
A Density Functional Theory-Based Scheme to Compute the Redox Potential of a Transition Metal Complex: Applications to Heme Compound


Abstract
:
1. Introduction
2. Theory and Computational Scheme
2.1. PCIS Scheme
2.2. LC-BOP12, LC-BOP12, LCgau-BOP, and LCgau-BOP12
2.3. Computational Details
3. Results and Discussion
3.1. Significance of the Correction Term
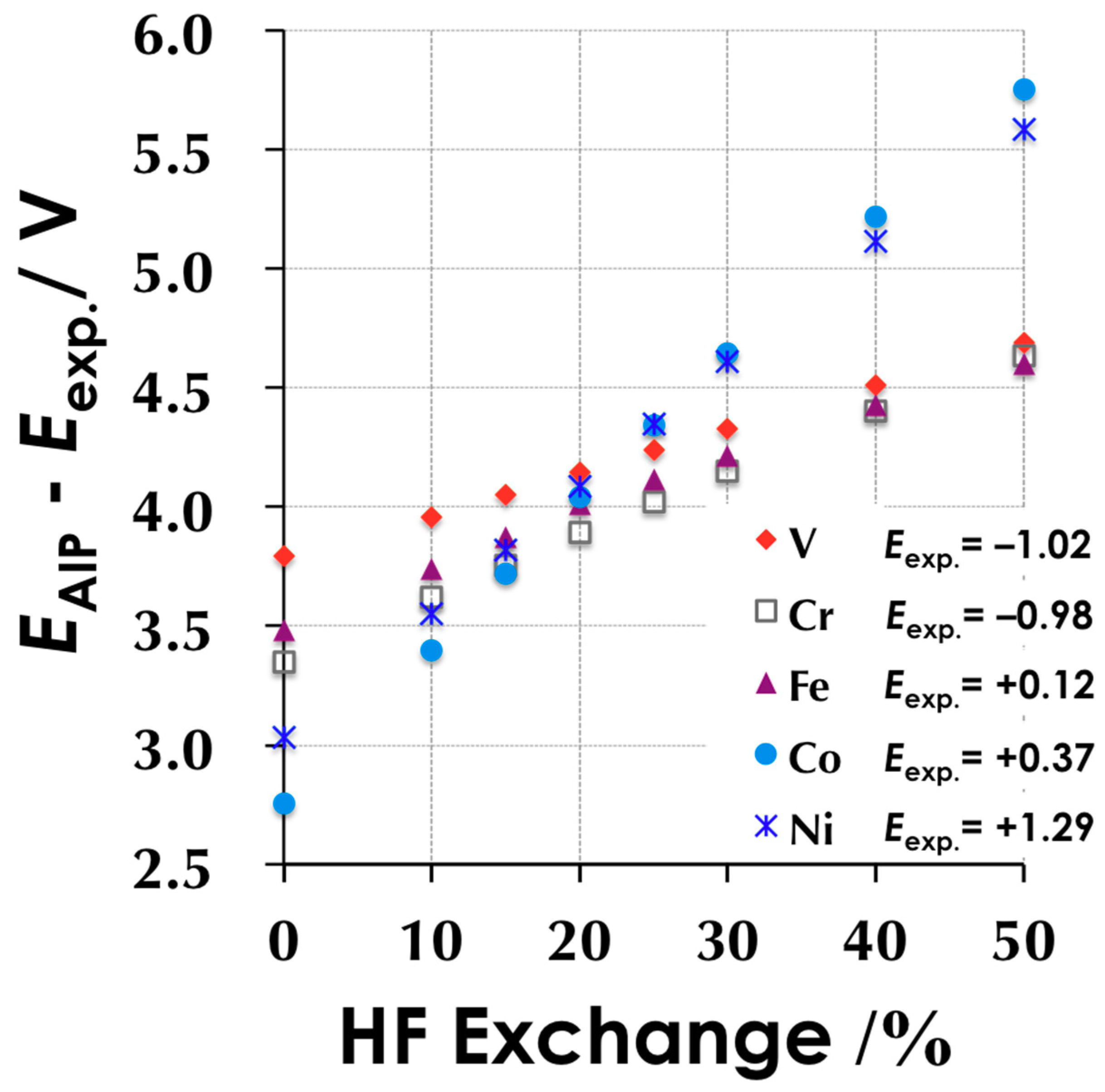
3.1.1. HF Exchange vs. Redox Potential
3.1.2. Solvation Model Dependencies
3.2. Functional Dependencies on Redox Potential
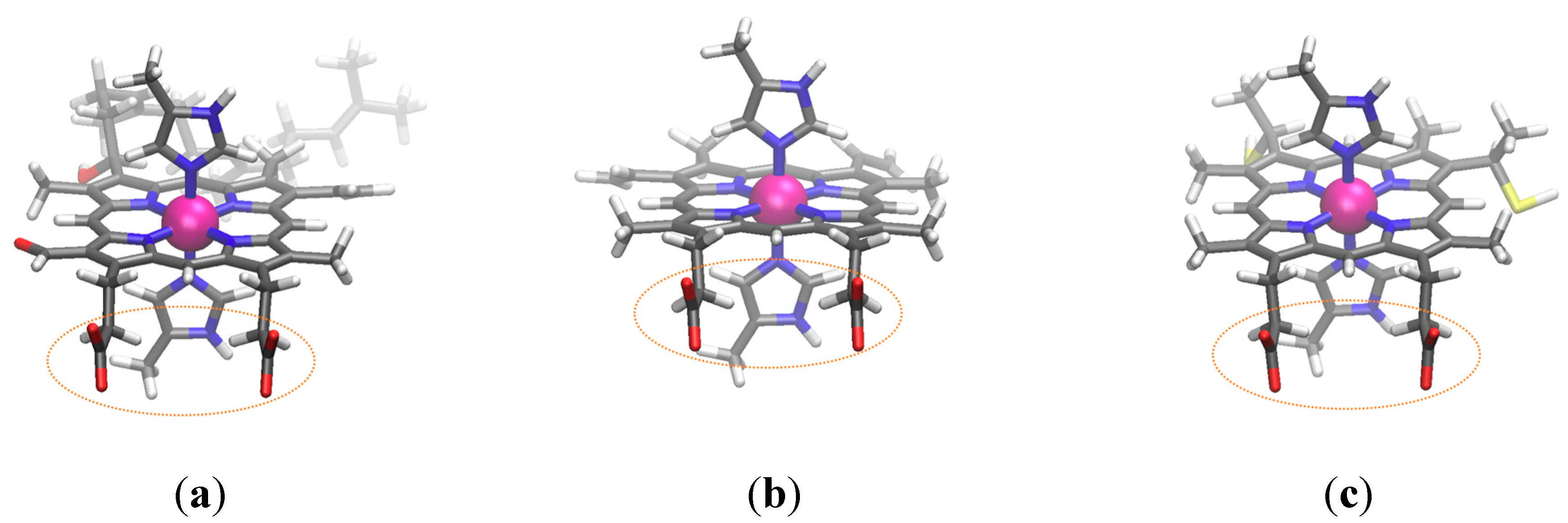
3.3. Application to Heme Products
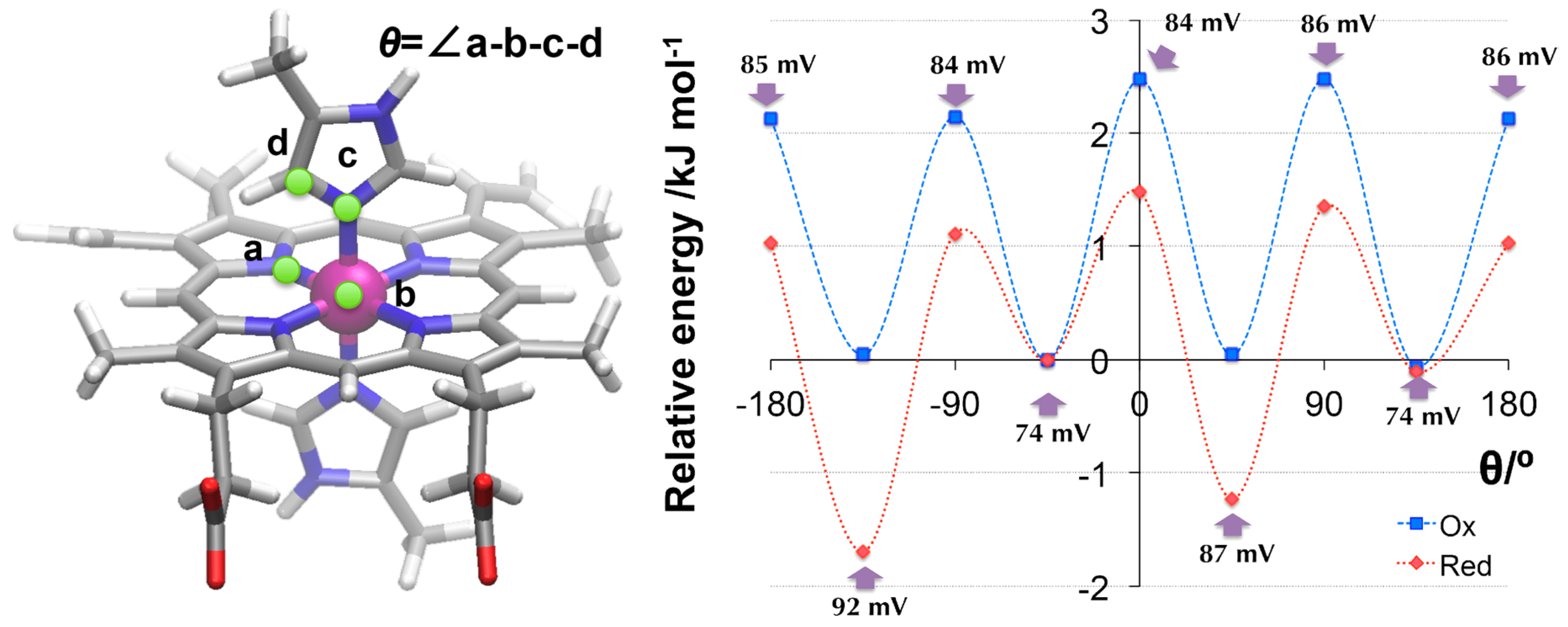
3.3.1. Flipping of the Model Histidine
3.3.2. Kind of Heme vs. Redox Potential
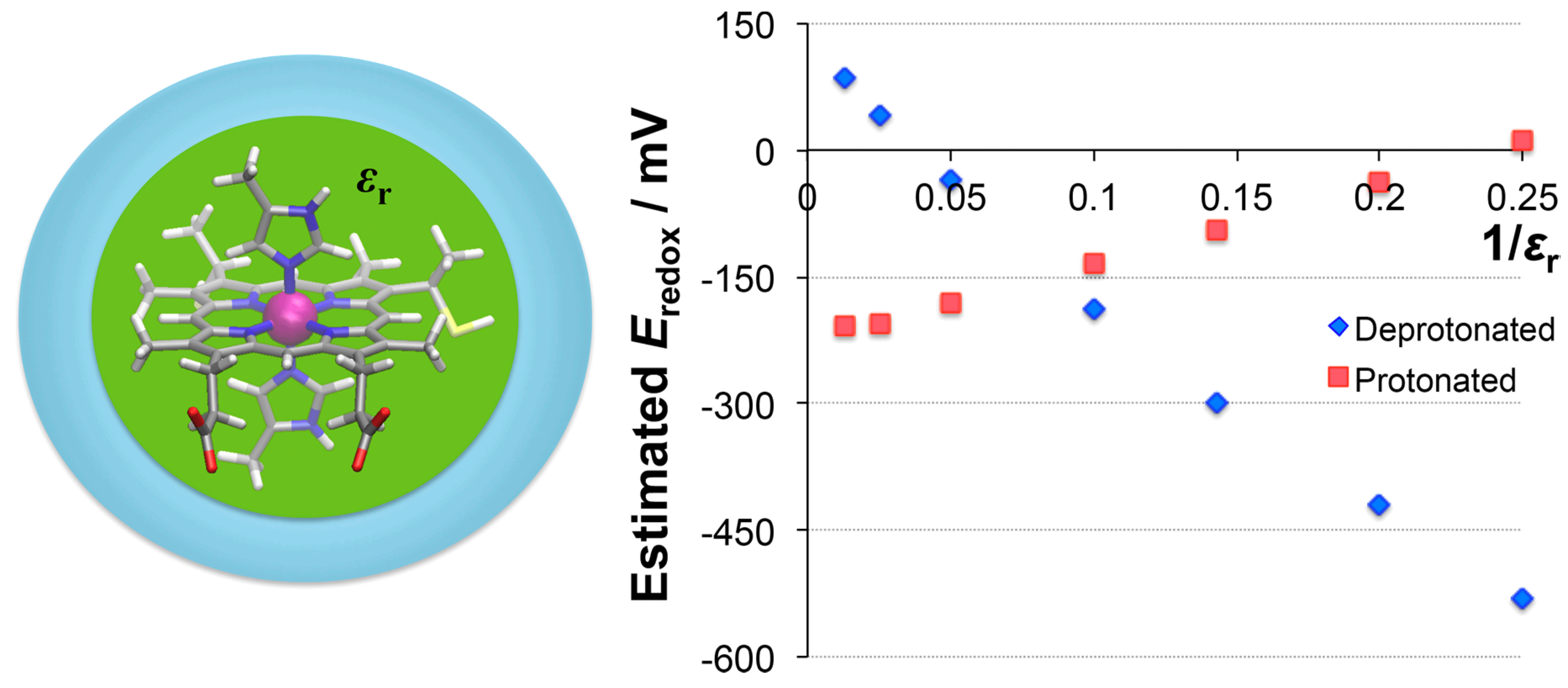
3.3.3. Dielectric Constants vs. Redox Potential
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shifman, J.M.; Gibney, B.R.; Sharp, R.E.; Dutton, P.L. Heme Redox Potential Control in de Novo Designed Four-R-Helix Bundle Protein. Biochemistry 2000, 39, 14813–14821. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-W. Structure and Function of Heme Proteins Regulatedd by Diverse Post-translational Modifications. Arch. Biochem. Biophys. 2018, 641, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Battistuzzi, G.; Brsari, M.; Cowan, J.A.; Ranieri, A.; Sola, M. Control of Cytochrome c Redox Potential: Axial Ligation and Protein Environment Effects. J. Am. Chem. Soc. 2002, 124, 5315–5324. [Google Scholar] [CrossRef] [PubMed]
- Voigt, P.; Knapp, E.-W. Tuning Heme Redox Potentials in the Cytochrome c Subunit of Photosynthetic Reaction Centers. J. Biol. Chem. 2003, 278, 51993–52001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, Y.; Nakamura, H. Density Functional Study of Roles of Porphyrin Ring in Electronic Structures of Heme. Int. J. Quantum Chem. 2009, 109, 3583–3591. [Google Scholar] [CrossRef]
- Takano, Y.; Nakamura, H. Electronic Structures of Heme a of Cytochrome c Oxidase in the Redox States—Charge Density Migration to the Propionate Groups of Heme a. J. Comput. Chem. 2010, 31, 954–962. [Google Scholar] [PubMed]
- Jerome, S.V.; Hughes, T.F.; Friesner, R.A. Successful Application of DBLOC Method to the Hydroxylation of Camphor by Cytochrome p450. Protein Sci. 2016, 25, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Baik, M.-H.; Friesner, R.A. Computing Redox Potentials in Solution: Density Functional Theory as a Tool for Rational Design of Redox Agents. J. Phys. Chem. A 2002, 106, 7407–7412. [Google Scholar] [CrossRef]
- Uudsemaa, M.; Tamm, T. Density-Functional Theory Calculations of Aqueous Redox Potentials of Fourth-Period Transition Metals. J. Phys. Chem. A 2003, 107, 9997–10003. [Google Scholar] [CrossRef]
- Shimodaira, Y.; Miura, T.; Kudo, A.; Kobayashi, H. DFT Method Estimation of Standard Redox Potential of Metal Ions and Metal Complexes. J. Chem. Theory Comput. 2007, 3, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.E.; Batista, E.R.; Hay, P.J. Theoretical Studies on the Redox Potentials of Fe Dinuclear Complexes as Models for Hydrogenase. Inorg. Chem. 2008, 47, 9228–9237. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.J.; Blanford, C.F.; Warwicker, J.; de Visser, S.P. Prediction of Reduction Potentials of Copper Proteins with Continuum Electrostatics and Density Functional Theory. Chem. Eur. J. 2017, 23, 15346–15445. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Kitagawa, Y.; Okumura, M.; Shigeta, Y.; Sakaki, S. Consistent Scheme for Computing Standard Hydrogen Electrode and Redox Potentials. J. Comp. Chem. 2013, 34, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Kitagawa, Y.; Okumura, M.; Shigeta, Y. Accurate Standard Hydrogen Electrode Potential and Applications to the Redox Potentials of Vitamin C and NAD/NADH. J. Phys. Chem. A 2015, 119, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Kitagawa, Y.; Shigeta, Y.; Okumura, M. A Density Functional Theory Based Protocol to Compute the Redox Potential of Transition Metal Complex with the Correction of Pseudo-Counterion: General Theory and Applications. J. Chem. Theory Comput. 2013, 9, 2974–2980. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, S.; Matsui, T.; Hirao, K.; Shigeta, Y. Theoretical Study on Reaction Mechanisms of Nitrite Reduction by Copper Nitrite Complexes: Toward Understanding and Controlling Possible Mechanisms of Copper Nitrite Reductase. J. Phys. Chem. B 2015, 119, 5392–5403. [Google Scholar] [CrossRef] [PubMed]
- Kurniawan, I.; Kawaguchi, K.; Shoji, M.; Matsui, T.; Shigeta, Y.; Nagao, H. A Theoretical Study on Redox Potential and pKa of [2Fe-2S] Cluster Model from Iron-Sulfur Proteins. Bull. Chem. Soc. Jpn. 2018, 91, 1451–1456. [Google Scholar] [CrossRef]
- Savin, A. On degeneracy, near-degeneracy and density functional theory. In Recent Developments and Applications of Modern Density Functional Theory; Seminario, J.M., Ed.; Elsevier: Amsterdam, Netherlands, 1996; p. 327. [Google Scholar]
- Leininger, T.; Stoll, H.; Werner, H.J.; Savin, A. Combining long-range configuration interaction with short-range density functionals. Chem. Phys. Lett. 1997, 275, 151–160. [Google Scholar] [CrossRef]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-W.; Hirosawa, T.; Tsuneda, T.; Hirao, K. Long-range corrected density functional calculations of chemical reactions: Redetermination of parameter. J. Chem. Phys. 2007, 126, 154105. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. B 1964, 136, B864. [Google Scholar] [CrossRef]
- Kamiya, M.; Sekino, H.; Tsuneda, T.; Hirao, K. Nonlinear optical property calculations by the long-range-corrected coupled-perturbed Kohn–Sham method. J. Chem. Phys. 2005, 122, 234111. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Tsuneda, T.; Hirao, K. Long-range corrected density functional study on weakly bound systems: Balanced descriptions of various types of molecular interactions. J. Chem. Phys. 2007, 126, 234114. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Tsuneda, T.; Hirao, K. A density-functional study on pi-aromatic interaction: Benzene dimer and naphthalene dimer. J. Chem. Phys. 2005, 123, 104307. [Google Scholar] [CrossRef] [PubMed]
- Sekino, H.; Maeda, Y.; Kamiya, M.; Hirao, K. Polarizability and second hyperpolarizability evaluation of long molecules by the density functional theory with long-range correction. J. Chem. Phys. 2007, 126, 014107. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, M.; Tsuneda, T.; Hirao, K. A density functional study of van der Waals interactions. J. Chem. Phys. 2002, 117, 6010. [Google Scholar] [CrossRef]
- Song, J.-W.; Watson, M.A.; Sekino, H.; Hirao, K. Nonlinear optical property calculations of polyynes with long-range corrected hybrid exchange-correlation functionals. J. Chem. Phys. 2008, 129, 024117. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-W.; Watson, M.A.; Sekino, H.; Hirao, K. The effect of silyl and phenyl functional group end caps on the nonlinear optical properties of polyynes: A long-range corrected density functional theory study. Int. J. Quantum Chem. 2009, 109, 2012–2022. [Google Scholar] [CrossRef]
- Song, J.-W.; Watson, M.A.; Nakata, A.; Hirao, K. Core-excitation energy calculations with a long-range corrected hybrid exchange-correlation functional including a short-range Gaussian attenuation (LCgau-BOP). J. Chem. Phys. 2008, 129, 184113. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-W.; Tsuneda, T.; Sato, T.; Hirao, K. Calculations of Alkane Energies Using Long-Range Corrected DFT Combined with Intramolecular van der Waals Correlation. Org. Lett. 2010, 12, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-W.; Tsuneda, T.; Sato, T.; Hirao, K. An examination of density functional theories on isomerization energy calculations of organic molecules. Theor. Chem. Acc. 2011, 130, 851–857. [Google Scholar] [CrossRef]
- Song, J.-W.; Tokura, S.; Sato, T.; Watson, M.A.; Hirao, K. Erratum: “An improved long-range corrected hybrid exchange-correlation functional including a short-range Gaussian attenuation (LCgau-BOP)” [J. Chem. Phys. 2007, 127, 154109]. J. Chem. Phys. 2009, 131, 059901. [Google Scholar] [CrossRef]
- Tsuneda, T.; Suzumura, T.; Hirao, K. A new one-parameter progressive Colle-Salvetti-type correlation functional. J. Chem. Phys. 1999, 110, 10664–10678. [Google Scholar] [CrossRef]
- Colle, R.; Salvetti, O. Approximate Calculation of Correlation Energy for Closed Shells. Theor. Chim. Acta 1975, 37, 329–334. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Song, J.-W.; Hirao, K. Long-range corrected density functional theory with optimized one-parameter progressive correlation functional (LC-BOP12 and LCgau-BOP12). Chem. Phys. Lett. 2013, 563, 15–19. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Revision, D.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Song, J.-W.; Peng, D.; Hirao, K. A Semiempirical Long-range Corrected Exchange Correlation Functional Including a Short-range Gaussian Attenuation (LCgau-B97). J. Comput. Chem. 2011, 32, 3269–3275. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cancès, M.T.; Mennucci, B.; Tomasi, J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J. Continuum Solvation Models: A New Approach to the Problem of Solute’s Charge Distribution and Cavity Boundaries. J. Chem. Phys. 1997, 106, 5151–5158. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Galstyan, A.; Knapp, E.-W. Accurate Redox Potentials of Mononuclear Iron, Manganese, and Nickel Model Complexes. J. Comput. Chem. 2009, 30, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.F.; Friesner, R.A. Development of Accurate DFT Methods for Computing Redox Potentials of Transition Metal Complexes: Results for Model Complexes and Application to Cytochrome P450. J. Chem. Theory Comput. 2012, 8, 442–459. [Google Scholar] [CrossRef] [PubMed]
- Imada, Y.; Nakamura, H.; Takano, Y. Density Functional Study of Porphyrin Distortion Effects on Redox Potential of Heme. J. Comput. Chem. 2018, 39, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Kanematsu, Y.; Kondo, H.X.; Imada, Y.; Takano, Y. Statistical and Quantum-chemical Analysis of the Effect of Heme Porphyrindistortion in Heme Proteins: Differences between Oxidoreductases and Oxygen Carrier Proteins. Chem. Phys. Lett. 2018, 710, 108–112. [Google Scholar] [CrossRef]
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Charge at Ox | N 1 | Gas | SMD | C-PCM | |||
|---|---|---|---|---|---|---|---|
| Ave. | Dev. | Ave. | Dev. | Ave. | Dev. | ||
| −3 | 6 | −7.12 | 1.30 | 3.95 | 0.43 | 3.55 | 0.34 |
| −2 | 2 | −3.84 | 0.84 | 3.96 | 0.09 | 3.80 | 0.18 |
| −1 | 15 | −0.49 | 0.67 | 4.23 | 0.55 | 4.24 | 0.27 |
| 0 | 4 | 2.69 | 0.44 | 4.26 | 0.21 | 4.31 | 0.29 |
| 1 | 5 | 5.73 | 0.70 | 3.94 | 0.22 | 4.23 | 0.29 |
| 2 | 2 | 8.90 | 0.25 | 4.18 | 0.33 | 4.61 | 0.30 |
| 3 | 4 | 13.57 | 0.35 | 4.54 | 0.44 | 5.12 | 0.39 |
| a | μ | HF Mix/% | ESHE | MAE | |
|---|---|---|---|---|---|
| BLYP 1 | 14.99 | 0.1364 | 0 | 3.76 | 0.22 |
| PBE | 18.67 | 0.1213 | 0 | 3.80 | 0.22 |
| B3LYP 1 | 15.26 | 0.0383 | 20 | 4.26 | 0.17 |
| BHandHLYP | 6.21 | 0.0178 | 50 | 4.62 | 0.61 |
| B3PW91 | 12.58 | 0.0361 | 20 | 4.19 | 0.18 |
| M06 1 | 11.55 | 0.0293 | 27 | 4.20 | 0.33 |
| a | μ | RS Param. 1 | ESHE | MAE | |
|---|---|---|---|---|---|
| ωB97XD | 7.57 | 0.0153 | 0.20 | 4.33 | 0.22 |
| LC-BLYP 2 | 10.76 | 0.0226 | 0.47 | 4.46 | 0.20 |
| CAM-B3LYP | 11.83 | 0.0245 | 0.33 | 4.38 | 0.20 |
| LC-BOP | 11.53 | 0.0354 | 0.47 | 4.41 | 0.18 |
| LC-BOP12 | 12.13 | 0.0364 | 0.42 | 4.25 | 0.16 |
| LCgau-BOP | 11.55 | 0.0293 | 0.42 | 4.20 | 0.18 |
| LCgau-BOP12 | 8.97 | 0.0223 | 0.42 | 4.29 | 0.20 |
| LC-ωPBE | 12.07 | 0.0355 | 0.40 | 4.44 | 0.22 |
| LCgau-B97 | 12.13 | 0.0365 | 0.20 | 4.44 | 0.21 |
| Deprotonated | Protonated | |||
|---|---|---|---|---|
| EAIP1 | Eredox2 | EAIP1 | Eredox2 | |
| Heme a | 4.10 | +0.17 | 4.21 | −0.12 |
| Heme b | 3.98 | +0.07 | 4.10 | −0.23 |
| Heme c | 4.01 | +0.09 | 4.12 | −0.21 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsui, T.; Song, J.-W. A Density Functional Theory-Based Scheme to Compute the Redox Potential of a Transition Metal Complex: Applications to Heme Compound. Molecules 2019, 24, 819. https://doi.org/10.3390/molecules24040819
Matsui T, Song J-W. A Density Functional Theory-Based Scheme to Compute the Redox Potential of a Transition Metal Complex: Applications to Heme Compound. Molecules. 2019; 24(4):819. https://doi.org/10.3390/molecules24040819
Chicago/Turabian StyleMatsui, Toru, and Jong-Won Song. 2019. "A Density Functional Theory-Based Scheme to Compute the Redox Potential of a Transition Metal Complex: Applications to Heme Compound" Molecules 24, no. 4: 819. https://doi.org/10.3390/molecules24040819
APA StyleMatsui, T., & Song, J. -W. (2019). A Density Functional Theory-Based Scheme to Compute the Redox Potential of a Transition Metal Complex: Applications to Heme Compound. Molecules, 24(4), 819. https://doi.org/10.3390/molecules24040819