Synthesis, Physical Properties, and Reactivity of Stable, π-Conjugated, Carbon-Centered Radicals


Abstract
:1. Introduction
2. Phenalenyl Radical
2.1. Electronic Structure of Phenalenyl Radical
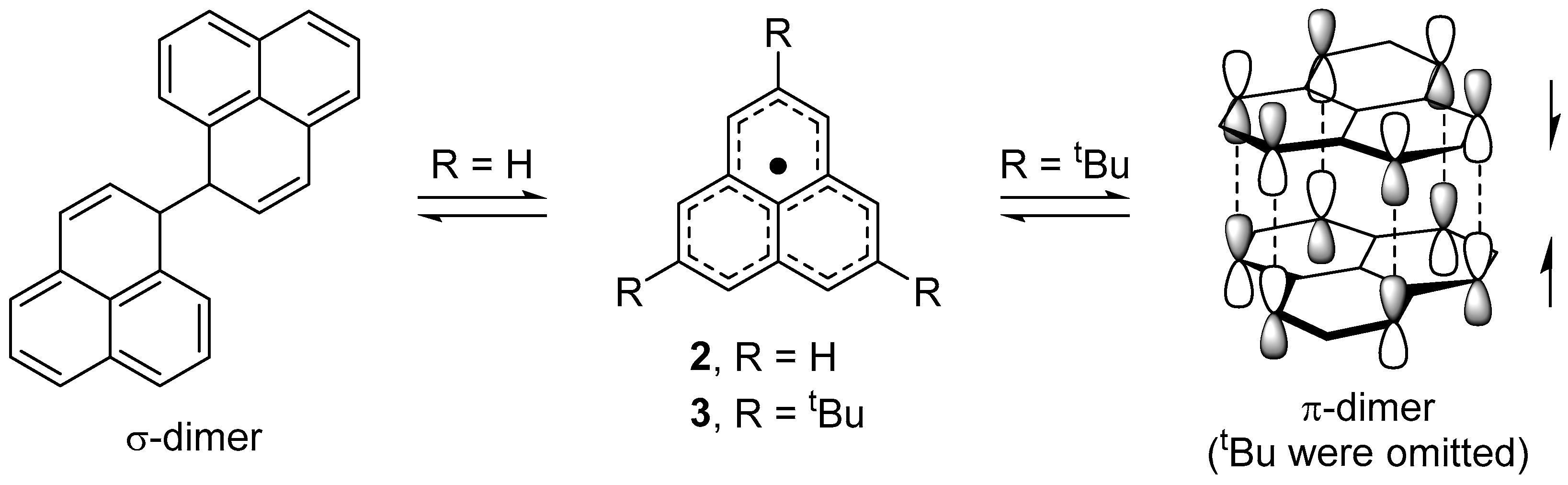
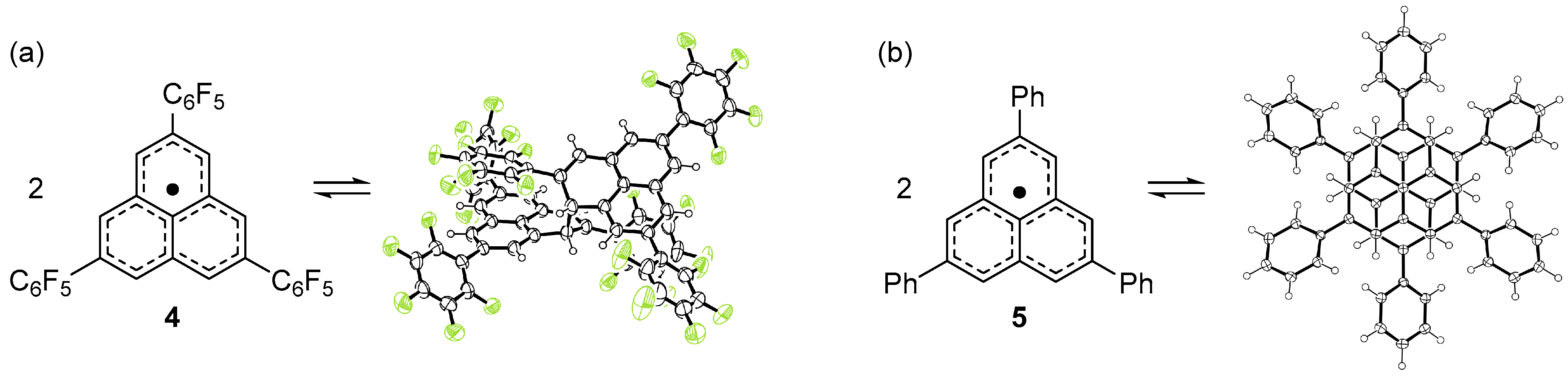
2.2. Dimerizaion Behavior of Phenalenyl Radicals
2.3. One-Dimensional Stack of Phenalenyl Radicals
3. Fluorenyl Radical
3.1. π-Extended Fluorenyl Radicals
3.2. Ring Expansion of Fluorenyl Radical
4. Trianthrylmethyl Radical
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Miller, J.S.; Glatzhofer, D.T.; Vazquez, C.; McLean, R.S.; Calabrese, J.C.; Marshall, W.J.; Raebiger, J.W. Electron-Transfer Salts of 1,2,3,4,5-Pentamethylferrocene, FeII(C5Me5)(C5H5). Structure and Magnetic Properties of Two 1:1 and Two 2:3 Fe(C5Me5)(C5H5) Electron-Transfer Salts of Tetracyanoethylene. Inorg. Chem. 2001, 40, 2058–2064. [Google Scholar] [CrossRef] [PubMed]
- Kothe, G.; Denkel, K.-H.; Sümmermann, W. Schlenk’s Biradical.—A Molecule in the Triplet Ground State. Angew. Chem. Int. Ed. Engl. 1970, 9, 906–907. [Google Scholar] [CrossRef]
- Hicks, R.G. What’s new in stable radical chemistry? Org. Biomol. Chem. 2007, 5, 1321–1338. [Google Scholar] [CrossRef] [PubMed]
- Rajca, A. Organic Diradicals and Polyradicals: From Spin Coupling to Magnetism? Chem. Rev. 1994, 94, 871–893. [Google Scholar] [CrossRef]
- Hicks, R.G. Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds; Wiley-Blackwell: Hoboken, NJ, USA, 2010; ISBN 9780470666975. [Google Scholar]
- Gaudenzi, R.; Burzurí, E.; Reta, D.; Moreira, I.d.P.R.; Bromley, S.T.; Rovira, C.; Veciana, J.; van der Zant, H.S.J. Exchange Coupling Inversion in a High-Spin Organic Triradical Molecule. Nano Lett. 2016, 16, 2066–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasini, D.; Takeuchi, D. Cyclopolymerizations: Synthetic Tools for the Precision Synthesis of Macromolecular Architectures. Chem. Rev. 2018, 118, 8983–9057. [Google Scholar] [CrossRef] [PubMed]
- Hioe, J.; Zipse, H. Radical stability and its role in synthesis and catalysis. Org. Biomol. Chem. 2010, 8, 3609–3617. [Google Scholar] [CrossRef] [PubMed]
- Gomberg, M. AN INSTANCE OF TRIVALENT CARBON: TRIPHENYLMETHYL. J. Am. Chem. Soc. 1900, 22, 757–771. [Google Scholar] [CrossRef]
- Reid, D.H. The chemistry of the phenalenes. Q. Rev. Chem. Soc. 1965, 19, 274–302. [Google Scholar] [CrossRef]
- Haddon, R.C. Design of organic metals and superconductors. Nature 1975, 256, 394–396. [Google Scholar] [CrossRef]
- Morita, Y.; Nishida, S. Phenalenyls, Cyclopentadienyls, and Other Carbon-Centered Radicals. In Stable Radicals; Hicks, R.G., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2010; pp. 81–145. [Google Scholar]
- Raman, K.V.; Kamerbeek, A.M.; Mukherjee, A.; Atodiresei, N.; Sen, T.K.; Lazić, P.; Caciuc, V.; Michel, R.; Stalke, D.; Mandal, S.K.; et al. Interface-engineered templates for molecular spin memory devices. Nature 2013, 493, 509–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, K.; Nakano, M.; Fukuda, K.; Matsui, H.; Takamuku, S.; Hirosaki, Y.; Kubo, T.; Kamada, K.; Champagne, B. Third-Order Nonlinear Optical Properties of One-Dimensional Open-Shell Molecular Aggregates Composed of Phenalenyl Radicals. Chem. A Eur. J. 2014, 20, 11129–11136. [Google Scholar] [CrossRef] [PubMed]
- Salustro, S.; Maschio, L.; Kirtman, B.; Rérat, M.; Dovesi, R. Third-Order Electric Field Response of Infinite Linear Chains Composed of Phenalenyl Radicals. J. Phys. Chem. C 2016, 120, 6756–6761. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sau, S.C.; Mandal, S.K. Exploring Closed-Shell Cationic Phenalenyl: From Catalysis to Spin Electronics. Acc. Chem. Res. 2017, 50, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.H. Stable π-electron systems and new aromatic structures. Tetrahedron 1958, 3, 339–352. [Google Scholar] [CrossRef]
- Goto, K.; Kubo, T.; Yamamoto, K.; Nakasuji, K.; Sato, K.; Shiomi, D.; Takui, T.; Kubota, M.; Kobayashi, T.; Yakusi, K.; et al. A Stable Neutral Hydrocarbon Radical: Synthesis, Crystal Structure, and Physical Properties of 2,5,8-Tri-tert-butyl-phenalenyl. J. Am. Chem. Soc. 1999, 121, 1619–1620. [Google Scholar] [CrossRef]
- Suzuki, S.; Morita, Y.; Fukui, K.; Sato, K.; Shiomi, D.; Takui, T.; Nakasuji, K. Aromaticity on the pancake-bonded dimer of neutral phenalenyl radical as studied by MS and NMR spectroscopies and NICS analysis. J. Am. Chem. Soc. 2006, 128, 2530–2531. [Google Scholar] [CrossRef]
- Small, D.; Zaitsev, V.; Jung, Y.; Rosokha, S.V.; Head-Gordon, M.; Kochi, J.K. Intermolecular pi-to-pi bonding between stacked aromatic dyads. Experimental and theoretical binding energies and near-IR optical transitions for phenalenyl radical/radical versus radical/cation dimerizations. J. Am. Chem. Soc. 2004, 126, 13850–13858. [Google Scholar] [CrossRef]
- Kolb, B.; Kertesz, M.; Thonhauser, T. Binding Interactions in Dimers of Phenalenyl and Closed-Shell Analogues. J. Phys. Chem. A 2013, 117, 3642–3649. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Chen, Y.; Cao, Y.; Best, T.P.; Itkis, M.E.; Beer, L.; Oakley, R.T.; Cordes, A.W.; Brock, C.P.; Haddon, R.C. Perchlorophenalenyl radical. J. Am. Chem. Soc. 2001, 123, 3864–3871. [Google Scholar] [CrossRef]
- Uchida, K.; Hirao, Y.; Kurata, H.; Kubo, T.; Hatano, S.; Inoue, K. Dual association modes of the 2,5,8-tris(pentafluorophenyl)phenalenyl radical. Chem. Asian J. 2014, 9, 1823–1829. [Google Scholar] [CrossRef]
- Blanchard, M.D.; Hughes, R.P.; Concolino, T.E.; Rheingold, A.L. π-Stacking between Pentafluorophenyl and Phenyl Groups as a Controlling Feature of Intra- and Intermolecular Crystal Structure Motifs in Substituted Ferrocenes. Observation of Unexpected Face-to-Face Stacking between Pentafluorophenyl Rings. Chem. Mater. 2000, 12, 1604–1610. [Google Scholar] [CrossRef]
- Smith, C.E.; Smith, P.S.; Thomas, R.L.; Robins, E.G.; Collings, J.C.; Dai, C.; Scott, A.J.; Borwick, S.; Batsanov, A.S.; Watt, S.W.; et al. Arene-perfluoroarene interactions in crystal engineering: Structural preferences in polyfluorinated tolans. J. Mater. Chem. 2004, 413–420. [Google Scholar] [CrossRef]
- Mou, Z.; Uchida, K.; Kubo, T.; Kertesz, M. Evidence of σ- and π-Dimerization in a Series of Phenalenyls. J. Am. Chem. Soc. 2014, 136, 18009–18022. [Google Scholar] [CrossRef]
- Uchida, K.; Mou, Z.; Kertesz, M.; Kubo, T. Fluxional σ-Bonds of the 2,5,8-Trimethylphenalenyl Dimer: Direct Observation of the Sixfold σ-Bond Shift via a π-Dimer. J. Am. Chem. Soc. 2016, 138, 4665–4672. [Google Scholar] [CrossRef] [PubMed]
- Rakus, K.; Verevkin, S.P.; Schätzer, J.; Beckhaus, H.-D.; Rüchardt, C. Thermolabile Hydrocarbons, 33. Thermochemistry and Thermal Decomposition of 9,9′-Bifluorenyl and 9,9′-Dimethyl-9,9′-bifluorenyl—The Stabilization Energy of 9-Fluorenyl Radicals. Berichte der Deutschen Chemischen Gesellschaft 1994, 127, 1095–1103. [Google Scholar] [CrossRef]
- Font-Sanchis, E.; Aliaga, C.; Focsaneanu, K.-S.; Scaiano, J.C. Greatly attenuated reactivity of nitrile-derived carbon-centered radicals toward oxygen. Chem. Commun. 2002, 2, 1576–1577. [Google Scholar] [CrossRef]
- Tian, Y.; Uchida, K.; Kurata, H.; Hirao, Y.; Nishiuchi, T.; Kubo, T. Design and Synthesis of New Stable Fluorenyl-Based Radicals. J. Am. Chem. Soc. 2014, 136, 12784–12793. [Google Scholar] [CrossRef]
- Nishiuchi, T.; Ito, R.; Takada, A.; Yasuda, Y.; Nagata, T.; Stratmann, E.; Kubo, T. Anthracene-Attached Persistent Tricyclic Aromatic Hydrocarbon Radicals. Chem. Asian J. 2019. [Google Scholar] [CrossRef]
- Frenette, M.; Aliaga, C.; Font-Sanchis, E.; Scaiano, J.C. Bond Dissociation Energies for Radical Dimers Derived from Highly Stabilized Carbon-Centered Radicals. Org. Lett. 2004, 6, 2579–2582. [Google Scholar] [CrossRef]
- Zeng, Z.; Sung, Y.M.; Bao, N.; Tan, D.; Lee, R.; Zafra, J.L.; Lee, B.S.; Ishida, M.; Ding, J.; López Navarrete, J.T.; et al. Stable tetrabenzo-Chichibabin’s hydrocarbons: Tunable ground state and unusual transition between their closed-shell and open-shell resonance forms. J. Am. Chem. Soc. 2012, 134, 14513–14525. [Google Scholar] [CrossRef] [PubMed]
- Sabacky, M.J.; Johnson, C.S.; Smith, R.G.; Gutowsky, H.S.; Martin, J.C. Triarylmethyl Radicals. Synthesis and Electron Spin Resonance Studies of Sesquixanthydryl Dimer and Related Compounds. J. Am. Chem. Soc. 1967, 89, 2054–2058. [Google Scholar] [CrossRef]
- Nishiuchi, T.; Aibara, S.; Kubo, T. Synthesis and Properties of a Highly Congested Tri(9-anthryl)methyl Radical. Angew. Chem. Int. Ed. 2018, 57, 16516–16519. [Google Scholar] [CrossRef]
- Vijaykumar, G.; Pariyar, A.; Ahmed, J.; Shaw, B.K.; Adhikari, D.; Mandal, S.K. Tuning the redox non-innocence of a phenalenyl ligand toward efficient nickel-assisted catalytic hydrosilylation. Chem. Sci. 2018, 9, 2817–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, X.; Evans, E.W.; Dong, S.; Gillett, A.J.; Guo, H.; Chen, Y.; Hele, T.J.H.; Friend, R.H.; Li, F. Efficient radical-based light-emitting diodes with doublet emission. Nature 2018, 563, 536–540. [Google Scholar] [CrossRef]
- Mas-Torrent, M.; Crivillers, N.; Rovira, C.; Veciana, J. Attaching Persistent Organic Free Radicals to Surfaces: How and Why. Chem. Rev. 2012, 112, 2506–2527. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radical | RSE/kJ mol−1 |
|---|---|
| Methyl (•CH3) | 0 (as standard) |
| tert-Butyl | −28.3 |
| Allyl | −77.5 |
| Benzyl | −50.4 |
| Cyclopentadienyl | −98.3 |
| Cycloheptatrienyl | −134.1 |
| Triphenylmethyl (1) | −103.4 |
| Phenalenyl (2) | −201.6 |
| 9-Fluorenyl (7) | −90.7 |
| Compounds | Eox/V | Ered/V |
|---|---|---|
| 11 | +0.28 | −1.11 |
| 12 | −0.10 | −1.76 |
| 13 | −0.30 | −1.78 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubo, T. Synthesis, Physical Properties, and Reactivity of Stable, π-Conjugated, Carbon-Centered Radicals. Molecules 2019, 24, 665. https://doi.org/10.3390/molecules24040665
Kubo T. Synthesis, Physical Properties, and Reactivity of Stable, π-Conjugated, Carbon-Centered Radicals. Molecules. 2019; 24(4):665. https://doi.org/10.3390/molecules24040665
Chicago/Turabian StyleKubo, Takashi. 2019. "Synthesis, Physical Properties, and Reactivity of Stable, π-Conjugated, Carbon-Centered Radicals" Molecules 24, no. 4: 665. https://doi.org/10.3390/molecules24040665
APA StyleKubo, T. (2019). Synthesis, Physical Properties, and Reactivity of Stable, π-Conjugated, Carbon-Centered Radicals. Molecules, 24(4), 665. https://doi.org/10.3390/molecules24040665