mucoadhesive nanoemulsion for intranasal administration reduces glioma growth in vitro. Int. J. Pharm. 2018, 543, 214–223. [Google Scholar] [CrossRef]
- Khan, A.; Aqil, M.; Imam, S.S.; Ahad, A.; Sultana, Y.; Ali, A.; Khan, K. Temozolomide loaded nano lipid based chitosan hydrogel for nose to brain delivery: Characterization, nasal absorption, histopathology and cell line study. Int. J. Biol. Macromol. 2018, 116, 1260–1267. [Google Scholar] [CrossRef]
- Sekerdag, E.; Lüle, S.; Bozdağ Pehlivan, S.; Öztürk, N.; Kara, A.; Kaffashi, A.; Vural, I.; Işıkay, I.; Yavuz, B.; Oguz, K.K.; et al. A potential non-invasive glioblastoma treatment: Nose-to-brain delivery of farnesylthiosalicylic acid incorporated hybrid nanoparticles. J. Control. Release 2017, 261, 187–198. [Google Scholar] [CrossRef]
- Shinde, R.L.; Devarajan, P.V. Docosahexaenoic acid-mediated, targeted and sustained brain delivery of curcumin microemulsion. Drug Deliv. 2017, 24, 152–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madane, R.G.; Mahajan, H.S. Curcumin-loaded nanostructured lipid carriers (NLCs) for nasal administration: Design, characterization, and in vivo study. Drug Deliv. 2016, 23, 1326–1334. [Google Scholar] [PubMed]
- Van Woensel, M.; Wauthoz, N.; Rosière, R.; Mathieu, V.; Kiss, R.; Lefranc, F.; Steelant, B.; Dilissen, E.; Van Gool, S.W.; Mathivet, T.; et al. Development of siRNA-loaded chitosan nanoparticles targeting Galectin-1 for the treatment of glioblastoma multiforme via intranasal administration. J. Control. Release 2016, 227, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Van Woensel, M.; Mathivet, T.; Wauthoz, N.; Rosière, R.; Garg, A.D.; Agostinis, P.; Mathieu, V.; Kiss, R.; Lefranc, F.; Boon, L.; et al. Sensitization of glioblastoma tumor micro-environment to chemo- and immunotherapy by Galectin-1 intranasal knock-down strategy. Sci. Rep. 2017, 7, 1217. [Google Scholar] [CrossRef]
- Jain, D.S.; Bajaj, A.N.; Athawale, R.B.; Shikhande, S.S.; Pandey, A.; Goel, P.N.; Gude, R.P.; Patil, S.; Raut, P. Thermosensitive PLA based nanodispersion for targeting brain tumor via intranasal route. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 63, 411–421. [Google Scholar] [CrossRef]
- Alex, A.T.; Joseph, A.; Shavi, G.; Rao, J.V.; Udupa, N. Development and evaluation of carboplatin-loaded PCL nanoparticles for intranasal delivery. Drug Deliv. 2016, 23, 2144–2153. [Google Scholar] [CrossRef]
- Mangraviti, A.; Tzeng, S.Y.; Gullotti, D.; Kozielski, K.L.; Kim, J.E.; Seng, M.; Abbadi, S.; Schiapparelli, P.; Sarabia-Estrada, R.; Vescovi, A.; et al. Non-virally engineered human adipose mesenchymal stem cells produce BMP4, target brain tumors, and extend survival. Biomaterials 2016, 100, 53–66. [Google Scholar] [CrossRef] [Green Version]
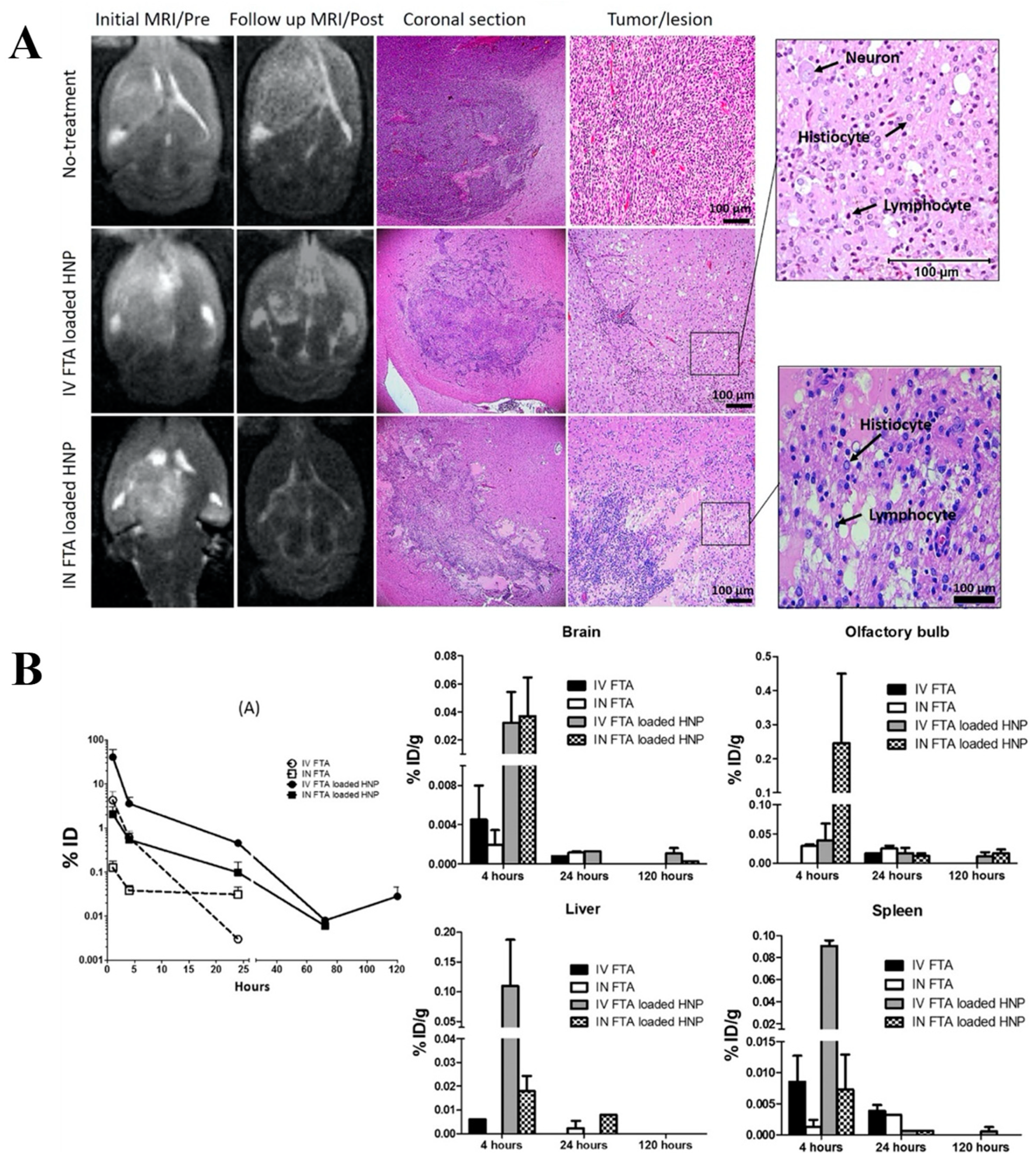
- Taki, H.; Kanazawa, T.; Akiyama, F.; Takashima, Y.; Okada, H. Intranasal Delivery of Camptothecin-Loaded Tat-Modified Nanomicells for Treatment of Intracranial Brain Tumors. Pharmaceuticals 2012, 5, 1092–1102. [Google Scholar] [CrossRef]
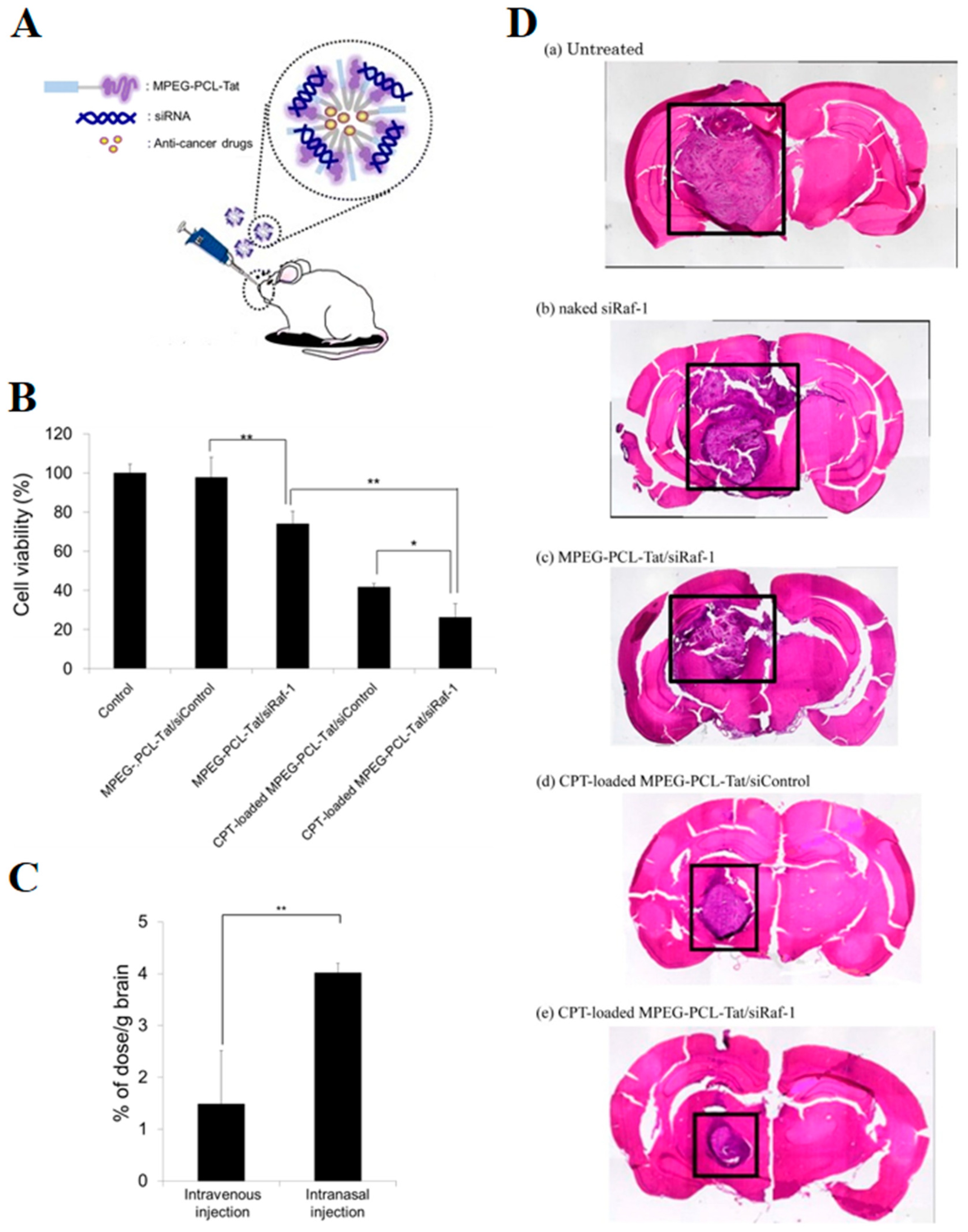
- Kanazawa, T.; Morisaki, K.; Suzuki, S.; Takashima, Y. Prolongation of life in rats with malignant glioma by intranasal siRNA/drug codelivery to the brain with cell-penetrating peptide-modified micelles. Mol. Pharm. 2014, 11, 1471–1478. [Google Scholar] [CrossRef]
- Zawilska, J.; Skene, D.; Arendt, J. Physiology and pharmacology of melatonin in relation to biological rhythms. Pharmacol. Rep. 2009, 61, 383–410. [Google Scholar] [CrossRef]
- Kostoglou-Athanassiou, I. Therapeutic applications of melatonin. Ther. Adv. Endocrinol. Metab. 2013, 4, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Menéndez, J.; Martínez-Campa, C. Melatonin: An Anti-Tumor Agent in Hormone-Dependent Cancers. Int. J. Endocrinol. 2018, 2018, 3271948. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.K.; Srivastava, A.K.; Dev, A.; Kaundal, B.; Choudhury, S.R.; Karmakar, S. Nanomelatonin triggers superior anticancer functionality in a human malignant glioblastoma cell line. Nanotechnology 2017, 28, 365102. [Google Scholar] [CrossRef] [PubMed]
- Hamelers, I.H.; Van Loenen, E.; Staffhorst, R.W.; De Kruijff, B.; de Kroon, A.I. Carboplatin nanocapsules: A highly cytotoxic, phospholipid-based formulation of carboplatin. Mol. Cancer Ther. 2006, 5, 2007–2012. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Chen, W.W.; Zhang, X. Glioblastoma multiforme: Effect of hypoxia and hypoxia inducible factors on therapeutic approaches. Oncol. Lett. 2016, 12, 2283–2288. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Weller, M.; Van den Bent, M.; Stupp, R. Bevacizumab and recurrent malignant gliomas: A European perspective. J. Clin. Oncol. 2010, 28, e188–e189. [Google Scholar] [CrossRef] [Green Version]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Sousa, F.; Cruz, A.; Fonte, P.; Pinto, I.M.; Neves-Petersen, M.T.; Sarmento, B. A new paradigm for Antiangiogenic therapy through controlled release of bevacizumab from PLGA nanoparticles. Sci. Rep. 2017, 7, 3736. [Google Scholar] [CrossRef]
- Rotblat, B.; Ehrlich, M.; Haklai, R.; Kloog, Y. The Ras inhibitor farnesylthiosalicylic acid (Salirasib) disrupts the spatiotemporal localization of active Ras: A potential treatment for cancer. Methods Enzymol. 2008, 439, 467–489. [Google Scholar]
- Goldberg, L.; Haklai, R.; Bauer, V.; Heiss, A.; Kloog, Y. New derivatives of farnesylthiosalicylic acid (salirasib) for cancer treatment: Farnesylthiosalicylamide inhibits tumor growth in nude mice models. J. Med. Chem. 2009, 52, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Ugwoke, M.I.; Verbeke, N.; Kinget, R. The biopharmaceutical aspects of nasal mucoadhesive drug delivery. J. Pharm. Pharmacol. 2001, 53, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Garg, T.; Rath, G.; Goyal, A.K. In situ nasal gel drug delivery: A novel approach for brain targeting through the mucosal membrane. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhai, Y.; Heng, X.; Che, F.Y.; Chen, W.; Sun, D.; Zhai, G. Oral bioavailability of curcumin: Problems and advancements. J. Drug Target. 2016, 24, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Chougule, M.B.; Shoyele, S.A.; Alexander, A. Nose-to-brain drug delivery: An update on clinical challenges and progress towards approval of anti-Alzheimer drugs. J. Control. Release 2018, 281, 139–177. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Pachner, A.; Menguy-Vacheron, F.; Kaplan, J.; Wiendl, H. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs 2014, 74, 659–674. [Google Scholar] [CrossRef] [Green Version]
- Xuan, J.; Ren, Z.; Qing, T.; Couch, L.; Shi, L.; Tolleson, W.H.; Guo, L. Mitochondrial dysfunction induced by leflunomide and its active metabolite. Toxicology 2018, 396–397, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Huang, O.; Zhang, W.; Zhi, Q.; Xue, X.; Liu, H.; Shen, D.; Geng, M.; Xie, Z.; Jiang, M. Teriflunomide, an immunomodulatory drug, exerts anticancer activity in triple negative breast cancer cells. Exp. Biol. Med. 2015, 240, 426–437. [Google Scholar] [CrossRef]
- Hail, N., Jr.; Chen, P.; Bushman, L.R. Teriflunomide (Leflunomide) Promotes Cytostatic, Antioxidant, and Apoptotic Effects in Transformed Prostate Epithelial Cells: Evidence Supporting a Role for Teriflunomide in Prostate Cancer Chemoprevention. Neoplasia 2010, 12, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Zhang, W.; Li, W.; Ling, C.; Jiang, M. Anti-inflammatory drug, leflunomide and its metabolite teriflunomide inhibit NSCLC proliferation in vivo and in vitro. Toxicol. Lett. 2018, 282, 154–165. [Google Scholar] [CrossRef]
- Calderón-Montaño, J.M.; Burgos-Morón, E.; Pérez-Guerrero, C.; López-Lázaro, M. A review on the dietary flavonoid kaempferol. Mini Rev. Med. Chem. 2011, 11, 298–340. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Nandakumar, N.; Palaniswami, R.; Nishigaki, Y.; Nishigaki, I. Kaempferol, a potential cytostatic and cure for inflammatory disorders. Eur. J. Med. Chem. 2014, 86, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.P.; Sun, J.; Chen, H.X.; Xiao, Y.Y.; Liu, D.; Chen, J.; Cai, H.; Cai, B.C. Comparative pharmacokinetics and bioavailability studies of quercetin, kaempferol and isorhamnetin after oral administration of Ginkgo biloba extracts, Ginkgo biloba extract phospholipid complexes and Ginkgo biloba extract solid dispersions in rats. Fitoterapia 2010, 81, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Vllasaliu, D.; Exposito-Harris, R.; Heras, A.; Casettari, L.; Garnett, M.; Illum, L.; Stolnik, S. Tight junction modulation by chitosan nanoparticles: Comparison with chitosan solution. Int. J. Pharm. 2010, 400, 183–193. [Google Scholar] [CrossRef]
- Casettari, L.; Illum, L. Chitosan in nasal delivery systems for therapeutic drugs. J. Control. Release 2014, 190, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Ujházy, P.; Berleth, E.S.; Pietkiewicz, J.M.; Kitano, H.; Skaar, J.R.; Ehrke, M.J.; Mihich, E. Evidence for the involvement of ecto-5’-nucleotidase (CD73) in drug resistance. Int. J. Cancer 1996, 68, 493–500. [Google Scholar] [CrossRef]
- Bavaresco, L.; Bernardi, A.; Braganhol, E.; Cappellari, A.R.; Rockenbach, L.; Farias, P.F.; Wink, M.R.; Cañedo, A.D.; Battastini, A.M.O. The role of ecto-5′-nucleotidase in glioma cell line proliferation. Mol. Cell Biochem. 2008, 319, 61–68. [Google Scholar] [CrossRef]
- Wink, M.R.; Lenz, G.; Braganhol, E.; Tamajusuku, A.S.; Schwartsmann, G.; Sarkis, J.J.; Battastini, A.M. Altered extracellular ATP, ADP and AMP catabolism in glioma cell lines. Cancer Lett. 2003, 198, 211–218. [Google Scholar] [CrossRef]
- Toussaint, L.G., 3rd; Nilson, A.E.; Goble, J.M.; Ballman, K.V.; James, C.D.; Lefranc, F.; Kiss, R.; Uhm, J.H. Galectin-1, a gene preferentially expressed at the tumor margin, promotes glioblastoma cell invasion. Mol. Cancer 2012, 11, 32. [Google Scholar] [CrossRef]
- Janes, P.W.; Slape, C.I.; Farnsworth, R.H.; Atapattu, L.; Scott, A.M.; Vail, M.E. EphA3 biology and cancer. Growth Factors 2014, 32, 176–189. [Google Scholar] [CrossRef]
- Kanazawa, T.; Taki, H.; Tanaka, K.; Takashima, Y.; Okada, H. Cell-Penetrating Peptide-Modified Block Copolymer Micelles Promote Direct Brain Delivery via Intranasal Administration. Pharm. Res. 2011, 28, 2130–2139. [Google Scholar] [CrossRef]
- Pescina, S.; Ostacolo, C.; Gomez-Monterrey, I.M.; Sala, M.; Bertamino, A.; Sonvico, F.; Padula, C.; Santi, P.; Bianchera, A.; Nicoli, S. Cell penetrating peptides in ocular delivery: State of the art. J. Control. Release 2018, 284, 84–102. [Google Scholar] [CrossRef]
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar] [PubMed]
- Martino, E.; Della Volpe, S.; Terribile, E.; Benetti, E.; Sakaj, M.; Centamore, A.; Sala, A.; Collina, S. The long story of camptothecin: From traditional medicine to drugs. Bioorgan. Med. Chem. Lett. 2017, 27, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Ahn, Y.; Kim, S.U.; Wang, K.C.; Cho, B.K.; Phi, J.H.; Park, I.H.; Black, P.M.; Carroll, R.S.; Lee, J.; et al. Targeting rat brainstem glioma using human neural stem cells and human mesenchymal stem cells. Clin. Cancer Res. 2009, 15, 4925–4934. [Google Scholar] [CrossRef] [Green Version]
- Nakamizo, A.; Marini, F.; Amano, T.; Khan, A.; Studeny, M.; Gumin, J.; Chen, J.; Hentschel, S.; Vecil, G.; Dembinski, J.; et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005, 65, 3307–3318. [Google Scholar] [CrossRef]
- Barish, M.E.; Herrmann, K.; Tang, Y.; Herculian, S.A.; Metz, M.; Aramburo, S.; Tirughana, R.; Gutova, M.; Annala, A.; Moats, R.A.; et al. Human neural stem cell biodistribution and predicted tumor coverage by a diffusible therapeutic in a mouse glioma model. Stem Cells Transl. Med. 2017, 6, 1522–1532. [Google Scholar] [CrossRef]
- Gutova, M.; Flores, L.; Adhikarla, V.; Tsaturyan, L.; Tirughana, R.; Aramburo, S.; Metz, M.; Gonzaga, J.; Annala, A.; Synold, T.W.; et al. Quantitative evaluation of intraventricular delivery of therapeutic neural stem cells to orthotopic glioma. Front. Oncol. 2019, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Klopp, A.H.; Spaeth, E.L.; Dembinski, J.L.; Woodward, W.A.; Munshi, A.; Meyn, R.E.; Cox, J.D.; Andreeff, M.; Marini, F.C. Tumor irradiation increases the recruitment of circulating mesenchymal stem cells into the tumor microenvironment. Cancer Res. 2007, 67, 11687–11695. [Google Scholar] [CrossRef]
- Karp, J.M.; Leng Teo, G.S. Mesenchymal stem cell homing: The devil is in the details. Cell Stem Cell. 2009, 4, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Aboody, K.S.; Najbauer, J.; Metz, M.Z.; D’Apuzzo, M.; Gutova, M.; Annala, A.J.; Synold, T.W.; Couture, L.A.; Blanchard, S.; Moats, R.A.; et al. Neural stem cell-mediated enzyme/prodrug therapy for glioma: Preclinical studies. Sci. Transl. Med. 2013, 5, 184ra59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, M.; Mao, J.; Duan, X.; Lu, L.; Zhang, F.; Lin, B.; Chen, M.; Zheng, C.; Zhang, X.; Shen, J. In vivo tracking of the tropism of mesenchymal stem cells to malignant gliomas using reporter gene-based MR imaging. Int. J. Cancer 2018, 142, 1033–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Sun, S.; Dangelmajer, S.; Zhang, Q.; Wang, J.; Hu, F.; Dong, F.; Kahlert, U.D.; Zhu, M.; Lei, T. Exploiting tumor-intrinsic signals to induce mesenchymal stem cell-mediated suicide gene therapy to fight malignant glioma. Stem Cell Res. Ther. 2019, 10, 88. [Google Scholar] [CrossRef] [PubMed]
- Young, J.S.; Morshed, R.A.; Kim, J.W.; Balyasnikova, I.V.; Ahmed, A.U.; Lesniak, M.S. Advances in stem cells, induced pluripotent stem cells, and engineered cells: Delivery vehicles for anti-glioma therapy. Expert Opin. Drug Deliv. 2014, 11, 1733–1746. [Google Scholar] [CrossRef]
- Bexell, D.; Svensson, A.; Bengzon, J. Stem cell-based therapy for malignant glioma. Cancer Treat. Rev. 2013, 39, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Sun, Z.; Han, Q.; Liao, L.; Wang, J.; Bian, C.; Li, J.; Yan, X.; Liu, Y.; Shao, C.; et al. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK1. Leukemia 2009, 23, 925–933. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Xu, Z.L.; Zhao, T.J.; Ye, L.H.; Zhang, X.D. DKK1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of Wnt signaling. Cancer Lett. 2008, 269, 67–77. [Google Scholar] [CrossRef]
- Ma, S.; Liang, S.; Jiao, H.; Chi, L.; Shi, X.; Tian, Y.; Yang, B.; Guan, F. Human umbilical cord mesenchymal stem cells inhibit C6 glioma growth via secretion of dickkopf-1 (DKK1). Mol. Cell Biochem. 2014, 385, 277–286. [Google Scholar] [CrossRef]
- An, J.; Yan, H.; Li, X.; Tan, R.; Chen, X.; Zhang, Z.; Liu, Y.; Zhang, P.; Lu, H.; Liu, Y. The inhibiting effect of neural stem cells on proliferation and invasion of glioma cells. Oncotarget 2017, 8, 76949–76960. [Google Scholar] [CrossRef] [Green Version]
- Reitz, M.; Demestre, M.; Sedlacik, J.; Meissner, H.; Fiehler, J.; Kim, S.U.; Westphal, M.; Schmidt, N.O. Intranasal delivery of neural stem/progenitor cells: A noninvasive passage to target intracerebral glioma. Stem Cells Transl. Med. 2012, 1, 866–873. [Google Scholar] [CrossRef]
- Danielyan, L.; Schäfer, R.; von Ameln-Mayerhofer, A.; Buadze, M.; Geisler, J.; Klopfer, T.; Klopfer, T.; Burkhardt, U.; Proksch, B.; Verleysdonk, S.; et al. Intranasal delivery of cells to the brain. Eur. J. Cell Biol. 2009, 88, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Yu, D.; Kanojia, D.; Li, G.; Sukhanova, M.; Spencer, D.A.; Pituch, K.C.; Zhang, L.; Han, Y.; Ahmed, A.U.; et al. Intranasal oncolytic virotherapy with CXCR4-enhanced stem cells extends survival in mouse model of glioma. Stem Cell Rep. 2016, 7, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulasov, I.V.; Zhu, Z.B.; Tyler, M.A.; Han, Y.; Rivera, A.A.; Khramtsov, A.; Curiel, D.T.; Lesniak, M.S. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum. Gene Ther. 2007, 18, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.U.; Tyler, M.A.; Thaci, B.; Alexiades, N.G.; Han, Y.; Ulasov, I.V.; Lesniak, M.S. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol. Pharm. 2011, 8, 1559–1572. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.U.; Thaci, B.; Tobias, A.L.; Auffinger, B.; Zhang, L.; Cheng, Y.; Kim, C.K.; Yunis, C.; Han, Y.; Alexiades, N.G.; et al. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J. Natl. Cancer Inst. 2013, 105, 968–977. [Google Scholar] [CrossRef]
- Morshed, R.A.; Gutova, M.; Juliano, J.; Barish, M.E.; Hawkins-Daarud, A.; Oganesyan, D.; Vazgen, K.; Yang, T.; Annala, A.; Ahmed, A.U.; et al. Analysis of glioblastoma tumor coverage by oncolytic virus-loaded neural stem cells using MRI based tracking and histological reconstruction. Cancer Gene Ther. 2015, 22, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Balyasnikova, I.V.; Prasol, M.S.; Ferguson, S.D.; Han, Y.; Ahmed, A.U.; Gutova, M.; Tobias, A.L.; Mustafi, D.; Rincón, E.; Zhang, L.; et al. Intranasal delivery of mesenchymal stem cells significantly extends survival of irradiated mice with experimental brain tumors. Mol. Ther. 2014, 22, 140–148. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Type of Nanocarrier | Surface Modification | Preparation Method | Size (nm) | Zeta Potential (mV) | In Vivo Model | Ref. |
|---|---|---|---|---|---|---|---|
| Bevacizumab | Polymeric NPs(PLGA) | - | Emulsification-evaporation | 185.0 ± 3.0 | ~−2 | U-87-luc mice | [116] |
| Ecto-5′-nucleotidase (CD73) | Nanoemulsion | - | Microfluidization | 262.7 ± 12.8 | +3.5 ± 3.0 | C6 rat glioma | [117] |
| Teriflunomide | Microemulsion | - | Progressive aqueous phase titration | 22.81 ± 0.48 | −22.62 ± 1.1 | - | [118] |
| Melatonin | Polymeric NPs(PCL) | - | Nanoprecipitation | 166.7 ± 6.3 | −34.0 ± 5.2 | - | [119] |
| Temozolomide | Polymeric NPs(PLGA) | Anti-EPHA3 | Emulsion-solvent evaporation | 125 to 146 | −21 to + 23 | C6 rat glioma | [120] |
| Kaempferol | Nanoemulsion | Chitosan | High-pressure homogenization | 180.53 ± 4.90 (coated) 145.07 ± 4.91 (uncoated) | +26.09 ± 2.67 (coated) −18.10 ± 2.55 (uncoated) | - | [121] |
| Temozolomide | Nanoemulsion | - | High-pressure homogenization | 134 nm | −13.11 | - | [122] |
| Farnesylthiosalicylic acid | Hybrid nanoparticles | - | Emulsion sonication | 164.3 ± 10.3 | −12.0 ± 1.3 | RG2 rat glioma | [123] |
| Curcumin | Microemulsion | - | Oil titration method | <20 | ~+10 | - | [124] |
| Curcumin | Nanostructured Lipid Carriers | - | High pressure homogenization | 146.8 | −21.4 ± 1.87 | - | [125] |
| siRNA | Chitosan nanoparticles | - | Ionic gelation | 141 ± 5 | +32 | GL261 tumor bearing mice | [126] |
| siRNA siRNA + TMZ or immunotherapy | Chitosan nanoparticles | - | Ionic gelation | 141 | +32 | GL261 tumor bearing mice | [127] |
| Methotrexate | Polymeric nanodispersion(PLA) | - | Emulsion/Solvent evaporation | 351 ± 13.4 | +25.1 ± 1.2 | - | [128] |
| Carboplatin | Polymericnanoparticles(PCL) | - | Double emulsion/solvent evaporation | 311.6 ± 4.7 | −16.3 ± 3.7 | - | [129] |
| BMP4 plasmid DNA | Polymeric nanoparticles(PBAE) | - | Self-assembly | 218 ± 7 | +17 ± 1 | U87 rat glioma | [130] |
| Camptothecin | Polymer micelles(MPEG-PCL) | Tat | Self-assembly | 88.5 ± 20.2 | +10.4 ± 2.84 | C6 rat glioma | [131] |
| siRaf-1/Camptothecin | Polymer micelles(MPEG-PCL) | Tat | Self-assembly | 60 to 200 | −2.86 to 15.9 | C6 rat glioma | [132] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruinsmann, F.A.; Richter Vaz, G.; de Cristo Soares Alves, A.; Aguirre, T.; Raffin Pohlmann, A.; Stanisçuaski Guterres, S.; Sonvico, F. Nasal Drug Delivery of Anticancer Drugs for the Treatment of Glioblastoma: Preclinical and Clinical Trials. Molecules 2019, 24, 4312. https://doi.org/10.3390/molecules24234312
Bruinsmann FA, Richter Vaz G, de Cristo Soares Alves A, Aguirre T, Raffin Pohlmann A, Stanisçuaski Guterres S, Sonvico F. Nasal Drug Delivery of Anticancer Drugs for the Treatment of Glioblastoma: Preclinical and Clinical Trials. Molecules. 2019; 24(23):4312. https://doi.org/10.3390/molecules24234312
Chicago/Turabian StyleBruinsmann, Franciele Aline, Gustavo Richter Vaz, Aline de Cristo Soares Alves, Tanira Aguirre, Adriana Raffin Pohlmann, Silvia Stanisçuaski Guterres, and Fabio Sonvico. 2019. "Nasal Drug Delivery of Anticancer Drugs for the Treatment of Glioblastoma: Preclinical and Clinical Trials" Molecules 24, no. 23: 4312. https://doi.org/10.3390/molecules24234312
APA StyleBruinsmann, F. A., Richter Vaz, G., de Cristo Soares Alves, A., Aguirre, T., Raffin Pohlmann, A., Stanisçuaski Guterres, S., & Sonvico, F. (2019). Nasal Drug Delivery of Anticancer Drugs for the Treatment of Glioblastoma: Preclinical and Clinical Trials. Molecules, 24(23), 4312. https://doi.org/10.3390/molecules24234312