
The Amyloid as a Ribbon-Like Micelle in Contrast to Spherical Micelles Represented by Globular Proteins



Abstract
:1. Introduction
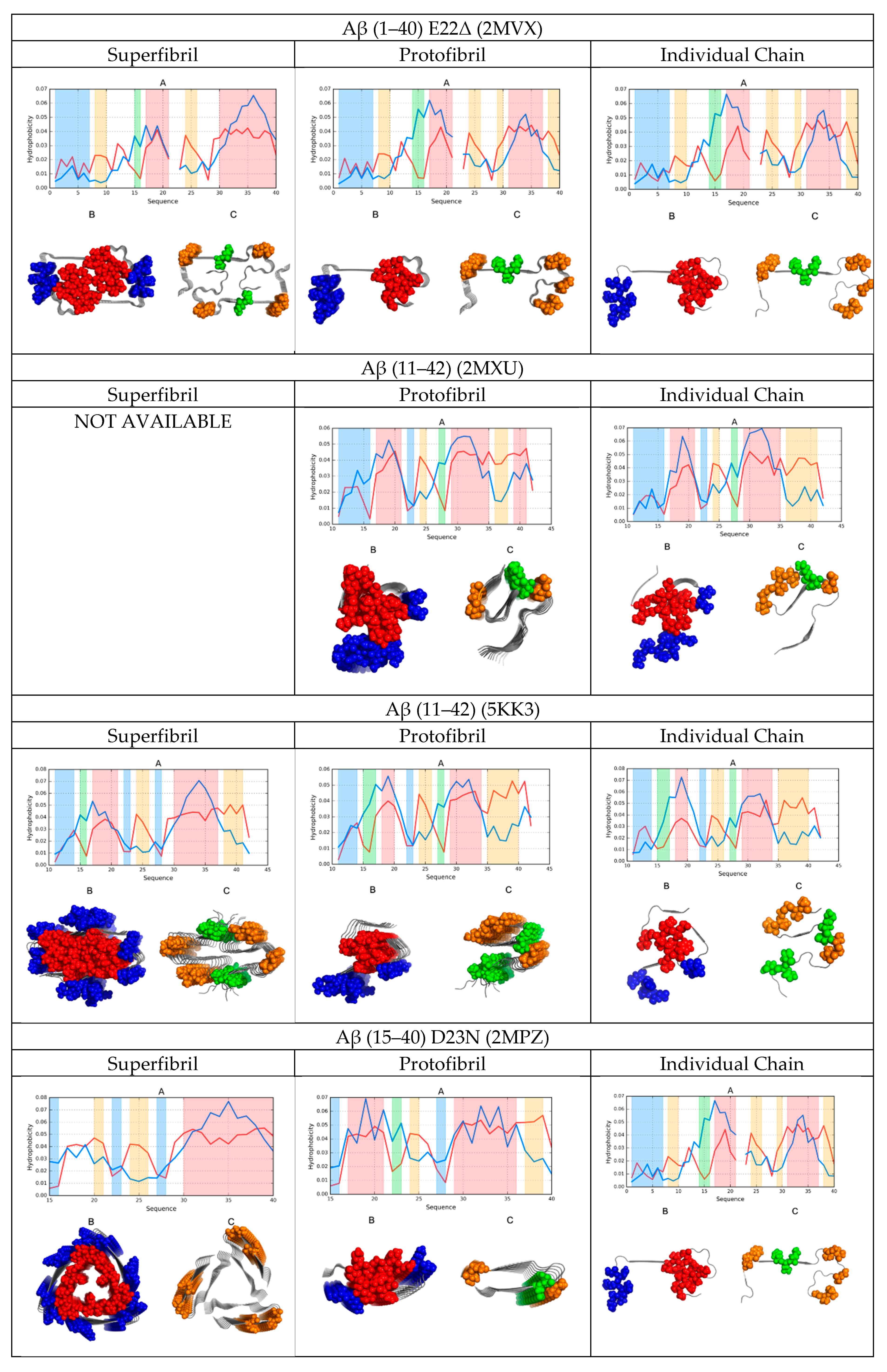
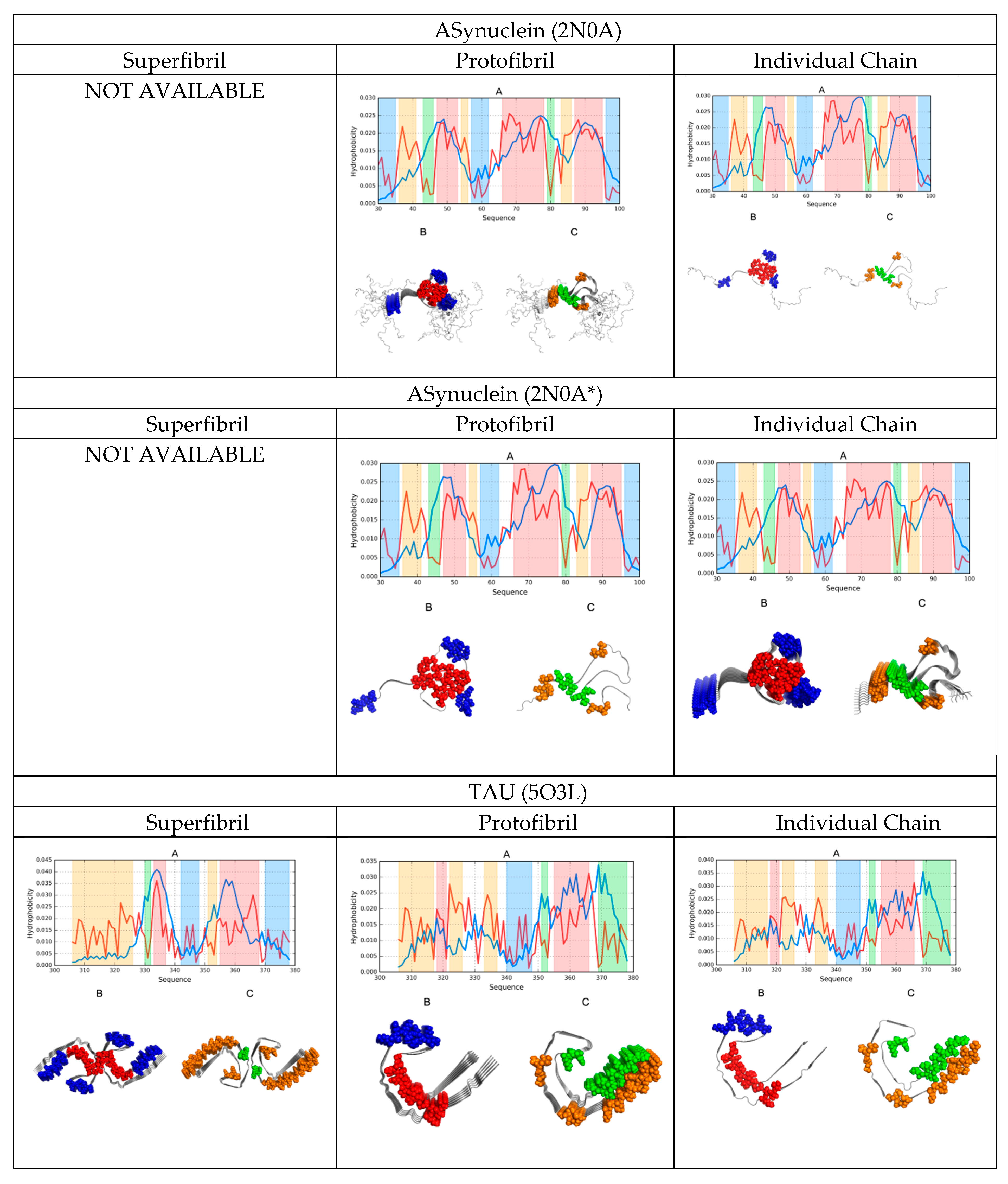
2. Results
2.1. FOD-Based Parameters to Interpret the Results
2.2. Comparative Analysis of Individual Chains
2.3. Protofibril Structure
2.4. Superfibril Structure
2.5. Status of the Inter-Fibril Interface
2.6. Comparative Analysis
2.7. Mathematical Differentiation of Globular Proteins and Amyloids
3. Discussion
4. Materials and Methods
4.1. Data
4.2. The fuzzy Oil Drop Model (FOD)
4.3. Comparative Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-synuclein | Asyn |
References
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; Chan, H.S. From Levinthal to pathways to funnels. Nat. Struct. Mol. Biol. 1997, 4, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M.; Šali, A.; Karplus, M. Protein Folding: A Perspective from Theory and Experiment. Angew. Chem. Int. Ed. 1998, 37, 868–893. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Knowles, T.P.J.; Tartaglia, G.G.; Fitzpatrick, A.W.; Devlin, G.L.; Shammas, S.L.; Dobson, C.M. Metastability of Native Proteins and the Phenomenon of Amyloid Formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Gazit, E. The “Correctly Folded” State of Proteins: Is It a Metastable State? Angew. Chem. Int. Ed. 2002, 41, 257. [Google Scholar] [CrossRef]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Knowles, T.P.J. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [Green Version]
- Gazit, E. A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman, I. Why do antifreeze proteins require a solenoid? Biochimie 2018, 144, 74–84. [Google Scholar] [CrossRef]
- Roterman, I.; Konieczny, L.; Jurkowski, W.; Prymula, K.; Banach, M. Two-intermediate model to characterize the structure of fast-folding proteins. J. Theor. Biol. 2011, 283, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Kalinowska, B.; Banach, M.; Wiśniowski, Z.; Konieczny, L.; Roterman, I. Is the hydrophobic core a universal structural element in proteins? J. Mol. Model. 2017, 23, 205. [Google Scholar] [CrossRef] [Green Version]
- Banach, M.; Konieczny, L.; Roterman, I. Secondary and supersecondary structure of proteins in light of the structure of hydrophobic cores. In Protein Supersecondary Structures; Humana Press: New York, NY, USA, 2019. [Google Scholar]
- Dygut, J.; Kalinowska, B.; Banach, M.; Piwowar, M.; Konieczny, L.; Roterman, I. Structural Interface Forms and Their Involvement in Stabilization of Multidomain Proteins or Protein Complexes. Int. J. Mol. Sci. 2016, 17, 1741. [Google Scholar] [CrossRef] [Green Version]
- Roterman, I.; Banach, M.; Konieczny, L. Application of the Fuzzy Oil Drop Model Describes Amyloid as a Ribbonlike Micelle. Entropy 2017, 19, 167. [Google Scholar] [CrossRef] [Green Version]
- Kalinowska, B.; Banach, M.; Konieczny, L.; Roterman, I. Application of Divergence Entropy to Characterize the Structure of the Hydrophobic Core in DNA Interacting Proteins. Entropy 2015, 17, 1477–1507. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Levinthal, C. Are there pathways for protein folding? J. Chim. Phys. 1968, 65, 44–45. [Google Scholar] [CrossRef]
- Levinthal, C. How to Fold Graciously. Mossbauer Spectroscopy in Biological Systems: Proceedings of a Meeting Held at Allerton House; University of Illinois Press: Monticello, IL, USA, 1969. [Google Scholar]
- Smith, R.; Tanford, C. The critical micelle concentration of l-α-dipalmitoylphosphatidylcholine in water and water/methanol solutions. J. Mol. Biol. 1972, 67, 75–83. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Anfinsen, C.B.; Scheraga, H.A. Experimental and theoretical aspects of protein folding. In Advances in Protein Chemistry; Academic Press: New York, NY, USA, 1975; pp. 205–300. [Google Scholar]
- Chothia, C. Hydrophobic bonding and accessible surface area in proteins. Nature 1974, 248, 338–339. [Google Scholar] [CrossRef]
- Chothia, C. Structural invariants in protein folding. Nature 1975, 254, 304–308. [Google Scholar] [CrossRef]
- Levitt, M.; Chothia, C. Structural patterns in globular proteins. Nature 1976, 261, 552–558. [Google Scholar] [CrossRef]
- Tanford, C. The hydrophobic effect and the organization of living matter. Science 1978, 200, 1012–1018. [Google Scholar] [CrossRef]
- Tanford, C. Interfacial free energy and the hydrophobic effect. Proc. Natl. Acad. Sci. USA 1979, 76, 4175–4176. [Google Scholar] [CrossRef] [Green Version]
- Lesk, A.M.; Chothia, C. Solvent accessibility, protein surfaces, and protein folding. Biophys. J. 1980, 32, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Dill, K.A.; Flory, P.J. Molecular organization in micelles and vesicles. Proc. Natl. Acad. Sci. USA 1981, 78, 676–680. [Google Scholar] [CrossRef] [Green Version]
- Chothia, C. Principles that Determine the Structure of Proteins. Annu. Rev. Biochem. 1984, 53, 537–572. [Google Scholar] [CrossRef]
- Miller, S.; Janin, J.; Lesk, A.M.; Chothia, C. Interior and surface of monomeric proteins. J. Mol. Biol. 1987, 196, 641–656. [Google Scholar] [CrossRef]
- Miller, S.; Lesk, A.M.; Janin, J.; Chothia, C. The accessible surface area and stability of oligomeric proteins. Nature 1987, 328, 834–836. [Google Scholar] [CrossRef]
- Creighton, T.E.; Chothia, C. Selecting buried residues. Nature 1989, 339, 14–15. [Google Scholar] [CrossRef]
- Zwanzig, R.; Szabo, A.; Bagchi, B. Levinthal’s paradox. Proc. Natl. Acad. Sci. USA 1992, 89, 20–22. [Google Scholar] [CrossRef] [Green Version]
- Ripoll, D.R.; Piela, L.; Váasquez, M.; Scheraga, H.A. On the multiple-minima problem in the conformational analysis of polypeptides. V. Application of the self-consistent electrostatic field and the electrostatically driven monte carlo methods to bovine pancreatic trypsin inhibitor. Proteins Struct. Funct. Bioinf. 1991, 10, 188–198. [Google Scholar] [CrossRef]
- Sali, A.; Shakhnovich, E.; Karplus, M. How does a protein fold? Nature 1994, 369, 248–251. [Google Scholar] [CrossRef]
- Gerstein, M.; Chothia, C. Packing at the protein-water interface. Proc. Natl. Acad. Sci. USA 1996, 93, 10167–10172. [Google Scholar] [CrossRef] [Green Version]
- Karplus, M. The Levinthal paradox: Yesterday and today. Folding Des. 1997, 2, S69–S75. [Google Scholar] [CrossRef] [Green Version]
- Yon, J.M. Protein folding: Concepts and perspectives. Cell Mol Life Sci. 1997, 53, 557–567. [Google Scholar] [CrossRef]
- Tanford, C. How protein chemists learned about the hydrophobic factor. Protein Sci. 1997, 6, 1358–1366. [Google Scholar] [CrossRef] [Green Version]
- Durup, J. On “Levinthal paradox” and the theory of protein folding. J. Mol. Struct. THEOCHEM 1998, 424, 157–169. [Google Scholar] [CrossRef]
- Dill, K.A. Polymer principles and protein folding. Protein Sci. 1999, 8, 1166–1180. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y. Hidden intermediates and levinthal paradox in the folding of small proteins. Biochem. Biophys. Res. Commun. 2003, 305, 785–788. [Google Scholar] [CrossRef]
- Tanford, C. My debt to Walter Kauzmann. Biophys. Chem. 2003, 105, 159–160. [Google Scholar] [CrossRef]
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. [Google Scholar] [CrossRef]
- Hunter, P. Into the fold. EMBO Rep. 2006, 7, 249–252. [Google Scholar] [CrossRef]
- Dill, K.A.; Ozkan, S.B.; Weikl, T.R.; Chodera, J.D.; Voelz, V.A. The protein folding problem: When will it be solved? Curr. Opin. Struct. Biol. 2007, 17, 342–346. [Google Scholar] [CrossRef]
- Ben-Naim, A. Levinthal’s question revisited, and answered. J. Biomol. Struct. Dyn. 2012, 30, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Englander, S.W.; Mayne, L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. USA 2014, 111, 15873–15880. [Google Scholar] [CrossRef] [Green Version]
- Cruzeiro, L.; Degrève, L. Exploring the Levinthal limit in protein folding. J. Biol. Physics. 2016, 43, 15–30. [Google Scholar] [CrossRef] [Green Version]
- Hirata, F.; Sugita, M.; Yoshida, M.; Akasaka, K. Perspective: Structural fluctuation of protein and Anfinsen’s thermodynamic hypothesis. J. Chem. Phys. 2018, 148, 020901. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Jouet, M.; MacFarlane, J.; Du, J.S.; Kenwrick, S.; Chothia, C. Outline structure of the human L1 cell adhesion molecule and the sites where mutations cause neurological disorders. EMBO J. 1996, 15, 6050–6059. [Google Scholar] [CrossRef]
- Kauzmann, W. Some Factors in the Interpretation of Protein Denaturation. Adv. Protein Chem. 1959, 14, 1–63. [Google Scholar] [CrossRef]
- Konieczny, L.; Brylinski, M.; Roterman, I. Gauss-function-Based model of hydrophobicity density in proteins. In Silico Biol. 2006, 6, 15–22. [Google Scholar]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Sałapa, K.; Kalinowska, B.; Jadczyk, T.; Roterman, I. Measurement of hydrophobicity distribution in protein-non-redundant Protein Data Bank. BAMS 2012, 8, 327–337. [Google Scholar] [CrossRef]
- Swuec, P.; Lavatelli, F.; Tasaki, M.; Paissoni, C.; Rognoni, P.; Maritan, M.; Bolognesi, M. Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Banach, M.; Konieczny, L.; Roterman, I. The fuzzy oil drop model, based on hydrophobicity density distribution, generalizes the influence of water environment on protein structure and function. J. Theor. Biol. 2014, 359, 6–17. [Google Scholar] [CrossRef] [Green Version]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The Function of α-Synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [Green Version]
- Konieczny, L.; Roterman, I. Introduction. From Globular Proteins to Amyloids. 2020. [Google Scholar] [CrossRef]
- Dułak, D.; Banach, M.; Gadzała, M.; Konieczny, L.; Roterman, I. Structural analysis of the Aβ (15–40) amyloid fibril based on hydrophobicity distribution. Acta Biochim. Pol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Dułak, D.; Gadzała, M.; Banach, M.; Ptak, M.; Wisniowski, Z.; Konieczny, L.; Roterman, I. Filamentous Aggregates of Tau Proteins Fulfil Standard Amyloid Criteria Provided by the Fuzzy Oil Drop (FOD) Model. Int. J. Mol. Sci. 2018, 19, 2910. [Google Scholar] [CrossRef] [Green Version]
- Banach, M.; Konieczny, L.; Roterman, I. Fuzzy Oil Drop Model Application—From Globular Proteins to Amyloids. In Computational Methods to Study the Structure and Dynamics of Biomolecules and Biomolecular Processes; Springer: Cham, Switzerland, 2019; pp. 639–658. [Google Scholar]
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. Biochim. Biophys. Acta Mol. Basis Dis. 2000, 1502, 16–30. [Google Scholar] [CrossRef] [Green Version]
- Belli, M.; Ramazzotti, M.; Chiti, F. Prediction of amyloid aggregation in vivo. EMBO Rep. 2011, 12, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Cecchini, M.; Curcio, R.; Pappalardo, M.; Melki, R.; Caflisch, A. A Molecular Dynamics Approach to the Structural Characterization of Amyloid Aggregation. J. Mol. Biol. 2006, 357, 1306–1321. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. The CamSol Method of Rational Design of Protein Mutants with Enhanced Solubility. J. Mol. Biol. 2015, 427, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Bemporad, F.; Ramazzotti, M. From the evolution of protein sequences able to resist self-assembly to the prediction of aggregation propensity. In International Review of Cell and Molecular Biology; Academic Press: Cambridge, MA, USA, 2017; pp. 1–47. [Google Scholar]
- Tsolis, A.C.; Papandreou, N.C.; Iconomidou, V.A.; Hamodrakas, S.J. A Consensus Method for the Prediction of “Aggregation-Prone” Peptides in Globular Proteins. PLoS ONE 2013, 8, e54175. [Google Scholar] [CrossRef] [Green Version]
- Tycko, R. Structure of aggregates revealed. Nature 2016, 537, 492–493. [Google Scholar] [CrossRef] [Green Version]
- Tycko, R. Molecular Structure of Aggregated Amyloid-β: Insights from Solid-State Nuclear Magnetic Resonance. Cold Spring Harbor Perspect. Med. 2016, 6, a024083. [Google Scholar] [CrossRef]
- Sgourakis, N.G.; Yau, W.-M.; Qiang, W. Modeling an In-Register, Parallel “Iowa” Aβ Fibril Structure Using Solid-State NMR Data from Labeled Samples with Rosetta. Structure 2015, 23, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Ishii, Y. Aβ (1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Griffin, R.G. Atomic Resolution Structure of Monomorphic Aβ 42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Rienstra, C.M. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Biol. 1976, 104, 59–107. [Google Scholar] [CrossRef]
- Huang, D.B.; Ainsworth, C.F.; Stevens, F.J.; Schiffer, M. Three quaternary structures for a single protein. Proc. Natl. Acad. Sci. USA 1996, 93, 7017–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roterman, I.; Dułak, D.; Gadzała, M.; Banach, M.; Konieczny, L. Structural analysis of the Aβ (11–42) amyloid fibril based on hydrophobicity distribution. J. Comput.-Aided Mol. Des. 2019, 33, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| RD | Correlation Coefficient | Amyloid | |||
|---|---|---|---|---|---|
| T–O–R | T–O–H | HvT | TvO | HvO | |
| Chain as Part of Superfibril | |||||
| 0.554 | 0.475 | 0.452 | 0.558 | 0.843 | Aβ (15–40) D23N (2MPZ) |
| 0.607 | 0.620 | 0.459 | 0.664 | 0.784 | Aβ (1–40) E22Δ (2MVX) |
| 0.565 | 0.594 | 0.395 | 0.466 | 0.782 | Aβ (11–42) (5KK3) |
| 0.747 | 0.697 | 0.012 | 0.265 | 0.696 | TAU (5O3L) |
| 0.761 | 0.722 | 0.010 | 0.224 | 0.731 | TAU (5O3O) |
| 0.728 | 0.666 | −0.019 | 0.136 | 0.785 | TAU (5O3T) |
| Chain as Part of Protofibril | |||||
| 0.491 | 0.487 | 0.382 | 0.458 | 0.839 | Aβ (15–40) D23N (2MPZ) |
| 0.649 | 0.686 | 0.310 | 0.322 | 0.779 | Aβ (1–40) E22Δ (2MVX) |
| 0.513 | 0.620 | 0.404 | 0.471 | 0.849 | Aβ (11–42) (2MXU) |
| 0.569 | 0.600 | 0.299 | 0.286 | 0.789 | Aβ (11–42) (5KK3) |
| 0.664 | 0.595 | −0.022 | 0.083 | 0.767 | TAU (5O3L) |
| 0.661 | 0.607 | −0.012 | 0.089 | 0.772 | TAU (5O3O) |
| 0.688 | 0.618 | −0.029 | 0.098 | 0.782 | TAU (5O3T) |
| 0.506 | 0.588 | 0.285 | 0.506 | 0.823 | ASYN (2N0A *) |
| Chain as Individual Unit | |||||
| 0.626 | 0.467 | 0.355 | 0.351 | 0.615 | Aβ (15–40) D23N (2MPZ) |
| 0.635 | 0.562 | 0.295 | 0.363 | 0.615 | Aβ (1–40) E22Δ (2MVX) |
| 0.536 | 0.519 | 0.408 | 0.567 | 0.697 | Aβ (11–42) (2MXU) |
| 0.660 | 0.555 | 0.355 | 0.263 | 0.698 | Aβ (11–42) (5KK3) |
| 0.674 | 0.410 | −0.039 | 0.091 | 0.545 | TAU (5O3L) |
| 0.679 | 0.430 | −0.027 | 0.095 | 0.548 | TAU (5O3O) |
| 0.683 | 0.415 | −0.033 | 0.096 | 0.551 | TAU (5O3T) |
| 0.784 | 0.727 | 0.099 | 0.208 | 0.725 | ASYN (2N0A *) |
| RD | Correlation Coefficient | Amyloid | |||
|---|---|---|---|---|---|
| T–O–R | T–O–H | HvT | TvO | HvO | PDB ID |
| 0.614 | 0.600 | 0.262 | 0.412 | 0.786 | Aβ (15–40) D23N (2MPZ) |
| 0.639 | 0.659 | 0.280 | 0.365 | 0.718 | Aβ (1–40) E22Δ (2MVX) |
| 0.680 | 0.756 | 0.246 | 0.363 | 0.821 | Aβ (11–42) (2MXU) |
| 0.608 | 0.623 | 0.235 | 0.335 | 0.750 | Aβ (11–42) (5KK3) |
| 0.652 | 0.564 | −0.022 | 0.145 | 0.705 | TAU (5O3L) |
| 0.652 | 0.0.577 | −0.012 | 0.151 | 0.712 | TAU (5O3O) |
| 0.674 | 0.584 | −0.028 | 0.152 | 0.720 | TAU (5O3T) |
| 0.531 | 0.598 | 0.241 | 0.492 | 0.798 | ASYN (2N0A *) |
| RD | Correlation Coefficient | Amyloid | |||
|---|---|---|---|---|---|
| T–O–R | T–O–H | HvT | TvO | HvO | |
| 0.578 | 0.494 | 0.394 | 0.554 | 0.790 | Aβ (15–40) D23N (2MPZ) |
| 0.590 | 0.592 | 0.438 | 0.674 | 0.727 | Aβ (1–40) E22Δ (2MVX) |
| 0.620 | 0.652 | 0.330 | 0.440 | 0.756 | Aβ (11–42) (5KK3) |
| 0.730 | 0.662 | 0.014 | 0.297 | 0.643 | TAU (5O3L) |
| 0.745 | 0.687 | 0.012 | 0.259 | 0.675 | TAU (5O3O) |
| 0.724 | 0.641 | 0.008 | 0.301 | 0.716 | TAU (5O3T) |
| RD | Correlation Coefficient | Amyloid | ||||
|---|---|---|---|---|---|---|
| T–O–R | T–O–H | HvT | TvO | HvO | Amyloid | Residues in Interface |
| 0.424 | 0.155 | 0.452 | 0.717 | 0.768 | Aβ (15–40) D23N | 17, 28, 29, 31, 38, 40 |
| 0.454 | 0.313 | 0.375 | 0.709 | 0.623 | Aβ (1–40) E22Δ | 3, 4, 13, 28–30, 37–40 |
| 0.595 | 0.707 | 0.35 | 0.425 | 0.591 | Aβ (11–42)) | 11, 13, 15, 17, 34–38 |
| 0.388 | 0.532 | 0.612 | 0.828 | 0.811 | TAU (5O3L) | 331–336, 338 |
| 0.401 | 0.550 | 0.666 | 0.754 | 0.950 | TAU (5O3O) | 331–336 |
| 0.538 | 0.314 | −0.189 | −0.069 | 0.809 | TAU (5O3T) | 321, 323/313, 15, 317 |
| Name | PDB ID | Chain Length | Fragment | Mutation | Beta-Structure (%) | NCPxNPF | Ref. |
|---|---|---|---|---|---|---|---|
| Aβ (15–40) | 2MPZ | 26 aa | 15–40 | D23N | 56 | 9 × 3 | [73] |
| Aβ (11–42) | 2MXU | 32 aa | 11–42 | 68 | 12 × 1 | [74] | |
| Aβ (1–40) | 2MVX | 39aa | 1–40 | E22Δ | 35 | 5 × 2 | [73] |
| Aβ (11–42) | 5KK3 | 32 aa | 11–42 | 59 | 9 × 2 | [75] | |
| TAU | 5O3L | 73 aa | 306–378 | 13 | 5 × 2 | [76] | |
| 5O3O | 73 aa | 306–378 | 13 | 5 × 2 | [77] | ||
| 5O3T | 73 aa | 306–378 | 13 | 5 × 2 | [78] | ||
| ASyn | 2N0A | 140/70 aa* | 1–140 * | 92 | 10 × 1 | [79] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banach, M.; Konieczny, L.; Roterman, I. The Amyloid as a Ribbon-Like Micelle in Contrast to Spherical Micelles Represented by Globular Proteins. Molecules 2019, 24, 4395. https://doi.org/10.3390/molecules24234395
Banach M, Konieczny L, Roterman I. The Amyloid as a Ribbon-Like Micelle in Contrast to Spherical Micelles Represented by Globular Proteins. Molecules. 2019; 24(23):4395. https://doi.org/10.3390/molecules24234395
Chicago/Turabian StyleBanach, Mateusz, Leszek Konieczny, and Irena Roterman. 2019. "The Amyloid as a Ribbon-Like Micelle in Contrast to Spherical Micelles Represented by Globular Proteins" Molecules 24, no. 23: 4395. https://doi.org/10.3390/molecules24234395
APA StyleBanach, M., Konieczny, L., & Roterman, I. (2019). The Amyloid as a Ribbon-Like Micelle in Contrast to Spherical Micelles Represented by Globular Proteins. Molecules, 24(23), 4395. https://doi.org/10.3390/molecules24234395