
Convenient Synthesis of 6,7,12,13-Tetrahydro-5H-Cyclohepta[2,1-b:3,4-b’]diindole Derivatives Mediated by Hypervalent Iodine (III) Reagent


Abstract
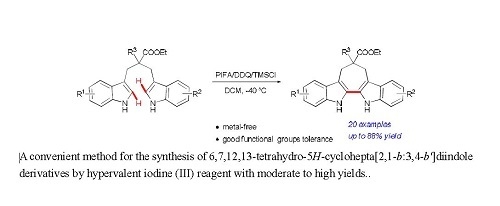
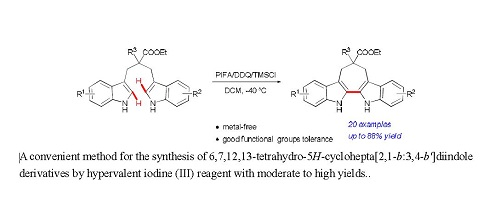
:
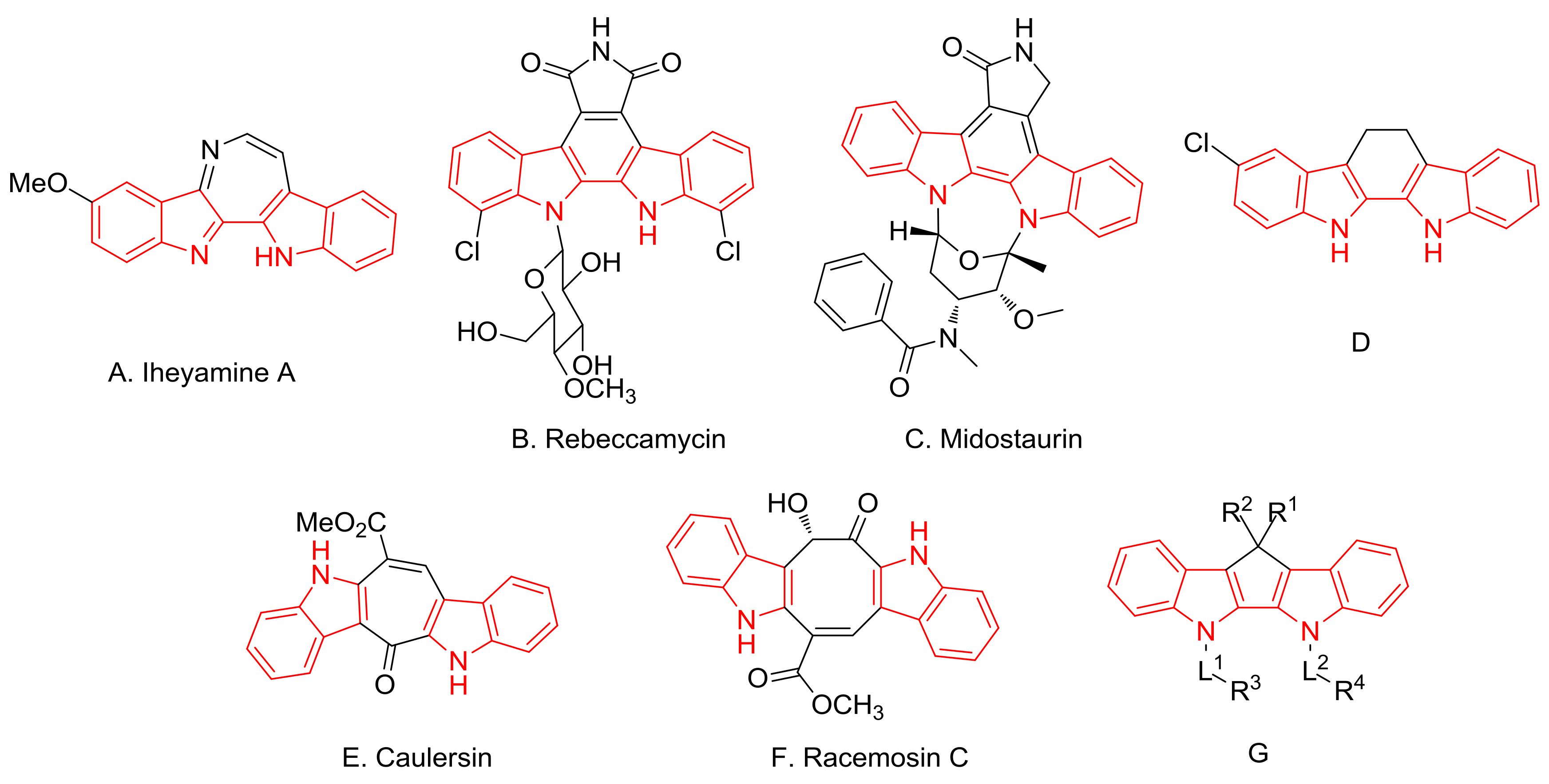
1. Introduction
2. Results and Discussion
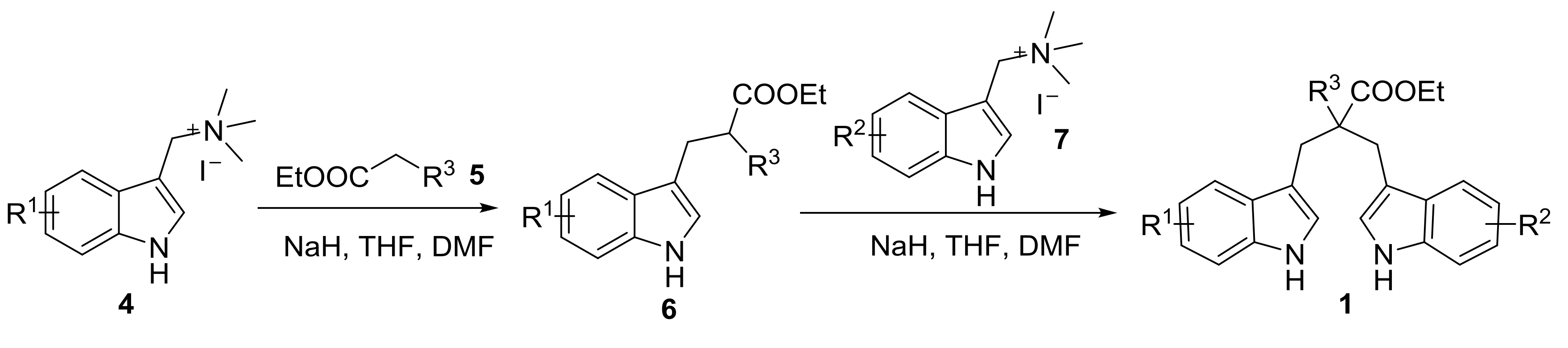
2.1. Preparation of 1,3-di(1H-indol-3-yl)propanes
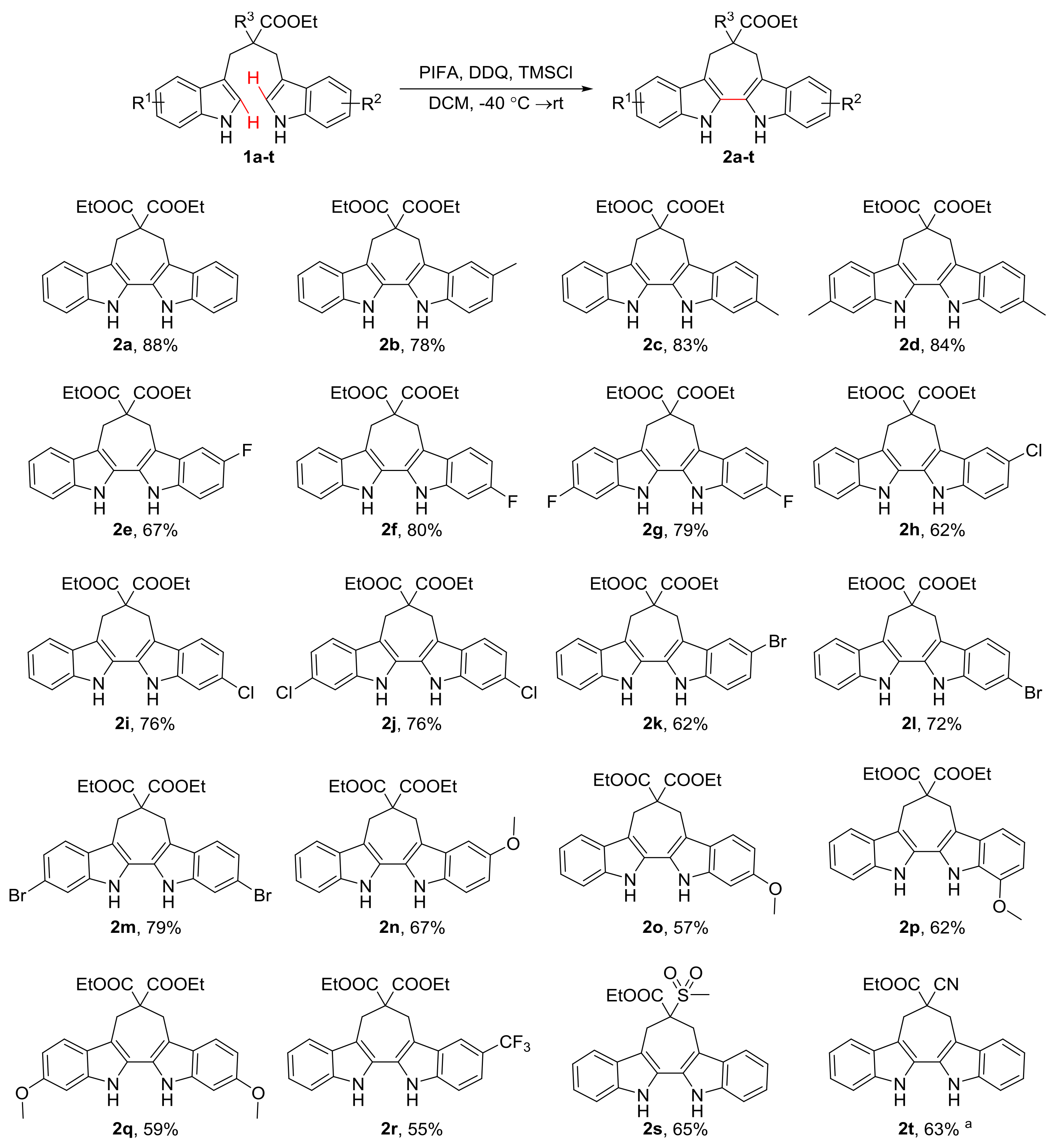
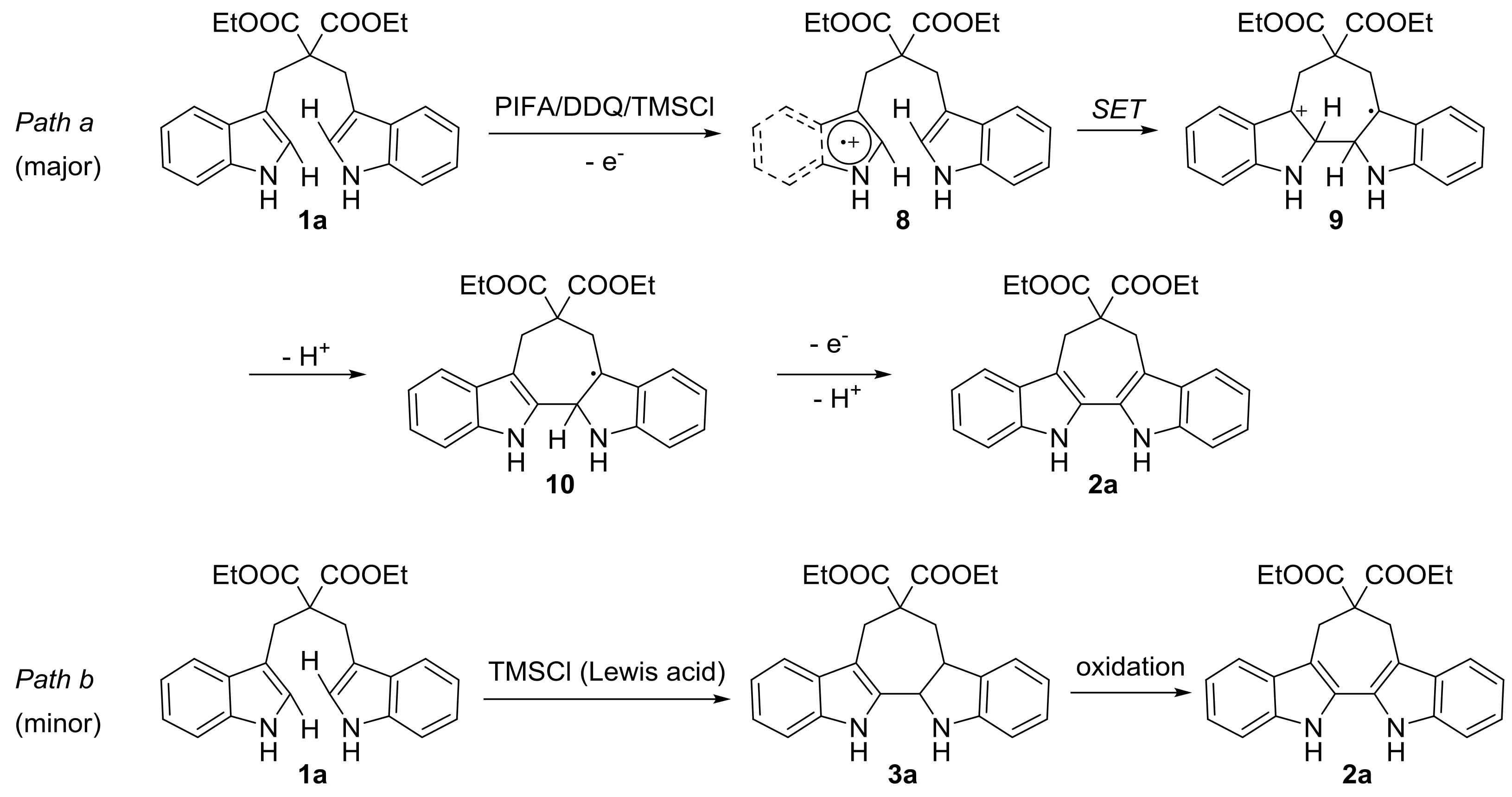
2.2. Intramolecular Oxidative Coupling of 1,3-di(1H-indol-3-yl)propanes
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of 6,7,12,13-Tetrahydro-5H-Cyclohepta[2,1-b:3,4-b’]diindoles (2a–t)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Praveen, P.; Parameswaran, P.; Majik, M. Bis(indolyl)methane Alkaloids: Isolation, Bioactivity, and Syntheses. Synthesis 2015, 47, 1827–1837. [Google Scholar] [CrossRef]
- Damodiran, M.; Muralidharan, D.; Perumal, P.T. Regioselective synthesis and biological evaluation of bis(indolyl)methane derivatized 1,4-disubstituted 1,2,3-bistriazoles as anti-infective agents. Bioorg. Med. Chem. Lett. 2009, 19, 3611–3614. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Mei, W.; Guo, Z.; Liu, S.; Zhao, Y.; Yang, D.; Zeng, Y.; Jiang, B.; Dai, H. Two New Types of Bisindole Alkaloid from Trigonostemon lutescens. Org. Lett. 2013, 15, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Ling, F.; Xiao, L.; Fang, L.; Feng, C.; Xie, Z.; Lv, Y.; Zhong, W. B(C6F5)3-catalyzed Markovnikov addition of indoles to aryl alkynes: An approach toward bis(indolyl)alkanes. Org. Biomol. Chem. 2018, 16, 9274–9278. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Jiang, F.; Sheng, F.T.; Jiao, Y.; Mei, G.J.; Shi, F. Design and Catalytic Asymmetric Construction of Axially Chiral 3,3’-Bisindole Skeletons. Angew. Chem. Int. Ed. 2018, 57, 1–7. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, X.; Wu, Y.; Zhang, W.; Chen, X.; You, X.; Hu, L. Synthesis and structure-activity relationship of novel bisindole amidines active against MDR Gram-positive and Gram-negative bacteria. Eur. J. Med. Chem. 2018, 150, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Ohtani, I.I.; Tanaka, J.; Higa, T. Iheyamines, new cytotoxic bisindole pigments from a colonial ascidian, Polycitorella sp. Tetrahedron Lett. 1999, 40, 303–306. [Google Scholar] [CrossRef]
- Nettleton, D.E.; Doyle, T.W.; Krishnan, B. Isolation and structure of rebeccamycin—A new antitumor antibiotic from nocardia aerocoligenes. Tetrahedron Lett. 1985, 26, 4011–4014. [Google Scholar] [CrossRef]
- Bush, J.A.; Long, B.H.; Catino, J.J.; Bradner, W.T. Production and biological activity of rebeccamycin, a novel antitumor agent. J. Antibiot. 1987, 40, 668–678. [Google Scholar] [CrossRef]
- Yamashita, Y.; Fujii, N.; Murakata, C.; Ashizawa, T.; Okabe, M.; Nakano, H. Induction of Mammalian DNA Topoisomerase I Mediated DNA Cleavage by Antitumor Indolocarbazole Derivatives. Biochemistry 1992, 31, 12069–12075. [Google Scholar] [CrossRef]
- Wu, M.; Li, C.; Zhu, X. FLT3 inhibitors in acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Balamurali, R.; Prasad, K.J.R. Synthesis, characterization and pharmacological activities of 5,6,11,12-tetrahydroindolo[2,3-a]carbazole derivatives. II Farmaco 2001, 56, 229–232. [Google Scholar] [CrossRef]
- Su, J.; Zhu, Y.; Zeng, L.; Xu, X. A New Bisindole from Alga Caulerpa serrulata. J. Nat. Prod. 1997, 60, 1043–1044. [Google Scholar] [CrossRef]
- Wahlström, N.; Stensland, B.; Bergman, J. Synthesis of the marine alkaloid caulersin. Tetrahedron 2004, 60, 2147–2153. [Google Scholar] [CrossRef]
- Miki, Y.; Aoki, Y.; Miyatake, H.; Minematsu, T.; Hibino, H. Synthesis of caulersin and its isomers by reaction of indole-2,3-dicarboxylic anhydrides with methyl indoleacetates. Tetrahedron Lett. 2006, 47, 5215–5218. [Google Scholar] [CrossRef]
- Yang, H.; Liu, D.Q.; Liang, T.J.; Li, J.; Liu, A.H.; Yang, P.; Lin, K.; Yu, X.Q.; Guo, Y.W.; Mao, S.C.; et al. Racemosin C, a novel minor bisindole alkaloid with protein tyrosine phosphatase-1B inhibitory activity from the green alga Caulerpa racemosa. J. Asian Nat. Prod. Res. 2014, 16, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Lee, D.H.; Park, S.H.; Ham, J.S.; Nam, H.G.; Jang, S.H.; Baek, Y.G.; Cho, G.O. Novel Organic Electroluminescent Compound and Organic Electroluminescent Element Including the Same; KR 2016126150; 2 November 2016. Available online: https://worldwide.espacenet.com/publicationDetails/originalDocument?CC=KR&NR=20160126150A&KC=A&FT=D&ND=3&date=20161102&DB=EPODOC&locale=en_EP# (accessed on 8 March 2019).
- Kuckländer, U.; Töberich, H. Eine neue Synthese von 7,8,9,10-Tetrahydrocyclohept[b]indol-6(5H)-onen. Chem. Ber. 1981, 114, 2238–2244. [Google Scholar] [CrossRef]
- Thummel, R.P.; Hegde, V. Polyaza-Cavity Shaped Molecules. 14. Annelated 2-(2’-Pyridyl)indoles, 2,2’-Biindoles, and Related Systems. J. Org. Chem. 1989, 54, 1720–1725. [Google Scholar] [CrossRef]
- Kavitha, C.; Prasad, K.J.R. Synthesis, Characterization and Biological Activity of Some Indolo[2′,3′:7,6] cyclohept[b]indoles and 1,2,3-Selenadiazolo[4′,5′:6,7]cyclohept[b]indoles. Asian J. Chem. 2004, 16, 40–48. [Google Scholar]
- Wahlström, N.; Slätt, J.; Stensland, B.; Ertan, A.; Bergman, J.; Janosik, T. Synthetic Applications of Cyanoacetylated Bisindoles: Synthesis of Novel Cycloheptadiindoles, Indolocarbazoles, and Related Aza Analogues. J. Org. Chem. 2007, 72, 5886–5889. [Google Scholar] [CrossRef]
- Yang, X.; Sun, G.; Yang, C.; Wang, B. Novel Rhein Analogues as Potential Anticancer Agents. ChemMedChem 2011, 6, 2294–2301. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Xiong, B.; Shen, Y.; Yang, C. Design, synthesis, and preliminary biological evaluation of novel ketone derivatives of shikimic acid. RSC Adv. 2013, 3, 20599. [Google Scholar] [CrossRef]
- Zhang, X.; He, Q.; Xiang, H.; Song, S.; Miao, Z.; Yang, C. Rapid access to α-carbolines via a one-pot tandem reaction of α,β-unsaturated ketones with 2-nitrophenylacetonitrile and the anti-proliferative activities of the products. Org. Biomol. Chem. 2014, 12, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Chen, H.; Xu, X.; Zhu, F.; Guo, L.; Jiang, M.; Yang, C.; Deng, L. Synthesis of Rigid Analogues of Flavone by Intramolecular Heck Reaction. Eur. J. Org. Chem. 2015, 2015, 3040–3043. [Google Scholar] [CrossRef]
- Xu, X.; Qi, X.; Yan, Y.; Qi, J.; Qian, N.; Guo, L.; Li, C.; Wang, F.; Huang, P.; Zhou, H.; et al. Synthesis and biological evaluation of rhein amides as inhibitors of osteoclast differentiation and bone resorption. Eur. J. Med. Chem. 2016, 123, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Dohi, T.; Kita, Y. Metal-free Oxidative Cross-Coupling Reaction of Aromatic Compounds Containing Heteroatoms. Synlett 2017, 28, 1680–1694. [Google Scholar] [CrossRef]
- Tohma, H.; Iwata, M.; Maegawa, T.; Kiyono, Y.; Maruyama, A.; Kita, Y. A novel and direct synthesis of alkylated 2,2′-bithiophene derivatives using a combination of hypervalent iodine(III) reagent and BF3·Et2O. Org. Biomol. Chem. 2003, 1, 1647–1649. [Google Scholar] [CrossRef]
- Dohi, T.; Morimoto, K.; Maruyama, A.; Kita, Y. Direct Synthesis of Bipyrroles Using Phenyliodine Bis(trifluoroacetate) with Bromotrimethylsilane. Org. Lett. 2006, 8, 2007–2010. [Google Scholar] [CrossRef]
- Morimoto, K.; Yamaoka, N.; Ogawa, C.; Nakae, T.; Fujioka, H.; Dohi, T.; Kita, Y. Metal-Free Regioselective Oxidative Biaryl Coupling Leading to Head-to-Tail Bithiophenes: Reactivity Switching, a Concept Based on the Iodonium(III) Intermediate. Org. Lett. 2010, 12, 3804–3807. [Google Scholar] [CrossRef]
- Rana, A.; Panda, P.K. β-Octamethoxyporphycenes. Org. Lett. 2014, 16, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Lee, S.; Kim, D.; Panda, P.K. β-Octakis(methylthio)porphycenes: Synthesis, characterisation and third order nonlinear optical studies. Chem. Commun. 2015, 51, 7705–7708. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.S.; Tsai, M.T.; Tsai, M.H.; Ong, C.W. The regioselective homocoupling of meta-hydroxypyridines with hypervalent iodine(III). Chem. Asian. J. 2015, 10, 849–852. [Google Scholar] [CrossRef]
- Jean, A.; Cantat, J.; Bérard, D.; Bouchu, D.; Canesi, S. Novel Method of Aromatic Coupling between N-Aryl Methanesulfonamide and Thiophene Derivatives. Org. Lett. 2007, 9, 2553–2556. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Ito, M.; Yamaoka, N.; Morimoto, K.; Fujioka, H.; Kita, Y. Unusual ipso substitution of diaryliodonium bromides initiated by a single-electron-transfer oxidizing process. Angew. Chem. Int. Ed. Engl. 2010, 49, 3334–3337. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Ohnishi, Y.; Koseki, D.; Nakamura, A.; Dohi, T.; Kita, Y. Stabilized pyrrolyl iodonium salts and metal-free oxidative cross-coupling. Org. Biomol. Chem. 2016, 14, 8947–8951. [Google Scholar] [CrossRef]
- Kantak, A.A.; Potavathri, S.; Barham, R.A.; Romano, K.M.; DeBoef, B. Metal-free intermolecular oxidative C-N bond formation via tandem C-H and N-H bond functionalization. J. Am. Chem. Soc. 2011, 133, 19960–19965. [Google Scholar] [CrossRef]
- Parumala, S.K.R.; Peddinti, R.K. Reversal of Polarity in Masked o-Benzoquinones: Rapid Access to Unsymmetrical Oxygenated Biaryls. Org. Lett. 2013, 15, 3546–3549. [Google Scholar] [CrossRef]
- Morimoto, K.; Sakamoto, K.; Ohshika, T.; Dohi, T.; Kita, Y. Organo-Iodine(III)-Catalyzed Oxidative Phenol-Arene and Phenol-Phenol Cross-Coupling Reaction. Angew. Chem. Int. Ed. 2016, 55, 3652–3656. [Google Scholar] [CrossRef]
- Sharma, S.; Parumala, S.K.R.; Peddinti, R.K. Lewis Acid-Mediated Site-Selective Synthesis of Oxygenated Biaryls from Methoxyphenols and Electron-Rich Arenes. J. Org. Chem. 2017, 82, 9367–9383. [Google Scholar] [CrossRef]
- Arisawa, M.; Ramesh, N.G.; Nakajima, M.; Tohma, H.; Kita, Y. Hypervalent Iodine(III)-Induced Intramolecular Cyclization of α-(Aryl)alkyl-β-dicarbonyl Compounds: A Convenient Synthesis of Benzannulated and Spirobenzannulated Compounds. J. Org. Chem. 2001, 66, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, H.; Anilkumar, G.; Tohma, H.; Kita, Y. A Novel and Useful Oxidative Intramolecular Coupling Reaction of Phenol Ether Derivatives on Treatment with a Combination of Hypervalent Iodine(III) Reagent and Heteropoly Acid. Chem. Eur. J. 2002, 8, 5377–5383. [Google Scholar] [CrossRef]
- Hamamoto, H.; Shiozaki, Y.; Hata, K.; Tohma, H.; Kita, Y. A Novel and Concise Synthesis of Spirodienone Alkaloids Using Hypervalent Iodine(III) Reagents. Chem. Pharm. Bull. 2004, 52, 1231–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, I.; Tellitu, I.; Domínguez, E.; SanMartín, R.l. A Simple Route to New Phenanthro- and Phenanthroid-Fused Thiazoles by a PIFA-Mediated (Hetero)biaryl Coupling Reaction. Eur. J. Org. Chem. 2002, 2126–2135. [Google Scholar] [CrossRef]
- Barrett, T.N.; Braddock, D.C.; Monta, A.; Webb, M.R.; White, A.J. Total synthesis of the marine metabolite (±)-polysiphenol via highly regioselective intramolecular oxidative coupling. J. Nat. Prod. 2011, 74, 1980–1984. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, S.; Andrez, J.C.; Canesi, S. A Stereoselective Oxidative Polycyclization Process Mediated by a Hypervalent Iodine Reagent. Org. Lett. 2011, 13, 3406–3409. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, X.; Zhang, Y.; Ruan, L.; Zhang, J.; Zhang-Negrerie, D.; Du, Y. Iodocyclization of N-Arylpropynamides Mediated by Hypervalent Iodine Reagent: Divergent Synthesis of Iodinated Quinolin-2-ones and Spiro[4,5]trienones. Org. Lett. 2017, 19, 150–153. [Google Scholar] [CrossRef]
- Suvorov, N.N.; Velezheva, V.S.; Vampilova, V.V. Indole derivatives. LXXXVII. Improved methods for the synthesis of skatyl- and substituted skatylmalonic esters. Chem. Heterocycl. Compd. 1973, 9, 1367–1369. [Google Scholar] [CrossRef]
- Pelcman, B.; Gribble, G.W. Total Synthesis of The Marine Sponge Pigment Fascaplysin. Tetrahedron Lett. 1990, 31, 2381–2384. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1a–t and 2a–t are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Hypervalent iodine Reagent | Additive | Yield/% (2a/3a) b |
| 1 | PIFA (1 eq) | None | 55/trace |
| 2 | PIFA (0.5 eq) | None | 38/19 |
| 3 | PIFA (0.3 eq) | None | 36/22 |
| 4 a | PIFA (0.3 eq) | None | 21/25 |
| 5 | PIFA (0.3 eq) | DDQ (1 eq) | 43/trace |
| 6 | PIFA (0.3 eq) | DDQ (0.7 eq) | 56/trace |
| 7 | PIFA (0.3 eq) | DDQ (0.5 eq) | 60/trace |
| 8 | PIFA (0.3 eq) | BQ (0.5 eq) | 53/trace |
| 9 | PIFA (0.3 eq) | CAN (0.5 eq) | 50/trace |
| 10 | None | DDQ (1 eq) | 48/trace |
 | ||||
|---|---|---|---|---|
| Entry | Hypervalent Iodine Reagent | Lewis Acid | Temperature | Yield/% a |
| 1 | PIFA | TMSBr | 0 °C | 60 |
| 2 | PIFA | TMSCl | 0 °C | 61 |
| 3 | PIFA | TMSOTf | 0 °C | 23 |
| 4 | PIFA | BF3 OEt2 | 0 °C | 49 |
| 5 | PIFA | TMSCl | −40 °C | 88 |
| 6 | PIFA | TMSCl | −78 °C | 70 |
| 7 | PIDA | TMSCl | −40 °C | 85 |
| 8 | IBX | TMSCl | −40 °C | 58 |
| 9 | DMP | TMSCl | −40 °C | 75 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, L.; Zhang, X.; Yang, C. Convenient Synthesis of 6,7,12,13-Tetrahydro-5H-Cyclohepta[2,1-b:3,4-b’]diindole Derivatives Mediated by Hypervalent Iodine (III) Reagent. Molecules 2019, 24, 960. https://doi.org/10.3390/molecules24050960
Peng L, Zhang X, Yang C. Convenient Synthesis of 6,7,12,13-Tetrahydro-5H-Cyclohepta[2,1-b:3,4-b’]diindole Derivatives Mediated by Hypervalent Iodine (III) Reagent. Molecules. 2019; 24(5):960. https://doi.org/10.3390/molecules24050960
Chicago/Turabian StylePeng, Lei, Xiaofei Zhang, and Chunhao Yang. 2019. "Convenient Synthesis of 6,7,12,13-Tetrahydro-5H-Cyclohepta[2,1-b:3,4-b’]diindole Derivatives Mediated by Hypervalent Iodine (III) Reagent" Molecules 24, no. 5: 960. https://doi.org/10.3390/molecules24050960
APA StylePeng, L., Zhang, X., & Yang, C. (2019). Convenient Synthesis of 6,7,12,13-Tetrahydro-5H-Cyclohepta[2,1-b:3,4-b’]diindole Derivatives Mediated by Hypervalent Iodine (III) Reagent. Molecules, 24(5), 960. https://doi.org/10.3390/molecules24050960