
Synthesis and Biological Evaluation of Novel Thiazolyl-Coumarin Derivatives as Potent Histone Deacetylase Inhibitors with Antifibrotic Activity

Abstract
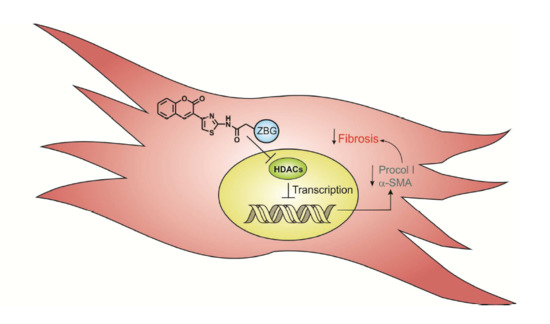
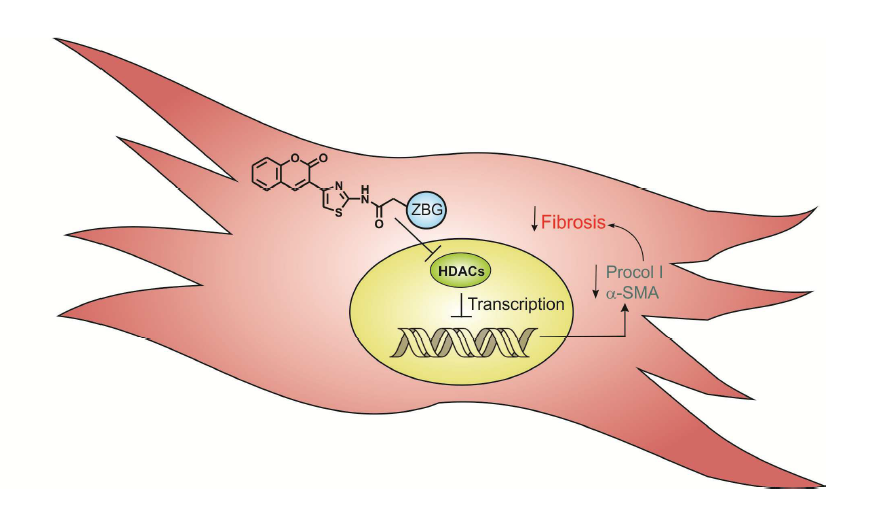
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
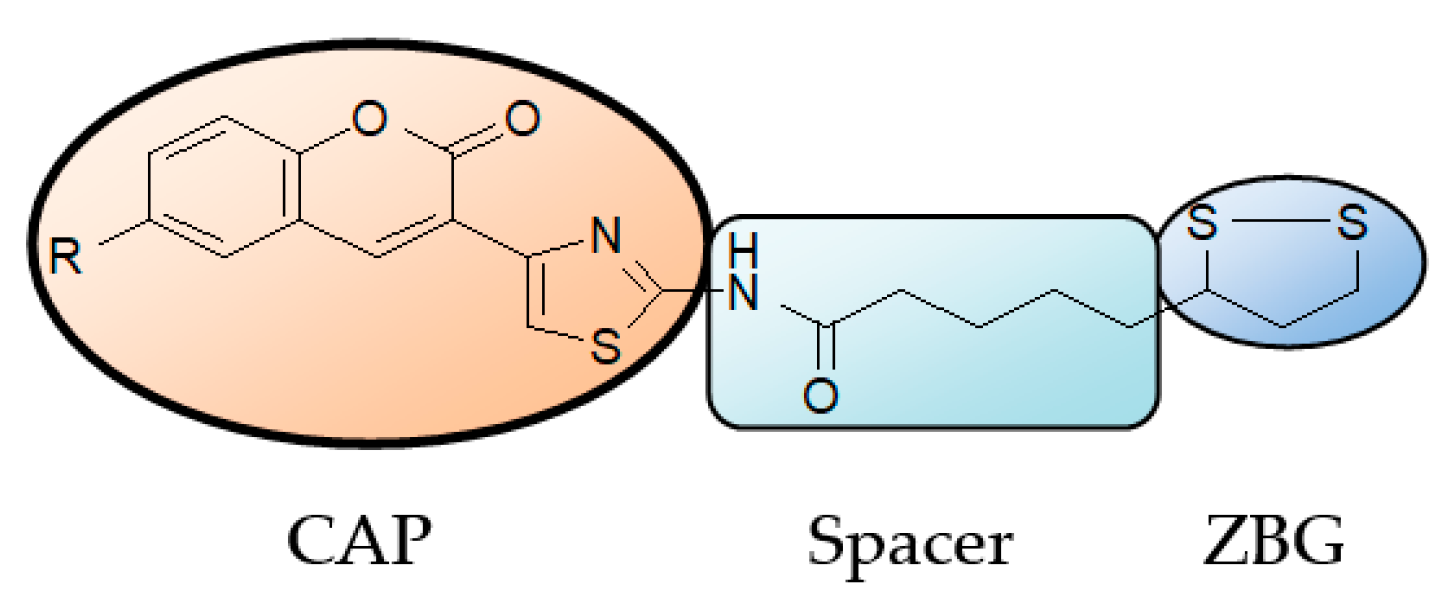
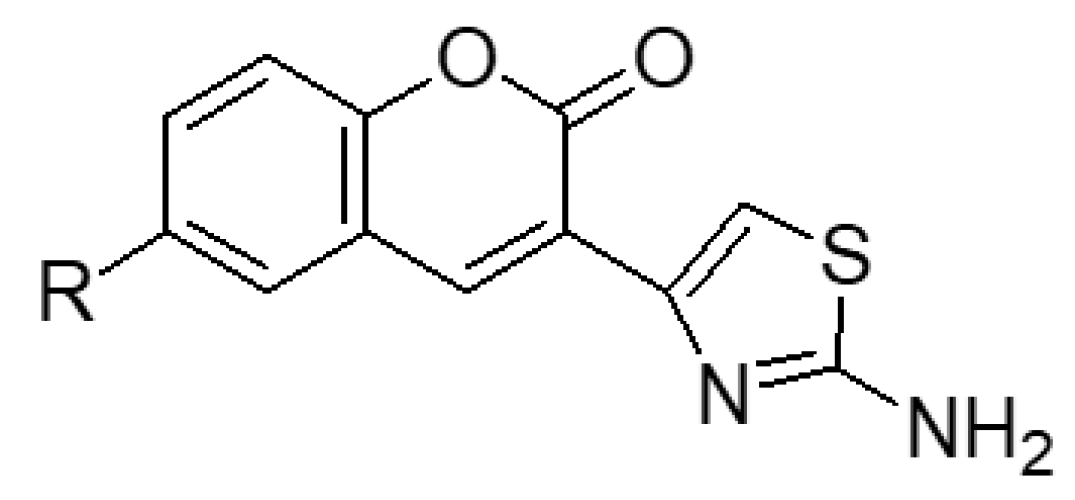
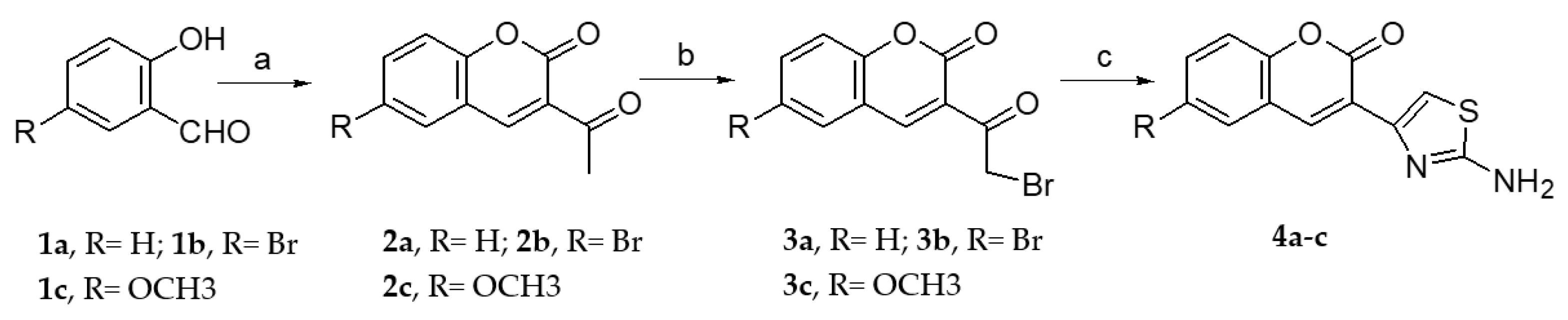
2.1.1. Scaffold CAP Synthesis
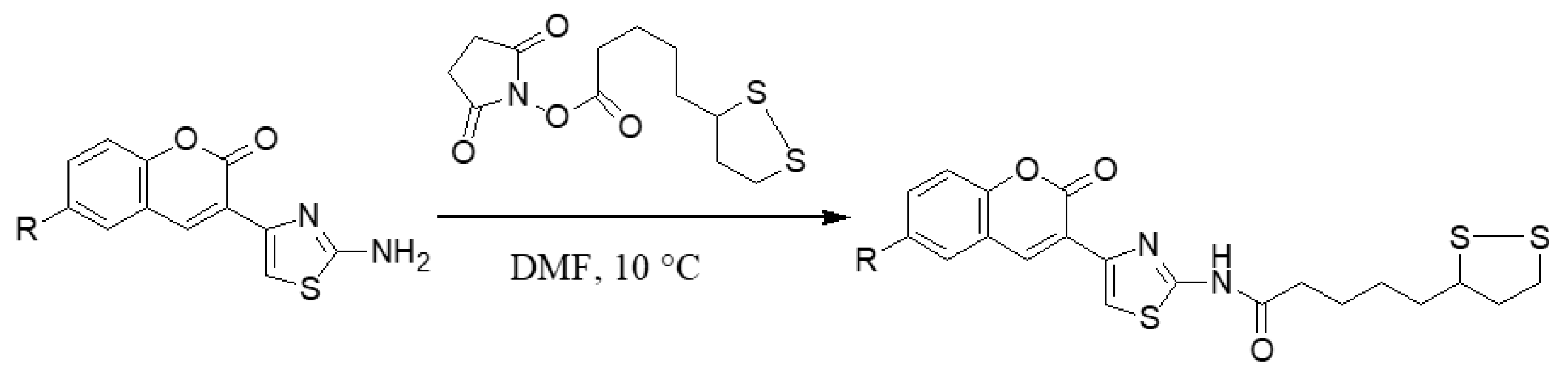
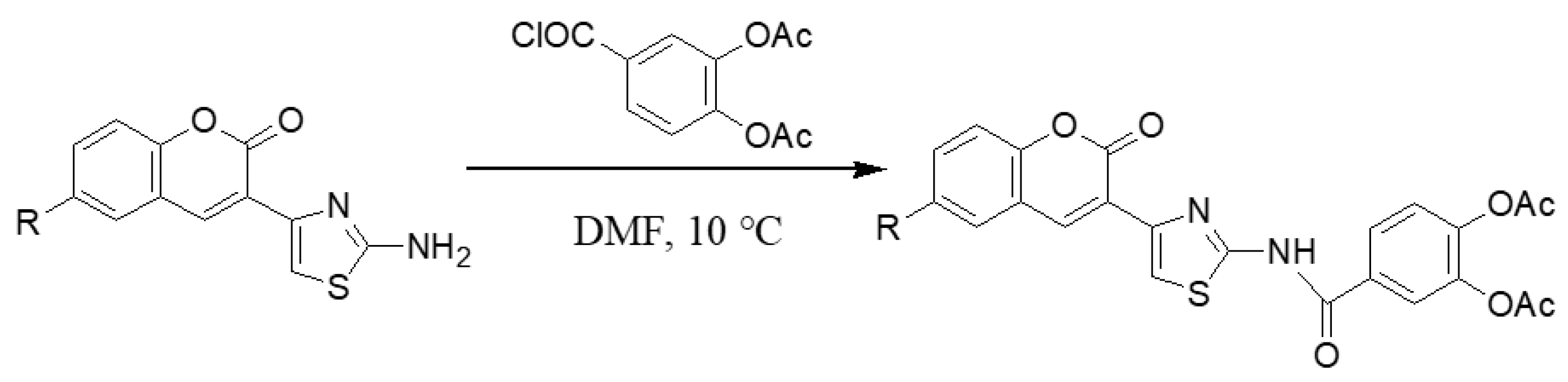
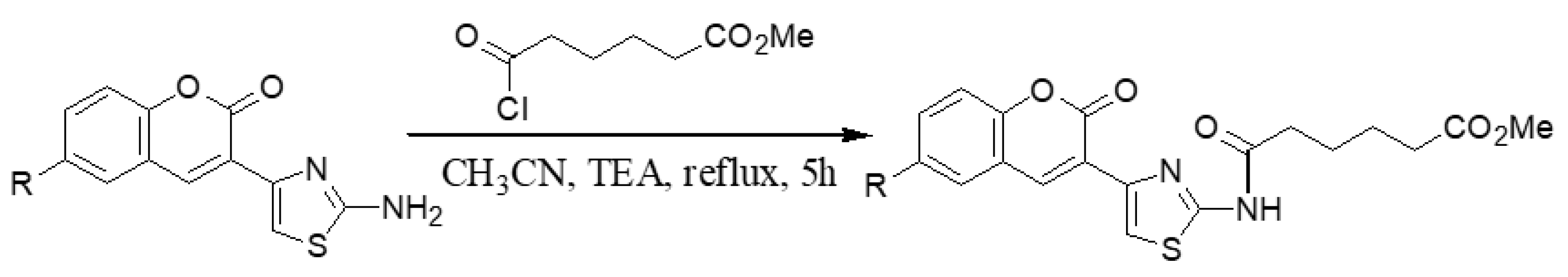
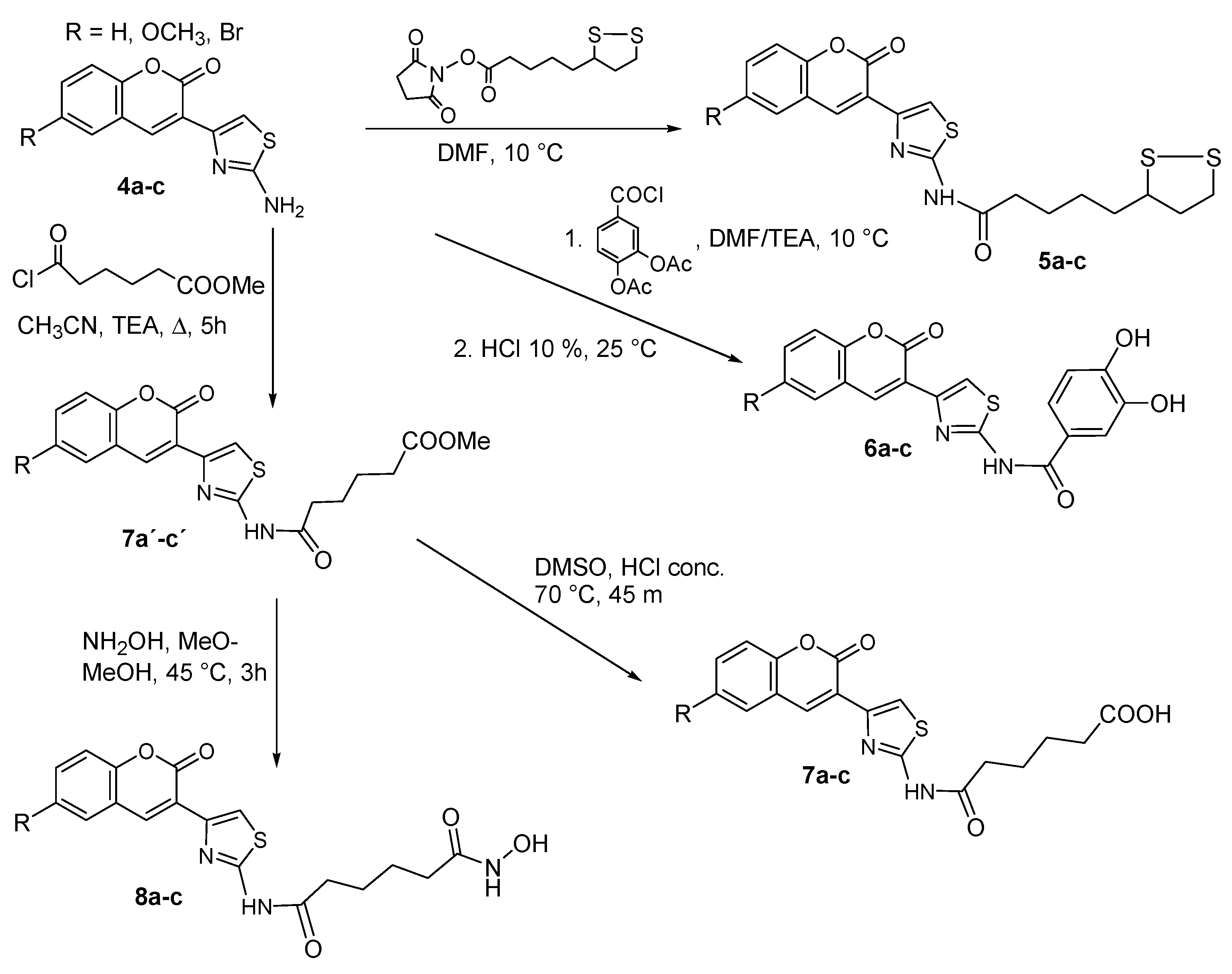
2.1.2. Adding the Spacer and the ZBG
2.2. HDACs Enzyme Inhibition
2.3. Cellular Assays
2.3.1. Cytotoxicity Assay
2.3.2. Cell Proliferation
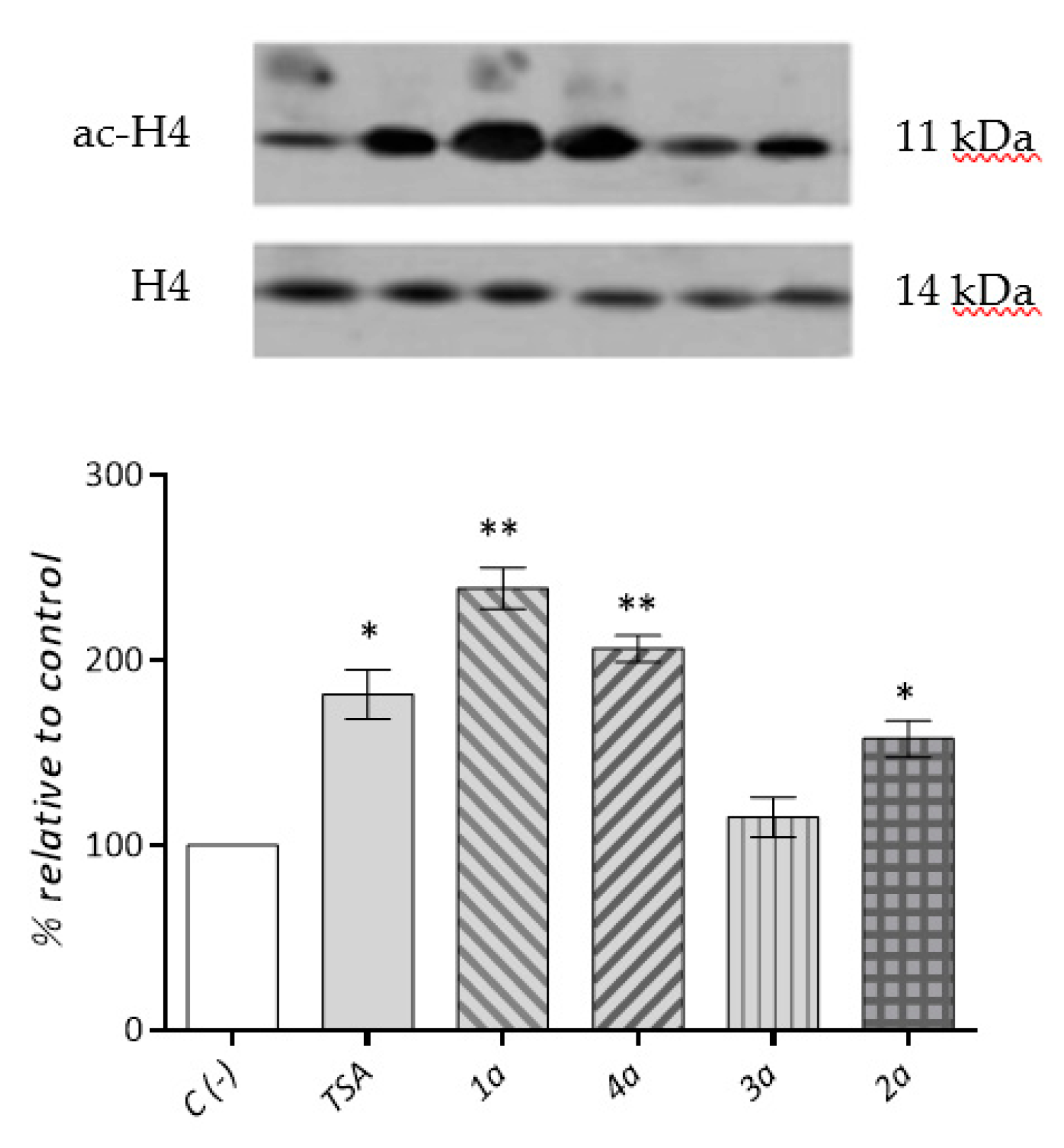
2.3.3. Cardiac Fibroblast HDACs Inhibition
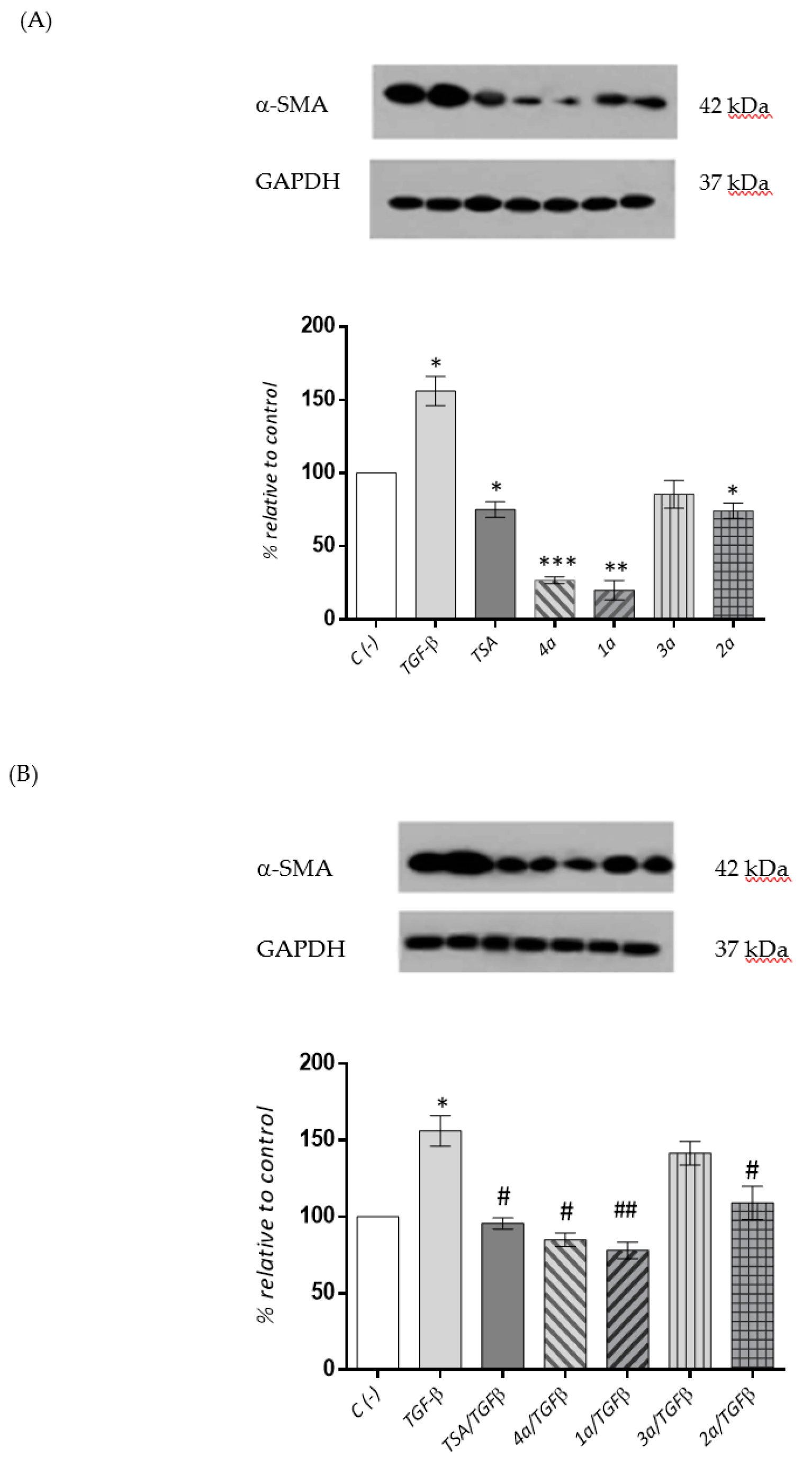
2.3.4. Cardiac Fibroblast α-SMA Expression Levels
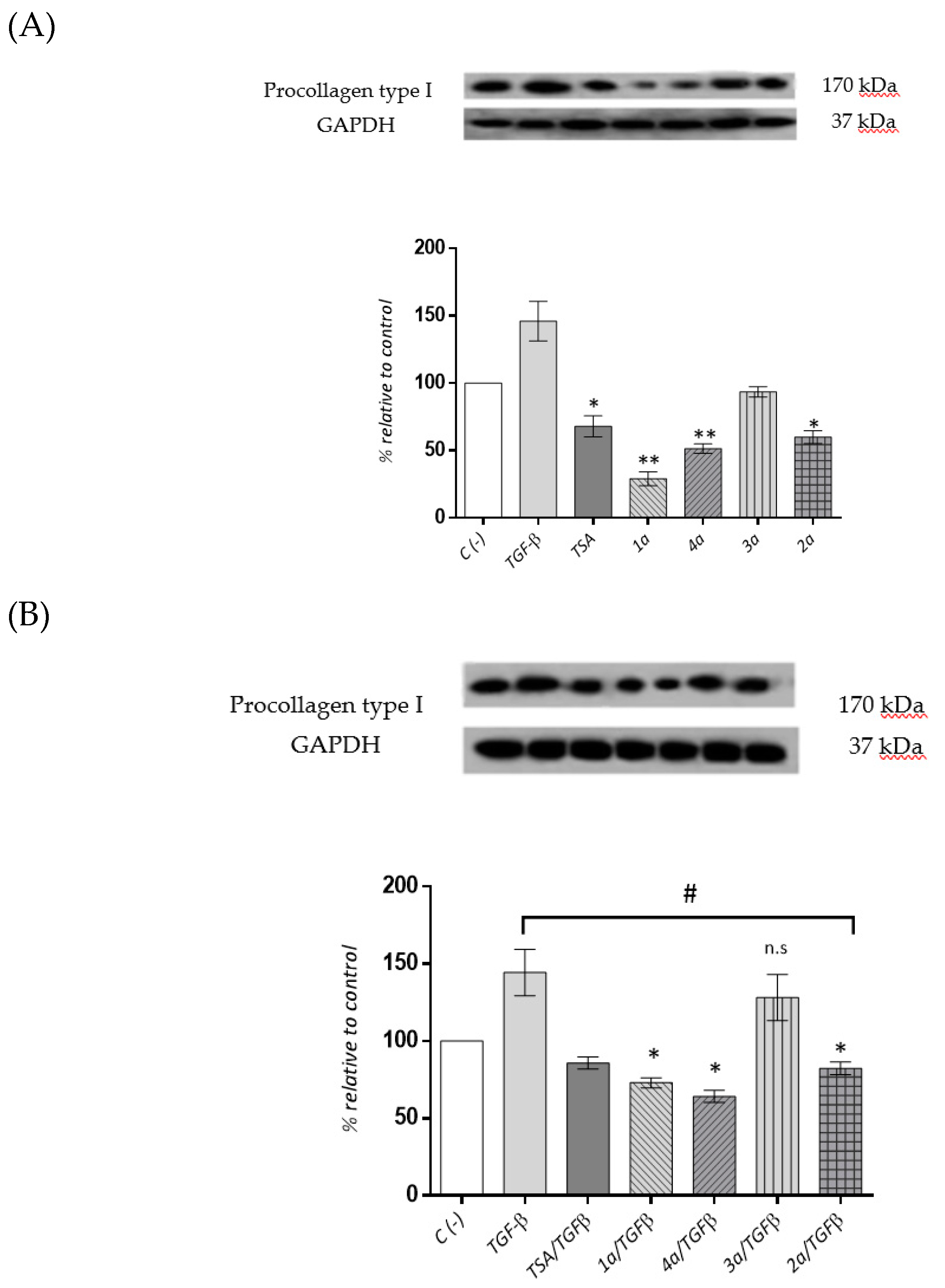
2.3.5. Cardiac Fibroblast Procollagen Type I Expression Levels
2.4. ADMET Analysis
3. Experimental Section
3.1. Chemistry
3.1.1. General Information
3.1.2. General Synthesis of 6-substituted-3-acetylcoumarins 2a–c
3.1.3. General Synthesis of 3-bromoacetylcoumarins 3a–c
3.1.4. General Synthesis of 6-substituted-3-(2-amino-thiazol-4-yl)-coumarins 4a–c
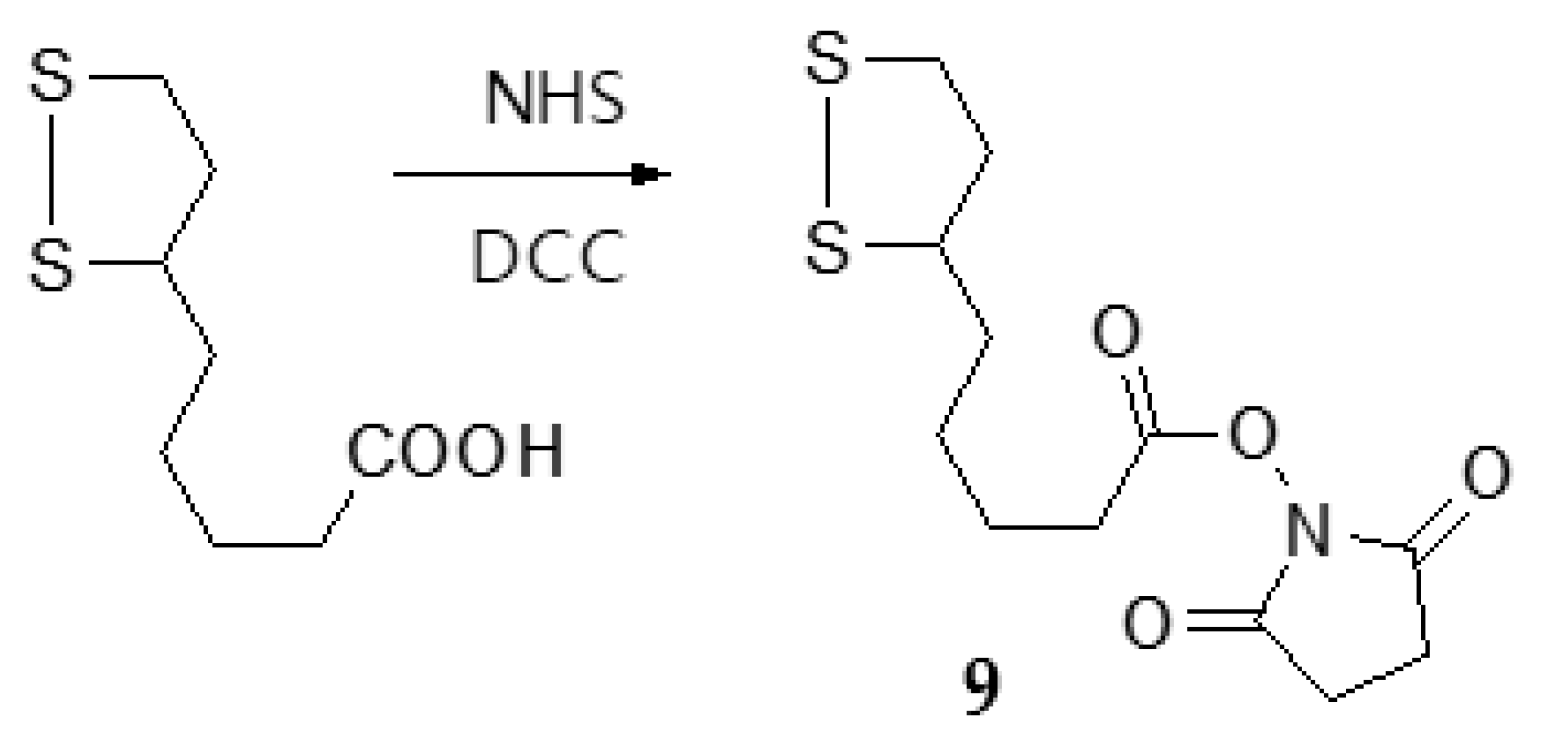
3.1.5. Synthesis of N-hydroxysuccinimide (NHS)-lipoate (9)
3.1.6. General Synthesis of 6-substituted-5-[1,2]-dithiolan-3-yl-pentanoic acid [4-(2-oxo-2H-chromen-3-yl)-thiazol-2-yl]-amides 5a–c
3.1.7. General Synthesis of 6-substituted-N-[4-(2-oxo-2H-chromen-3-yl)-thiazol-2-yl]-3,4-dihydroxy-acetylbenzamide Coumarins 6a′–c′
3.1.8. General Synthesis of 6-substituted N-[4-(2-oxo-2H-chromen-3-yl)-thiazol-2-yl]-3,4-dihydroxy-benzamide Coumarins 6a–c
3.1.9. General Synthesis of 6-substituted methyl 5-[4-(2-oxo-2H-chromen-3-yl)-thiazol-2-yl-carbamoyl]-pentanoate Coumarins 7a′–c′
3.1.10. General synthesis of 6-substituted5-[4-(2-oxo-2H-chromen-3-yl)-thiazol-2-yl-carbamoyl]-pentanoicacidcoumarins 7a–c
3.1.11. General synthesis of 6-substituted-5-[4-(2-oxo-2H-chromen-3-yl)-thiazol-2-yl-carbamoyl] –pentanoate hydroxamic acid derivatives 8a–c
3.1.12. Physical Characterization of the Main Precursors and Final Synthesized Compounds
3.2. Biology
3.2.1. Reagents
3.2.2. Cell Culture.
3.2.3. Cytotoxicity and Cell Proliferation Assay
3.2.4. HDACs Inhibitory Activity Assay
3.2.5. Preparation of the Cell Lysate
3.2.6. Western Blot Analysis
3.2.7. Prediction of ADMET Properties
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Weber, K.T.; Sun, Y.; Díez, J. Fibrosis: A living tissue and the infarcted heart. J. Am. Coll. Cardiol. 2008, 52, 2029–2031. [Google Scholar] [CrossRef] [PubMed]
- Gourdie, R.G.; Dimmeler, S.; Kohl, P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat. Rev. Drug Discov. 2016, 15, 620–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuetze, K.B.; McKinsey, T.A.; Long, C.S. Targeting cardiac fibroblasts to treat fibrosis of the heart: Focus on HDACs. J. Mol. Cell. Cardiol. 2014, 70, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, H.; Shi, K.H.; Yang, J.J.; Huang, C.; Zhan, H.Y.; Li, J. Histone deacetylases in cardiac fibrosis: Current perspectives for therapy. Cell. Signal. 2014, 26, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Hill, J.A. HDAC-dependent ventricular remodeling. Trends Cardiovasc. Med. 2013, 23, 229–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiagalingam, S.; Cheng, K.H.; Lee, H.J.; Mineva, N.; Thiagalingam, A.; Ponte, J.F. Histone deacetylases: Unique players in shaping the epigenetic histone code. Ann. N. Y. Acad. Sci. 2003, 983, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Sadoul, K.; Boyault, C.; Pabion, M.; Khochbin, S. Regulation of protein turnover by acetyltransferases and deacetylases. Biochimie 2008, 90, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Krame, O.H. Acetylation of non-histone proteins modulates cellular signaling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A. Isoform-selective HDAC inhibitors: Closing in on translational medicine for the heart. J. Mol. Cell. Cardiol. 2011, 51, 491–496. [Google Scholar] [CrossRef] [PubMed]
- McKinsey, T.A. Therapeutic potential for HDAC inhibitors in the heart. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 303–319. [Google Scholar] [CrossRef]
- Tang, J.; Yan, H.; Zhuang, S. Histone deacetylases as targets for treatment of multiple diseases. Clin. Sci. 2013, 124, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, E.W.; McKinsey, T.A. Protein acetylation in the cardiorenal axis: The promise of histone deacetylase inhibitors. Circ. Res. 2010, 106, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Nural-Guvener, H.F.; Zakharova, L.; Nimlos, J.; Popovic, S.; Mastroeni, D.; Gaballa, M.A. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair 2014, 7, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.M.; Golden-Mason, L.; Ferguson, B.S.; Schuetze, K.B.; Cavasin, M.A.; Demos-Davies, K.; Yeager, M.E.; Stenmark, K.R.; McKinsey, T.A. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J. Mol. Cell. Cardiol. 2014, 67, 112–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratton, M.S.; McKinsey, T.A. Epigenetic regulation of cardiac fibrosis. J. Mol. Cell. Cardiol. 2016, 92, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Monneret, C. Histone deacetylase inhibitors. Eur. J. Med. Chem. 2005, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P. Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem. 2010, 45, 2095–2116. [Google Scholar] [CrossRef]
- DellaGreca, M.; Fiorentino, A.; Isidori, M.; Previtera, L.; Temussi, F.; Zarrelli, A. Benzocoumarins from the rhizomes of Juncus acutus. Tetrahedron 2003, 59, 4821–4825. [Google Scholar] [CrossRef]
- Iaroshenko, V.O.; Ali, S.; Babar, T.M.; Dudkin, S.; Mkrtchyan, S.; Rama, N.H.; Villinger, A.; Langer, P. 4-Chloro-3-(trifluoroacetyl)coumarin as a novel building block for the synthesis of 7-(trifluoromethyl)-6H-chromeno[4,3-b]quinolin-6-ones. Tetrahedron Lett. 2011, 52, 3373–3376. [Google Scholar] [CrossRef]
- Temitope Olomola, O.; Klein, R.; Kevin Lobb, A.; Sayed, Y.; Kaye Perry, T. Towards the synthesis of coumarin derivatives as potential dual-action HIV-1 protease and reverse transcriptase inhibitors. Tetrahedron Lett. 2010, 51, 6325–6328. [Google Scholar] [CrossRef]
- Huang, W.J.; Chen, C.C.; Chao, S.W.; Yu, C.C.; Yang, C.Y.; Guh, J.H.; Lin, Y.C.; Kuo, C.I.; Yang, P.; Chang, C.I. Synthesis and evaluation of aliphatic-chain hydroxamates capped with osthole derivatives as histone deacetylase inhibitors. Eur. J. Med. Chem. 2011, 46, 4042–4049. [Google Scholar] [CrossRef] [PubMed]
- Abdizadeh, T.; Kalani, M.R.; Abnous, K.; Tayarani-Najaran, Z.; Khashyarmanesh, B.Z.; Abdizadeh, R.; Ghodsi, R.; Hadizadeh, F. Design, synthesis and biological evaluation of novel coumarin-based benzamides as potent histone deacetylase inhibitors and anticancer agents. Eur. J. Med. Chem. 2017, 132, 42–62. [Google Scholar] [CrossRef]
- Kumar, P.V.; Reddy, K.M.; Rao, V.R. Synthesis of Some 7-Methyl3-(2-oxo-2H-chromen-3-yl)-5H-[1,3]thiazolo-[3,2-a]pyrimidin-5-ones. Indian, J. Chem. Sect. B 2008, 47, 759–763. [Google Scholar] [CrossRef]
- Siddiqui, N.; Arshad, M.; Khan, S.A. Synthesis of some new coumarin incorporated thiazolylsemicarbazones as anticonvulsants. Acta Pol. Pharm. Drug Res. 2009, 66, 161–167. [Google Scholar]
- Arshad, A.; Osman, H.; Bagley, M.C.; Lam, C.K.; Mohamad, S.; Zahariluddin, A.S. Synthesis and antimicrobial properties of some new thiazolyl-coumarin derivatives. Eur. J. Med. Chem. 2011, 46, 3788–3794. [Google Scholar] [CrossRef]
- Kalkhambkar, R.; Kulkarni, G.; Shivkumar, H.; Rao, R. Synthesis of novel tri heterocyclic thiazoles as anti-inflammatory and analgesic agents. Eur. J. Med. Chem. 2007, 42, 1272–1276. [Google Scholar] [CrossRef]
- Kamal, A.; Adil, S.; Tamboli, J.; Siddardha, B.; Murthy, U. Synthesis of coumarin linked naphthalimide conjugates as potential anticancer and antimicrobial agents. Lett. Drug Des. Discov. 2009, 6, 201–209. [Google Scholar] [CrossRef]
- Suhaily, F.; Rahman, A.; Yusufzai, S.K.; Osman, H.; Mohamad, D. Synthesis, Characterisation and Cytotoxicity Activity of Thiazole Substitution of Coumarin Derivatives. J. Phys. Sci. 2016, 27, 77–87. [Google Scholar]
- Mohareb, R.M.; Megally Abdo, N.Y. Synthesis and Cytotoxic Evaluation of Pyran, Dihydropyridine and Thiophene Derivatives of 3-Acetylcoumarin. Chem. Pharm. Bull. 2015, 63, 678–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, M.; Zhuang, S. Histone Deacetylase: A Potential Therapeutic Target for Fibrotic Disorders. J. Pharmacol. Exp. Ther. 2010, 335, 266–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulow, R.; Fitzner, B.; Sparmann, G.; Emmrich, J.; Liebe, S.; Jaster, R. Antifibrogenic effects of histone deacetylase inhibitors on pancreatic stellate cells. Biochem. Pharmacol. 2007, 74, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Hantzsch, A.; Weber, J.H. Ueber Verbindungen des Thiazols (Pyridins der Thiophenreihe). Ber. Dtsch. Chem. Ges. 1887, 20, 3118–3132. [Google Scholar] [CrossRef]
- Nichols, T.W. alpha-lipoic acid: Biological effects and Clinical Implications. Alt. Med. Rev. 1997, 2, 177–183. [Google Scholar]
- Islam, M.T. Antioxidant activities of dithiol alpha-lipoic acid. Bangladesh J. Med. Sci. 2009, 8, 46–51. [Google Scholar] [CrossRef]
- Goraca, A.; Huk-Kolega, H.; Piechota, A.; Kleniewska, P.; Skibska, B. Lipoic acid—Biological activity and therapeutic potential. Pharmacol. Rep. 2011, 63, 849–858. [Google Scholar] [CrossRef]
- Jia, L.; Zhang, Z.; Zhai, L.; Bai, Y. Prottective effect of lipoic acid against acrolein-induced cytotoxicity in IMR-90 human fibroblast. J. Nutr. Sci. Vitaminol. 2009, 55, 126–130. [Google Scholar] [CrossRef]
- Leppert, U.; Gillespie, A.; Orphal, M.; Böhme, K.; Plum, C.; Nagorsen, K.; Berkholz, J.; Kreutz, R.; Eisenreich, A. The impact of α-Lipoic acid on cell viability and expression of nephrin and ZNF580 in normal human podocytes. Eur. J. Pharmacol. 2017, 810, 1–8. [Google Scholar] [CrossRef]
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Hagen, T.M. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta 2009, 1790, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, X.; Chen, D.; Chen, S. Antioxidant Activity and Mechanism of Protocatechuic Acid in vitro. Funct. Foods Health Dis. 2011, 7, 232–244. [Google Scholar] [CrossRef]
- Masoud, M.S.; Hagagg, S.S.; Ali, A.E.; Nasr, N.M. Synthesis and spectroscopic characterization of gallic acid and some of its azo complexes. J. Mol. Struct. 2012, 1014, 17–25. [Google Scholar] [CrossRef]
- Schweigert, N.; Zehnder, A.J.; Eggen, R.I. Chemical properties of catechols and their molecular modes of toxic action in cells, from microorganisms to mammals. Environ. Microbiol. 2001, 3, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, F.E.; Lewis, J.A.; Cohen, S.M. The Design of Inhibitors for Medicinally Relevant Metalloproteins. Chem. Med. Chem. 2007, 2, 152–171. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone Deacetylase Inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef]
- Elaut, G.; Laus, G.; Alexandre, E.; Richert, L.; Bachellier, P.; Tourwe, D.; Rogiers, V.; Vanhaecke, T. A Metabolic Screening Study of Trichostatin A (TSA) and TSA Like Histone Deacetylase Inhibitors in Rat and Human Primary Hepatocyte Cultures. J. Pharmacol. Exp. Ther. 2007, 321, 400–408. [Google Scholar] [CrossRef]
- Tan, S.; Liu, Z.P. Natural products as zinc-dependent histone deacetylase inhibitors. Chem. Med. Chem. 2015, 10, 441–450. [Google Scholar] [CrossRef]
- Shen, S.; Kozikowski, A.P. Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors--What Some May Have Forgotten or Would Rather Forget? Chem. Med. Chem. 2016, 11, 15–21. [Google Scholar] [CrossRef]
- Suzuki, T.; Miyata, N. Non-hydroxamate histone deacetylase inhibitors. Curr. Med. Chem. 2005, 12, 2867–2880. [Google Scholar] [CrossRef]
- Bora-Tatar, G.; Dayangaç-Erden, D.; Demir, A.S.; Dalkara, S.; Yelekçi, K.; Erdem-Yurter, H. Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorg. Med. Chem. 2009, 17, 5219–5228. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Tannous, P.; Lu, G.; Berenji, K.; Rothermel, B.A.; Olson, E.N.; Hill, J.A. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 2006, 113, 2579–2588. [Google Scholar] [CrossRef] [PubMed]
- Rombouts, K.; Niki, T.; Greenwel, P.; Vandermonde, A.; Wielant, A.; Hellemans, K.; De Bleser, P.; Yoshida, M.; Schuppan, D.; Rojkind, M.; et al. Trichostatin A, a histone deacetylase inhibitor, suppresses collagen synthesis and prevents TGF-beta(1)-induced fibrogenesis in skin fibroblasts. Exp. Cell. Res. 2002, 278, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; He, S.; Ma, L.; Ponnusamy, M.; Tang, J.; Tolbert, E.; Bayliss, G.; Zhao, T.C.; Yan, H.; Zhuang, S. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-beta and EGFR signaling. PLoS ONE 2013, 8, e54001. [Google Scholar] [CrossRef] [PubMed]
- Brufola, G.; Fringuelli, F.; Piermatti, O.; Pizzo, F. Simple and Efficient One-Pot Preparation of 3-Substituted Coumarins in Water. Heterocycles 1996, 43, 1257–1266. [Google Scholar] [CrossRef]
- Zhuravel, I.O.; Kovalenko, M.S.; Vlasov, S.V.; Chernykh, P.V. Solution-phase Synthesis of a Combinatorial Library of 3-[4-(Coumarin-3-yl)-1,3-thiazol-2-ylcarbamoyl]propanoic acid Amides. Molecules 2005, 10, 444–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parameshwar, R.; Sri Ranganath, Y.; HarinadhaBabu, V.; Sandeep, G. Synthesis and Antifungal Screening of Some Novel Coumarin Linked Imidazole Derivatives. Res. J. Pharm. Biol. Chem. Sci. 2011, 2, 514–520. [Google Scholar]
- Boza, P.; Ayala, P.; Vivar, R.; Humeres, C.; Cáceres, F.T.; Muñoz, C.; García, L.; Hermoso, M.A.; Díaz-Araya, G. Expression and function of toll-like receptor 4 and inflammasomes in cardiac fibroblasts and myofibroblasts: IL-1β synthesis, secretion, and degradation. Mol. Immunol. 2016, 74, 96–105. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
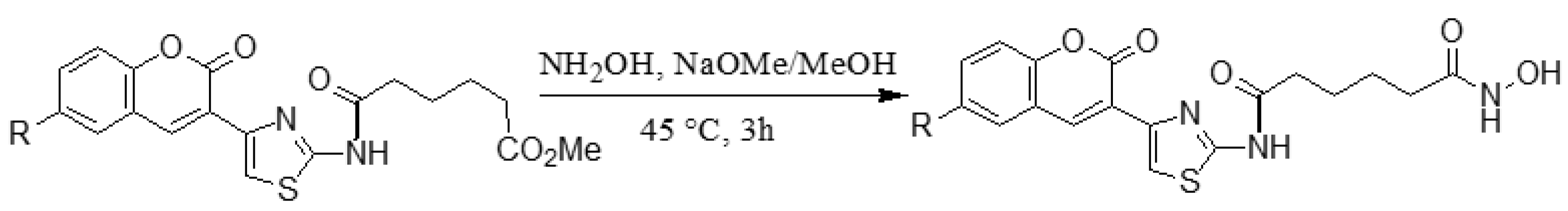{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Compound | R1 | R | Yield (%) |
| 5a |  | H | 80.6 |
| 5b |  | Br | 70.5 |
| 5c |  | OCH3 | 72.5 |
| 6a |  | H | 71.0 |
| 6b |  | Br | 82.0 |
| 6c |  | OCH3 | 78.0 |
| 7a |  | H | 81.0 |
| 7b |  | Br | 85.0 |
| 7c |  | OCH3 | 77.0 |
| 8a |  | H | 64.0 |
| 8b |  | Br | 57.0 |
| 8c |  | OCH3 | 68.0 |
| Compounds | Nuclear HeLa Cells Extract IC50 (nM) | Class I | Class II |
|---|---|---|---|
| HDAC1 IC50 (nM) | HDAC6 IC50 (nM) | ||
| 5a | 66.7 ± 2.9 | 60.4 ± 1.7 | 153.4 ± 5.3 |
| 5b | 62.1 ± 2.5 | 47.6 ± 2.3 | 122.8 ± 5.1 |
| 5c | 50.0 ± 1.7 | 39.7 ± 1.3 | 84.1 ± 2.9 |
| 6a | 573.5 ± 24.4 | 422.8 ± 30.2 | 276.1 ± 20.9 |
| 6b | 508.7 ± 23.3 | 612.7 ± 32.8 | 449.5 ± 20.7 |
| 6c | 476.7 ± 27.5 | 504.4 ± 31.4 | 580.5 ± 27.5 |
| 7a | 6100 ± 390 | 7689 ± 328 | 5589 ± 184 |
| 7b | 5400 ± 400 | 4666 ± 362 | 6336 ± 466 |
| 7c | 4700 ± 310 | 3382 ± 302 | 2427 ± 157 |
| 8a | 66.9 ± 1.8 | 50.6 ± 2.1 | 98.3 ± 3.5 |
| 8b | 57.1 ± 4.5 | 45.4 ± 2.6 | 80.2 ± 3.7 |
| 8c | 41.5 ± 2.7 | 46.5 ± 2.2 | 62.6 ± 2.3 |
| TSA | 5.0 ± 0.12 | 2.1 ± 0.06 | 48.0 ± 4.4 |
| Compound | CC50 (μM) |
|---|---|
| 5a | 200.9 ± 20.3 |
| 5b | 127.2 ± 19.2 |
| 5c | 170.7 ± 6.1 |
| 6a | 140.2 ± 11.7 |
| 6b | 102.2 ± 10.3 |
| 6c | 124.5 ± 12.8 |
| 7a | 111.4 ± 4.4 |
| 7b | 80.0 ± 23.6 |
| 7c | 99.5 ±6.3 |
| 8a | 90.5 ± 5.1 |
| 8b | 72.5 ± 11.6 |
| 8c | 86.5 ± 5.9 |
| Compound | 1 μM | 5 μM | 10 μM |
|---|---|---|---|
| 5a | 57.3 ± 6.7 | 70.7 ± 7.1 | 92.8 ± 6.4 |
| 5b | 71.5 ± 3.8 | 89.1 ± 9.5 | 96.4 ± 4.7 |
| 5c | 67.0 ± 8.3 | 83.2 ± 6.8 | 91.2 ± 3.2 |
| 6a | 3.0 ± 3.9 | 35.0 ± 1.9 | 60.6 ± 1.9 |
| 6b | 15.8 ± 3.6 | 40.0± 1.8 | 64.1 ± 1.7 |
| 6c | 10.9 ± 3.6 | 42.2 ± 1.8 | 64.9 ± 2.3 |
| 7a | 0.5 ± 0.1 | 26.8 ± 5.9 | 45.0 ± 5.1 |
| 7b | 2.4 ± 1.1 | 36.1 ± 5.4 | 48.0 ± 4.8 |
| 7c | 6.0 ± 0.4 | 33.0 ± 4.1 | 48.8 ± 5.2 |
| 8a | 59.4 ± 6.1 | 79.9 ± 9.7 | 94.4 ± 3.8 |
| 8b | 72.7 ± 3.9 | 94.4 ± 7.0 | 104.0 ± 2.8 |
| 8c | 69.7 ± 9.1 | 81.8 ± 5.5 | 97.7 ± 4.7 |
| Compounds | QlogPo/w a | QPPCaco b | QPlogBB c | Percent Human Oral Absorption d | QPlogKp e | #metab f |
|---|---|---|---|---|---|---|
| 5a | 4.4 | 1177 | −0.5 | 100 | −1.8 | 2 |
| 5b | 5.0 | 1173 | −0.4 | 100 | −2.0 | 2 |
| 5c | 4.5 | 1171 | −0.6 | 100 | −1.9 | 3 |
| 6a | 1.8 | 134 | −1.5 | 75 | −3.4 | 3 |
| 6b | 2.4 | 134 | −1.4 | 78 | −3.6 | 3 |
| 6c | 1.9 | 129 | −1.6 | 76 | −3.6 | 4 |
| 7a | 0.3 | 38 | −2.1 | 57 | −4.1 | 3 |
| 7b | 0.8 | 37 | −2.0 | 60 | −4.3 | 3 |
| 7c | 0.4 | 38 | −2.2 | 58 | −4.2 | 4 |
| 8a | 2.3 | 29 | −1.8 | 66 | −3.8 | 3 |
| 8b | 2.9 | 29 | −1.7 | 70 | −4.0 | 3 |
| 8c | 2.4 | 29 | −1.9 | 67 | −3.9 | 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pardo-Jiménez, V.; Navarrete-Encina, P.; Díaz-Araya, G. Synthesis and Biological Evaluation of Novel Thiazolyl-Coumarin Derivatives as Potent Histone Deacetylase Inhibitors with Antifibrotic Activity. Molecules 2019, 24, 739. https://doi.org/10.3390/molecules24040739
Pardo-Jiménez V, Navarrete-Encina P, Díaz-Araya G. Synthesis and Biological Evaluation of Novel Thiazolyl-Coumarin Derivatives as Potent Histone Deacetylase Inhibitors with Antifibrotic Activity. Molecules. 2019; 24(4):739. https://doi.org/10.3390/molecules24040739
Chicago/Turabian StylePardo-Jiménez, Viviana, Patricio Navarrete-Encina, and Guillermo Díaz-Araya. 2019. "Synthesis and Biological Evaluation of Novel Thiazolyl-Coumarin Derivatives as Potent Histone Deacetylase Inhibitors with Antifibrotic Activity" Molecules 24, no. 4: 739. https://doi.org/10.3390/molecules24040739
APA StylePardo-Jiménez, V., Navarrete-Encina, P., & Díaz-Araya, G. (2019). Synthesis and Biological Evaluation of Novel Thiazolyl-Coumarin Derivatives as Potent Histone Deacetylase Inhibitors with Antifibrotic Activity. Molecules, 24(4), 739. https://doi.org/10.3390/molecules24040739