
DesMol2, an Effective Tool for the Construction of Molecular Libraries and Its Application to QSAR Using Molecular Topology

 , , ,
, , ,
Abstract
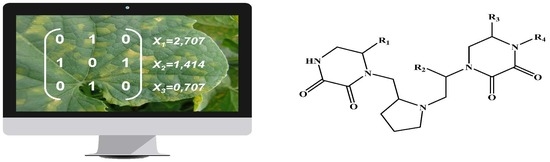
:
1. Introduction
- Using DesMol2, design a complete library of molecules from the SMILES codes of the bases and the substituents defined by Besalú [16].
- Calculate with DesMol2 the topological indices of each of the generated molecules.
- Build the topological model of activity prediction against the FPR receptors.
- Apply the topological model to the library and select the potentially most active molecules.
- Compare the results with those reported in other works.
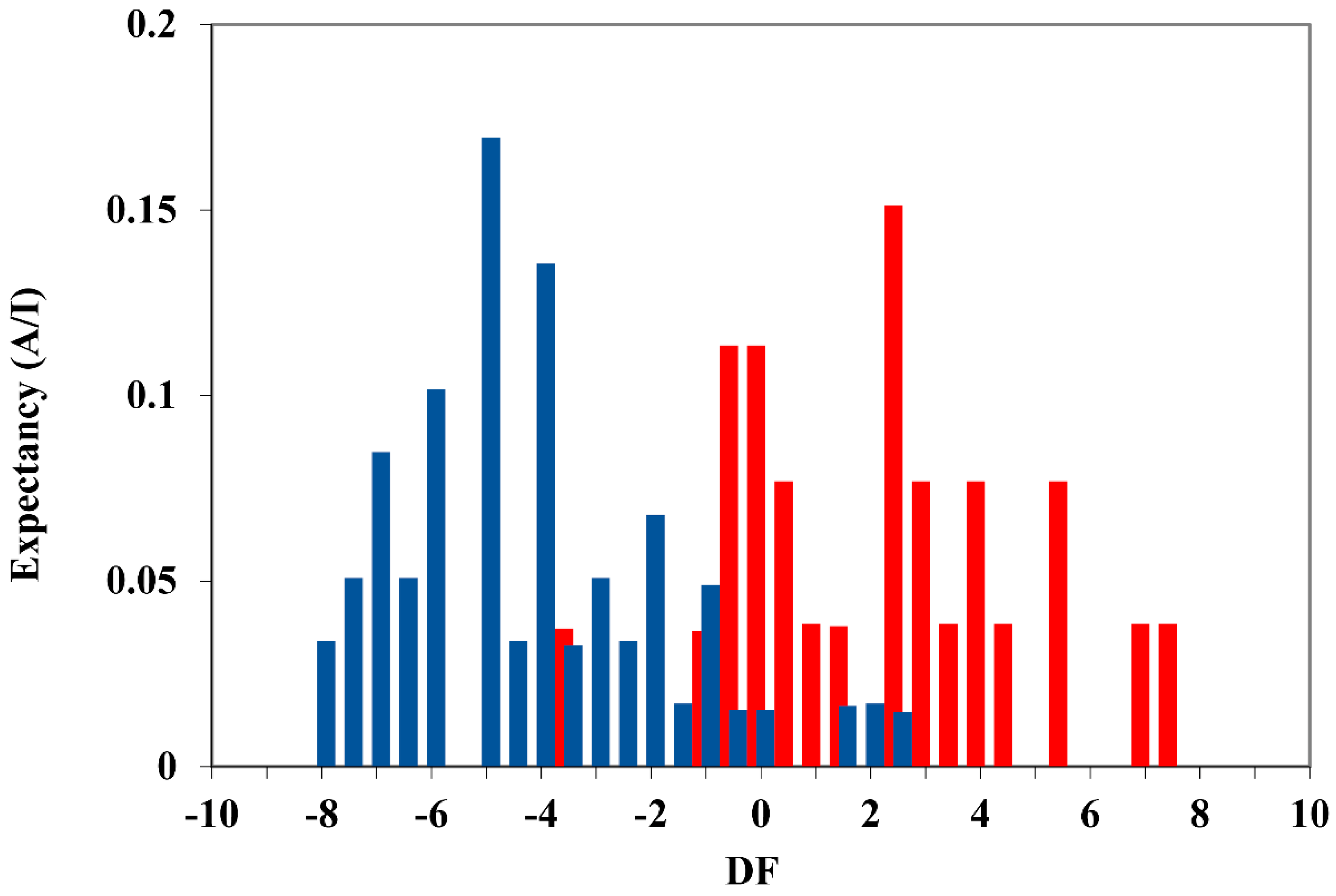
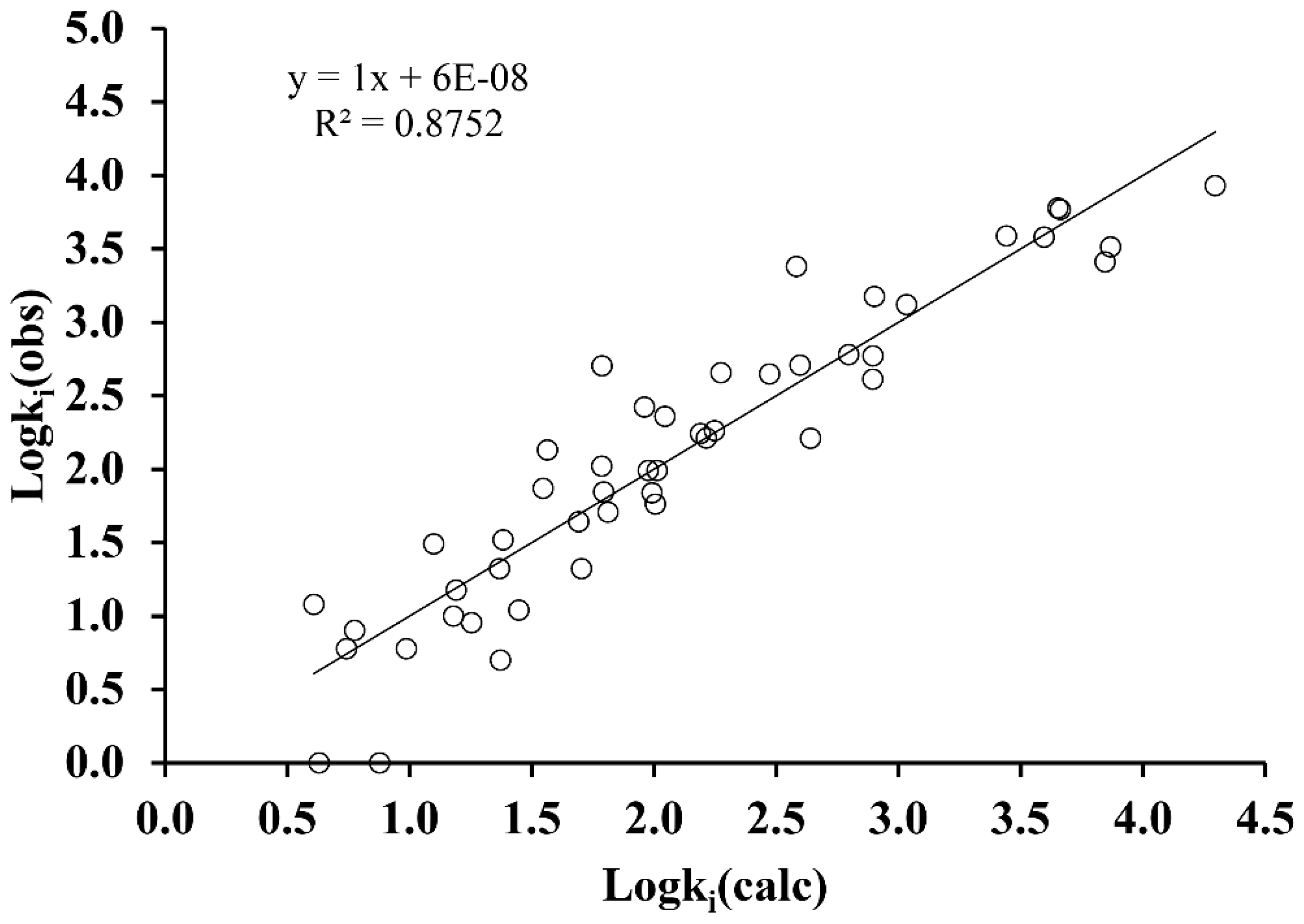
2. Results and Discussion
3. Materials and Methods
3.1. Data Set
3.2. Building of the Virtual Molecular Library
- The application is capable of generating libraries of molecules by combining different bases, and substituents provided by the user.
- You can enter one or more bases and each of them can have one or more anchor points. For each anchor point, zero or more substituents can be entered. The following syntax indicates the anchor points: [R]. For each substituent, the atom that goes at the anchor point is also indicated.
- All possible combinations are made between bases and substituents.
- The user can graphically visualize each molecular structure, be it a base, a substituent or the result of the combination of both.
- The user is informed when an error occurs when entering the information incorrectly.
3.3. Molecular Descriptors
3.4. Development of the QSAR Models
- (a)
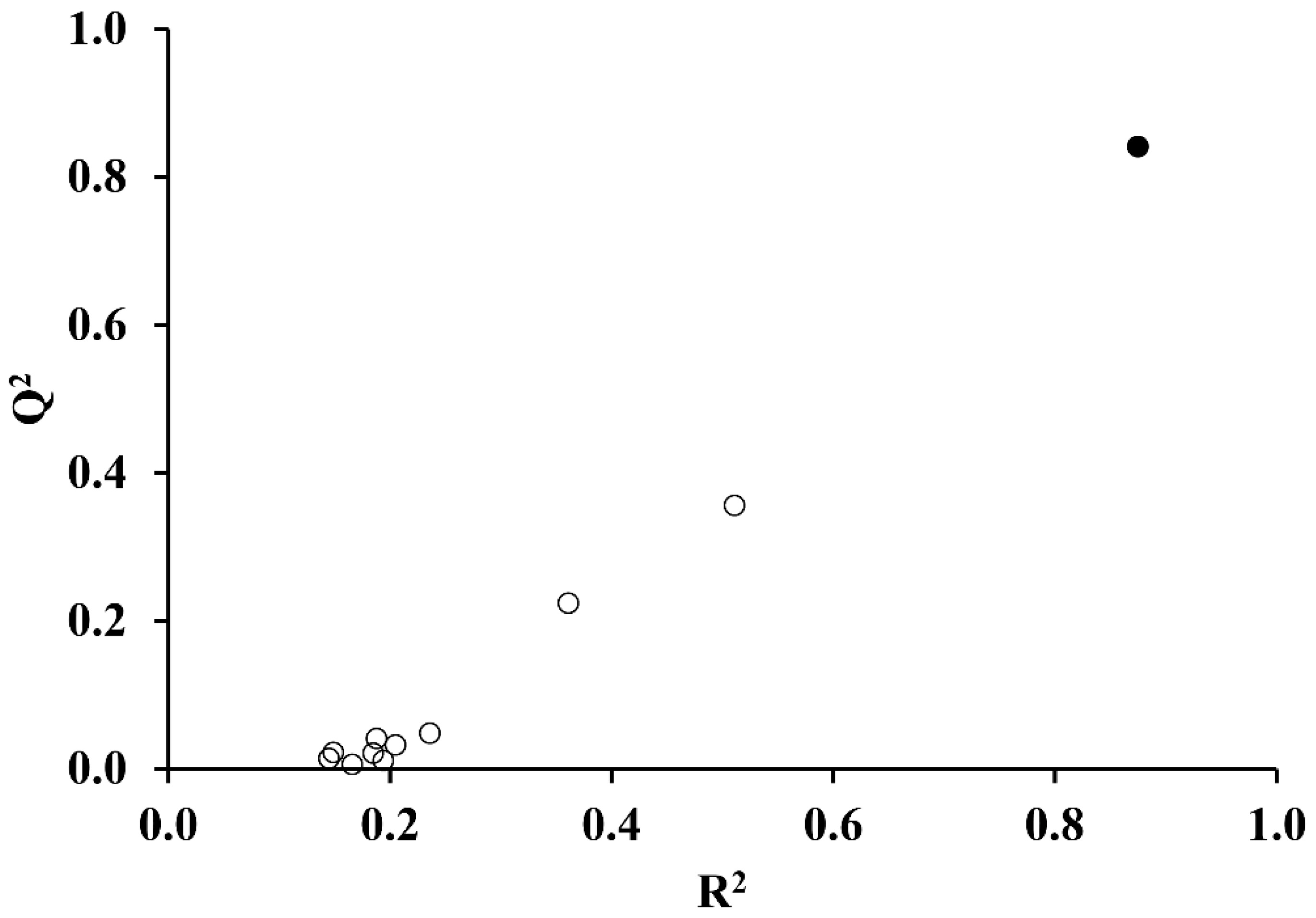
- Internal validation or cross-validation with leave-one-out (LOO) analysis. To do this, one compound of the set is extracted, and the model is recalculated using as a training set the remaining N-1 compounds. The property is then predicted for the removed element. This process is repeated for all the compounds of the set, obtaining a prediction for each of them. From the residual values obtained, the prediction coefficient, R2(cv) or Q2 is determined.
- (b)
- Data randomization or Y-scrambling. A randomization test can be analyzed to identify the possible existence of fortuitous correlations [30]. To do this, the values of the property of each compound are randomly permuted and linearly correlated with the topological descriptors.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chemical Abstracts Service. CAS Registry. The Gold Standard for Chemical Substance Information. Available online: http://www.cas.org/content/chemical-substances (accessed on 16 February 2019).
- Basak, S.C. Role of mathematical chemodescriptors and proteomics based biodescriptors in drug discovery. Drug Dev. Res. 2011, 72, 225–233. [Google Scholar] [CrossRef]
- Galvez, J.; Garcia-Domenech, R. On the contribution of molecular topology to drug design and discovery. Curr. Comput.-Aided Drug Des. 2010, 6, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Zanni, R.; Galvez-Llompart, M.; Morell, C.; Rodríguez-Henche, N.; Díaz-Laviada, I.; Recio-Iglesias, M.C.; Garcia-Domenech, R.; Galvez, J. Novel cancer chemotherapy hits by molecular topology: Dual akt and beta-catenin inhibitors. PLoS ONE 2015, 10, e0124244. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, N.; Garcia-Domenech, R.; Galvez, J.; Farhati, K.; Franetich, J.F.; Sauerwein, R.; Hannoun, L.; Derouin, F.; Danis, M.; Mazier, D. New active drugs against liver stages of Plasmodium predicted by molecular topology. Antimicrob. Agents Chemother. 2008, 52, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Land, D.; Ono, K.; Galvez, J.; Zhao, W.; Vempati, P.; Steele, J.W.; Cheng, A.; Yamada, M.; Levine, S.; et al. Molecular topology as novel strategy for discovery of drugs with ab lowering and anti-aggregation dual activities for Alzheimer’s disease. PLoS ONE 2014, 9, e92750. [Google Scholar]
- Garcia-Domenech, R.; Aguilera, J.; El Moncef, A.; Pocovi, S.; Galvez, J. Application of molecular topology to the prediction of mosquito repellents of a group of terpenoid compounds. Mol. Divers. 2010, 14, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Galvez, J.; Galvez-Llompart, M.; Garcia-Domenech, R. Application of molecular topology for the prediction of the reaction times and yields under solvent-free conditions. Green Chem. 2010, 12, 1056–1061. [Google Scholar] [CrossRef]
- Blay, V.; Gullon-Soleto, J.; Galvez-Llompart, M.; Galvez, J.; Garcia-Domenech, R. Biodegradability Prediction of Fragrant Molecules by Molecular Topology. ACS Sustain. Chem. Eng. 2016, 4, 4224–4231. [Google Scholar]
- SmiLib v2.0. Available online: http://melolab.org/smilib/ (accessed on 16 February 2019).
- ChemT. Available online: http://www.esa.ipb.pt/biochemcore/index.php/ds/c (accessed on 16 February 2019).
- Library Synthesizer. Available online: https://tripod.nih.gov/?p=370 (accessed on 16 February 2019).
- Virtual Combinatorial Library of cheminfo.org. Available online: http://www.cheminfo.org/Chemistry/Cheminformatics/Virtual_combinatorial_library/index.html (accessed on 16 February 2019).
- Combinatorial Library Design of e-LEA3D. Available online: http://chemoinfo.ipmc.cnrs.fr/eDESIGN/reagent.html (accessed on 16 February 2019).
- DesMol2 Software. Molecular Connectivity and Drug Design Research Unit. Department of Physical Chemistry. Faculty of Pharmacy. University of Valencia. Available online: http://desmol2.uv.es (accessed on 16 February 2019).
- Besalú, E. Fast Modeling of Binding Affinities by Means of Superposing Significant Interaction Rules (SSIR) Method. Int. J. Mol. Sci. 2016, 17, 827. [Google Scholar] [CrossRef] [PubMed]
- Galvez, J.; Garcia-Domenech, R.; de Gregorio Alapont, C.; de Julian-Ortiz, J.; Popa, L. Pharmacological distribution diagrams: A tool for de novo drug design. J. Mol. Graph. 1996, 14, 272–276. [Google Scholar] [CrossRef]
- Dragon for Windows (Software for Molecular Descriptor Calculations), version 5.4; Talete srl: Milan, Italy, 2006.
- Klein, D.J.; Lukovits, I.; Gutman, I. On the definition of the hyper–Wiener index for cycle–containing structures. J. Chem. Inf. Comput. Sci. 1995, 35, 50–52. [Google Scholar] [CrossRef]
- Kier, L.B.; Hall, L.H.; Frazer, J.W. An index of electrotopological state for atoms in molecules. J. Math. Chem. 1991, 7, 229–241. [Google Scholar] [CrossRef]
- Gramatica, P.; Corradi, M.; Consonni, V. Modelling and Prediction of Soil SorptionCoefficients of Non-ionic Organic Pesticides by Molecular Descriptors. Chemosphere 2000, 41, 763–777. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors, Methods and Principles in Medicinal Chemistry Series; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; Volume 41, pp. 719–721. [Google Scholar]
- Galvez, J.; Garcia-Domenech, R.; Salabert, M.T.; Soler, R. Charge indexes. New topological descriptors. J. Chem. Inf. Comput. Sci. 1994, 34, 520–525. [Google Scholar] [CrossRef]
- PaDEL-Descriptor. Available online: http://www.yapcwsoft.com/dd/padeldescriptor/ (accessed on 16 February 2019).
- Kier, L.B.; Hall, L.H. Molecular Connectivity in Structure-Activity Analysis; Research Studies Press: Letchworth, UK, 1986. [Google Scholar]
- Galvez, J.; Garcia-Domenech, R.; de Julian-Ortiz, J.V.; Soler, R. Topological Approach to Drug Design. J. Chem. Inf. Comput. Sci. 1995, 35, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Wiener, H. Structural determination of paraffin boiling points. J. Am. Chem. Soc. 1947, 69, 17–20. [Google Scholar] [CrossRef] [PubMed]
- StatSoft. Statistica (Data Analysis Software System), version 9; StatSoft: Tulsa, OK, USA, 2009. [Google Scholar]
- Roy, P.P.; Leonard, J.T.; Roy, K. Exploring the impact of size of training sets for the development of predictive QSAR models. Chemom. Intell. Lab. Syst. 2008, 90, 31–42. [Google Scholar] [CrossRef]
- Wold, S.; Eriksson, L.; Clementi, S. Statistical validation of QSAR results. In Chemometric Methods in Molecular Design; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1995; pp. 309–338. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

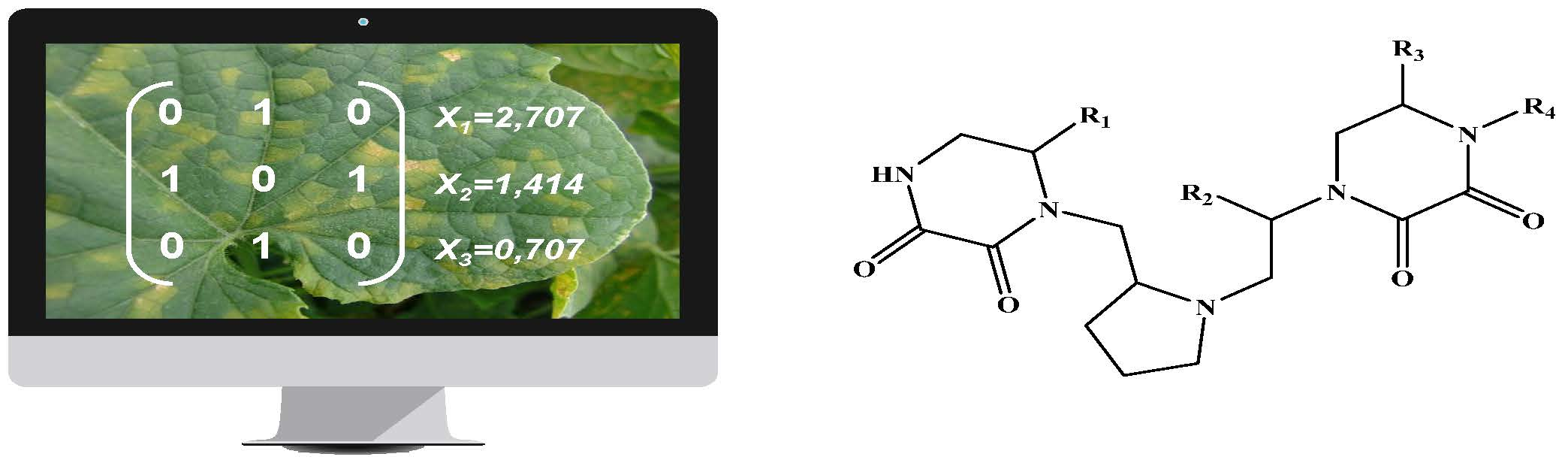
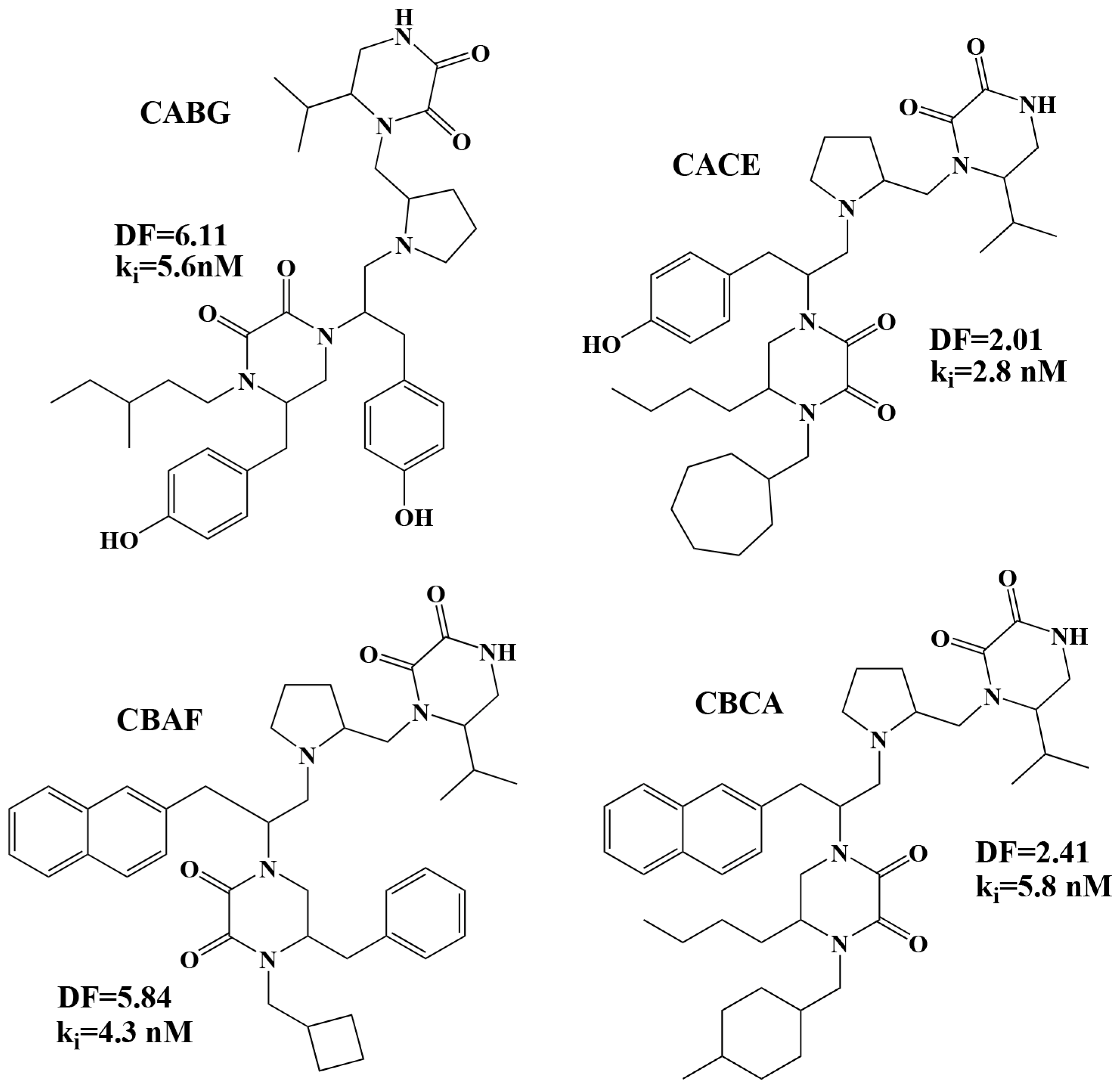
| Code | R1 | R2 | R3 | R4 |
|---|---|---|---|---|
| A | 2-naphthylmethyl | 4-hydroxybenzyl | benzyl | 4-Methyl-1-cyclohexyl-methyl |
| [R]C1=CC2=CC=CC=C2C=C1 | OC1=CC=C(C=C1)[R] | [R]C1=CC=CC=C1 | [R]C1CCC(C)CC1 | |
| B | propyl | 2-naphthylmethyl | 4-hydroxybenzyl | Cyclohexylpropyl |
| CC[R] | [R]C1=CC2=CC=CC=C2C=C1 | OC1=CC=C(C=C1)[R] | [R]CCC1CCCCC1 | |
| C | isopropyl | cyclohexyl | butyl | Cyclohexylmethyl |
| [R](C)(C) | C1CCCCC1[R] | [R]CCC | [R]C1CCCCC1 | |
| D | butyl | propyl | propyl | Cyclopentylmethyl |
| [R]CCC | CC[R] | CC[R] | [R]C1CCCC1 | |
| E | benzyl | hydroxymethyl | S-phenyl | Cycloheptylmethyl |
| [R]C1=CC=CC=C1 | O[R] | [R]1=CC=CC=C1 | [R]C1CCCCCC1 | |
| F | butyl | butyl | Cyclobutylmethyl | |
| [R]CCC | [R]CCC | [R]C1CCC1 | ||
| G | benzyl | cyclohexyl | 3-Methylpentyl | |
| [R]C1=CC=CC=C1 | C1CCCCC1[R] | [R]CC(C)CC | ||
| H | isobutyl | benzyl | 2-Biphenyl-4-yl-ethyl | |
| CC(C)[R] | [R]C1=CC=CC=C1 | [R]CC(C=C1)=CC=C1C2=CC=CC=C2 | ||
| I | propyl | 4-Tert-butyl-cyclohexylmethyl | ||
| CC[R] | [R]C1CCC(C(C)(C)C)CC1 | |||
| J | 2-(3-Methoxyphenyl)-ethyl | |||
| [R]CC1=CC(OC)=CC=C1 | ||||
| K | 2-(4-Isobutylphenyl)-propyl | |||
| [R]C(C1=CC=C(CC(C)C)C=C1)C | ||||
| L | m-Tolylethyl | |||
| [R]CC1=CC(C)=CC=C1 | ||||
| M | p-Tolylethyl | |||
| [R]CC1=CC=C(C)C=C1 | ||||
| N | 2-(4-Methoxyphenyl)-ethyl | |||
| [R]CC1=CC=C(OC)C=C1 | ||||
| O | 2-(4-Ethoxyphenyl)-ethyl | |||
| [R]CC1=CC=C(OCC)C=C1 | ||||
| P | Phenethyl | |||
| [R]CC1=CC=CC=C1 | ||||
| Q | 3-(3,4-Dimethoxyphenyl)-propyl | |||
| [R]CCC1=CC=C(OC)C(OC)=C1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Pereira, I.; Zanni, R.; Galvez-Llompart, M.; Galvez, J.; García-Domenech, R. DesMol2, an Effective Tool for the Construction of Molecular Libraries and Its Application to QSAR Using Molecular Topology. Molecules 2019, 24, 736. https://doi.org/10.3390/molecules24040736
García-Pereira I, Zanni R, Galvez-Llompart M, Galvez J, García-Domenech R. DesMol2, an Effective Tool for the Construction of Molecular Libraries and Its Application to QSAR Using Molecular Topology. Molecules. 2019; 24(4):736. https://doi.org/10.3390/molecules24040736
Chicago/Turabian StyleGarcía-Pereira, Inma, Riccardo Zanni, Maria Galvez-Llompart, Jorge Galvez, and Ramón García-Domenech. 2019. "DesMol2, an Effective Tool for the Construction of Molecular Libraries and Its Application to QSAR Using Molecular Topology" Molecules 24, no. 4: 736. https://doi.org/10.3390/molecules24040736
APA StyleGarcía-Pereira, I., Zanni, R., Galvez-Llompart, M., Galvez, J., & García-Domenech, R. (2019). DesMol2, an Effective Tool for the Construction of Molecular Libraries and Its Application to QSAR Using Molecular Topology. Molecules, 24(4), 736. https://doi.org/10.3390/molecules24040736