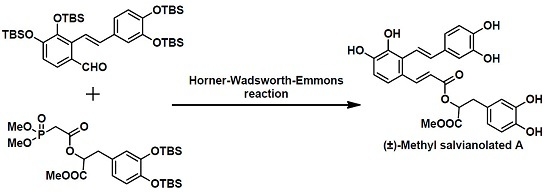
2. Results
To the best of our knowledge, most of the published formal syntheses of polyphenol compounds that are structurally similar to (±)-methyl salvianolate A involve the use of methyl ether protecting groups, which effectively avoids the influence of the phenol groups during the synthesis. However, the use of the methyl ether protecting group results in significant side reactions and low yields during its deprotection, in which a strong Lewis acid, such as BBr
3, is usually used. Although an allylic protective group which is more easily removed was employed by Zhang’s group, only a moderate yield (41%) was achieved under the deprotection conditions (Pd(PPh
3)
4, morpholine) [
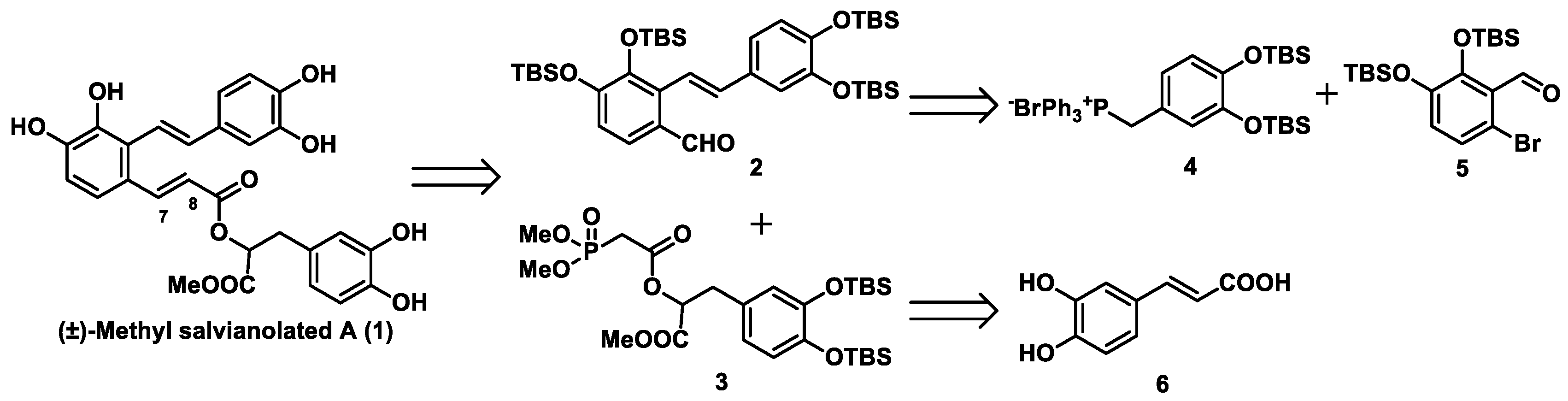
15]. As an alternative to using an alkyl group, we looked to install the readily removable tert-butyldimethylsilyl (TBS) group as the protecting group in our total synthesis. Our retrosynthetic analysis of methyl salvianolate A is shown in
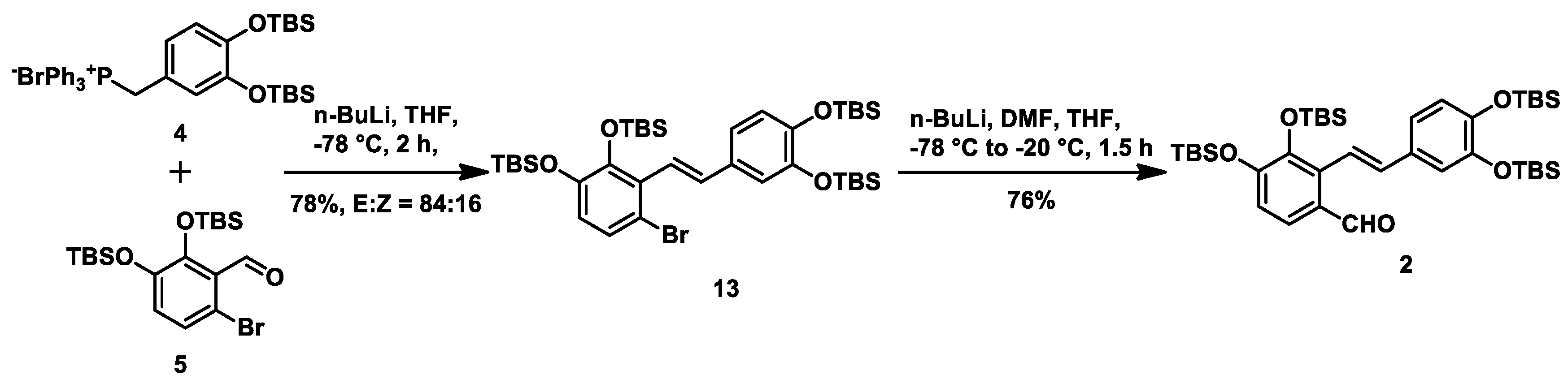
Scheme 1. (±)-Methyl salvianolate A was disconnected at the C7-C8 bond via a Horner–Wadsworth–Emmons (HWE) reaction to give two Fragments
2 and
3. We envisaged that fragment
2 could be obtained from fragments
4 and
5 via a Wittig reaction using Cotelle’s protocol [
16], and that fragment
3 could be prepared from caffeic acid (fragment
6).
As depicted in
Scheme 2, our synthesis of (±)-methyl salvianolate A commenced with the preparation of intermediate
4 from commercially available 4-methylcatechol (
7). 4-Methylcatechol was subjected to TBS protection under conventional conditions (TBSCl/imidazole in DMF) to give
8 in an almost quantitative yield. Compound
8 was then subjected to bromination at the benzylic position via a radical process using
N-bromosuccinimide (NBS) in the presence of 2,2′-azobis- (isobutyronictrile) (AIBN) in CCl
4 at 80 °C. Without further purification, the resultant bromide was reacted with triphenyl phosphine in anhydrous MeCN at 80 °C, followed by washing with toluene to give phosphonium salt
4 in an overall 76% yield from
8. Meanwhile, intermediate
5 was prepared from commercially available
o-vanillin (
9). To avoid the influence of the free phenol hydroxyl group, aldehyde
9 first reacted with acetic anhydride to afford
10, which was then converted to monobromide
11 in the presence of potassium bromide and bromine in water (64% over two steps). The acetyl group in
11 was removed under mild reaction conditions (sodium hydrogen carbonate in MeOH) followed by removal of the methyl group using BBr
3 at 0 °C and a second protection step using TBSCl in the presence of triethylamine and 4-dimethylaminopyridine (DMAP) to achieve intermediate
5 in 69% yield over three steps.
With intermediates
4 and
5 in hand, we then conducted the Wittig reaction. Treating salt
4 with n-BuLi gave the corresponding ylide that was then reacted with aldehyde
5 at −78 °C to afford alkene
13 in 78% yield with a
E/Z ratio of 84:16. On exposure to
n-BuLi at −78 °C, the resulting anion prepared in situ from the lithium-halogen exchange of alkene
13 was reacted with
N,
N-dimethylformamide at −20 °C to give aryl aldehyde
2 in 76% yield (
Scheme 3).
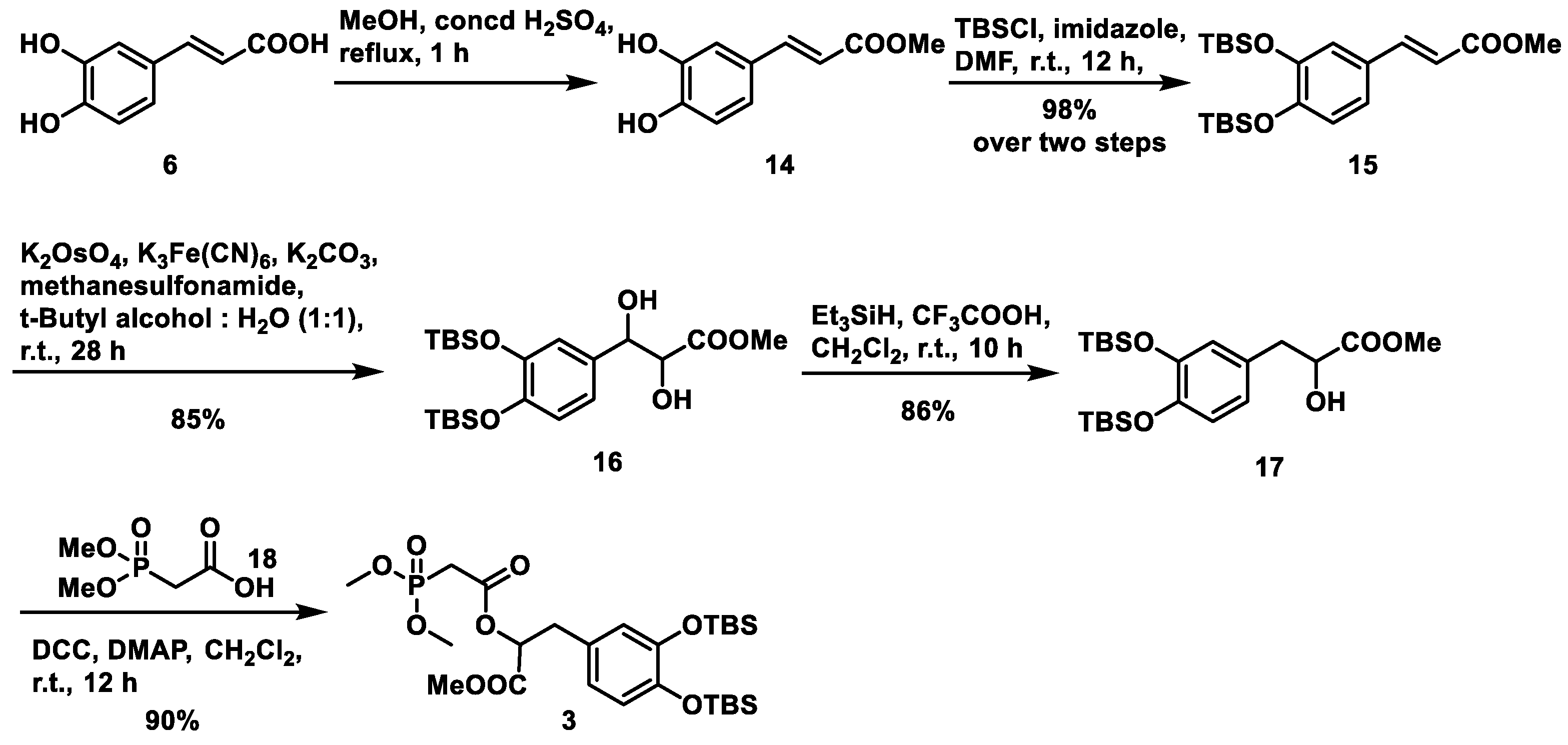
For fragment
3, we envisaged an esterification reaction between the commercially available acid
18 and alcohol
17. The latter was synthesized following an established procedure [
18] starting from caffeic acid
6, as shown in
Scheme 4, which included esterification, TBS protection, dihydroxylation and selective dehydroxylation steps. We obtained intermediate
17 in an overall 72% yield from
6. Subsequently, when treated with
N,
N′-dicyclohexylcarbodiimide (DCC) and catalytic DMAP in CH
2Cl
2 at room temperature,
17 and
18 were assembled into fragment 3 in 90% yield.
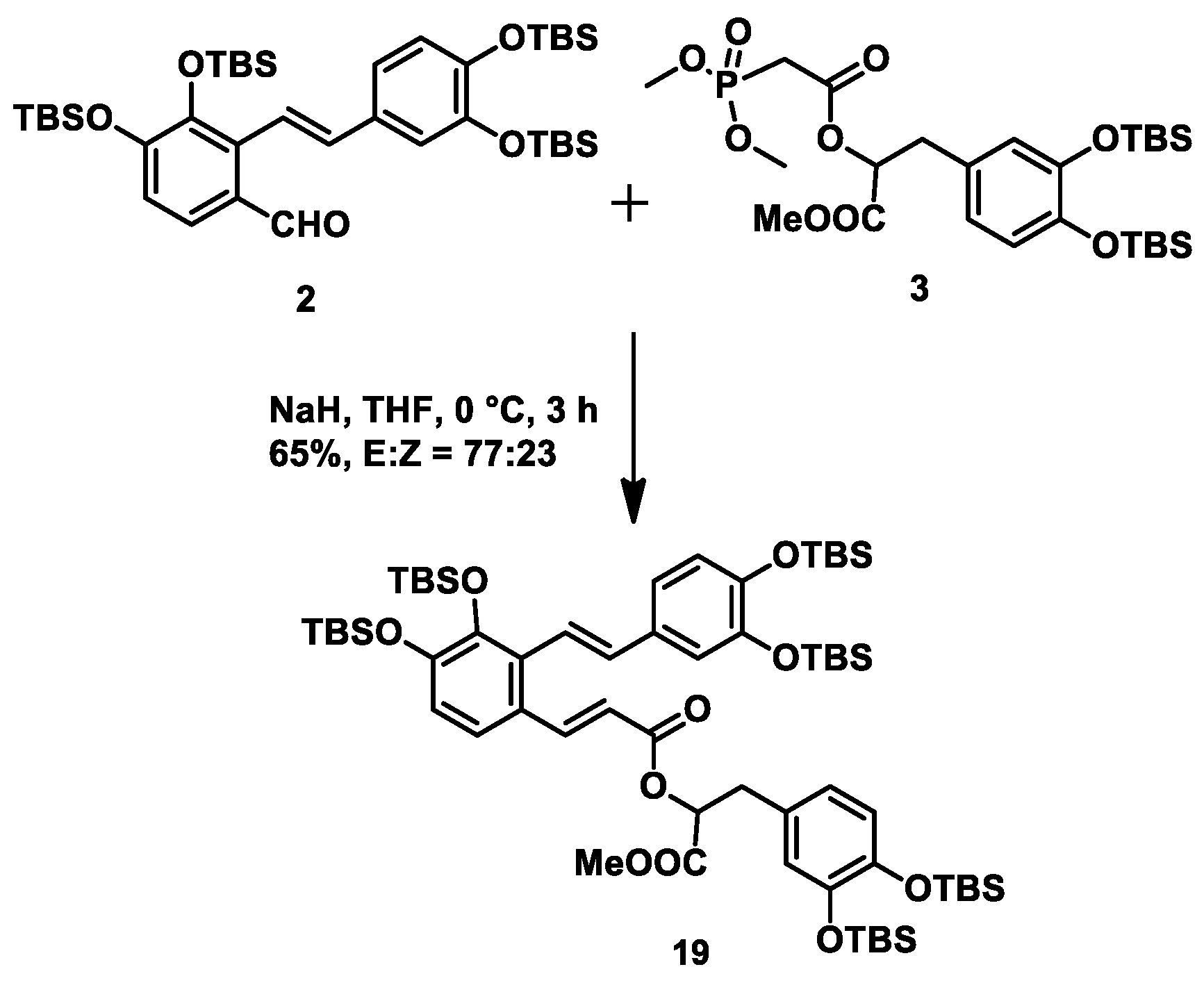
At this stage, we needed to connect fragments
2 and
3 to construct the α,β-unsaturated ester moiety via a HWE reaction. After conducting some experiments in different conditions (
n-BuLi, −78 °C; NaH, −78 °C; NaH, 0 °C), we successfully obtained intermediate
19 in 65% yield with an
E/Z ratio of ~4:1 when performing the HWE reaction at 0 °C for 3 h using NaH as base (
Scheme 5).
Finally, with
19 in hand, we turned to conduct the global deprotection step according to our retrosynthetic analysis. However, the deprotection proved surprisingly difficult. It was disappointing to find that the desired product was not obtained when treating
19 with the conventional reagent, tetrabutylammonium fluoride (TBAF). A great deal of TBAF and silicide accompanied
1 during the purification step, which hindered the isolation of the target product (
Table 1, Entry 1).
A similar phenomenon was observed when conducting the experiment under basic [
19,
20,
21] or acidic conditions, it gave either no reaction (Entry 2–4,6) or trace amounts of the isolated purified product mixed with a large amount of silicide (Entry 5,7,8). Fortunately, we finally obtained (±)-methyl salvianolate A (1) in excellent yield (92%) without any silicide after purification using triethylamine trihydrofluoride in anhydrous pyridine (Entry 9) [
18,
22].
Considering the low efficiency of the demethylation step in the endgame of the formally published total syntheses of the salvianolates, the deprotection of 19 employing the readily removable silyl ether protection group demonstrated that (±)-methyl salvianolate A and its derivatives can be prepared on a large scale using our strategy. The physical properties and
1H- and
13C-NMR spectra of compound 1 obtained using our synthetic route agreed with those reported in the literature [
11].
3. Experimental Section
3.1. General Methods
All air and water sensitive reactions were carried out under a nitrogen atmosphere with dry solvents under anhydrous conditions, unless otherwise noted. Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm silica gel plates (60F-254, Qingdao Ocean Chemical Factory, Qingdao, China) that were analyzed by fluorescence upon 254 nm irradiation or by staining with anisaldehyde (450 mL of 95% EtOH, 25 mL of con. H
2SO
4 (15 mL) of acetic acid, and 25 mL of anisaldehyde, phosphomolybdic acid (100 mL of 95% EtOH, 10 g phosphomolybdic acid) and dinitrophenylhydrazine (80 mL H
2O, 200 mL of 95% EtOH, 12 g dinitrophenylhydrazine and 60 mL concentrated sulfuric acid). Silica gel (60, particle size 0.040–0.063 mm) was used for flash column chromatography. All the chemicals were purchased commercially and used without further purification. Anhydrous THF was distilled from sodium-benzophenone. DMF, CH
3CN and CH
2Cl
2 were distilled from calcium hydride. Yields refer to chromatographically, unless otherwise stated. NMR spectra were recorded on an Avance 400 MHz instrument (
1H: 400 MHz,
13C: 100 MHz, Bruker Corporation, Karlsruhe, Germany). The following abbreviations were used to explain themultiplicities: s = singlet, d = doublet, m = multiplet. High-resolution mass spectra were obtained from a MALDI-TOF Mass Spectrometer (Applied Biosystems, Carlsbad, CA, USA). IR spectra were obtained using FT-IR Spectrometer (Bruker Vertex 70, Bruker Corporation, Karlsruhe, Germany). NMR and HRMS of compounds
1–5, 8, 10–17, 19 can be found in
Supplementary Materials.
3.2. 3,4-Bis(tert-butyldimethylsilyloxy)toluene (8)
Imidazole (13.7 g, 201.39 mmol) and tert-butyldimethylchlorosilane (18.2 g, 120.83 mmol) were added to a solution of 7 (5.0 g, 40.28 mmol), which was dissolved in dry DMF (200 mL), at 0 °C under a nitrogen atmosphere. The resulting mixture was warmed to room temperature and stirred overnight. The reaction was quenched with brine, extracted with ethyl acetate, and the combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue was purified by silica gel chromatography to afford compound 8 (14.20 g, 99%) as a colorless liquid. 1H-NMR (CDCl3): δ 6.72 (d, J = 8.0 Hz, 1H), 6.65 (s, 1H), 6.61 (d, J = 8.0 Hz, 1H), 2.24 (s, 3H), 1.01 (s, 9H), 1.00 (s, 9H), 0.21 (s, 6H), 0.20 (s, 6H).; HRMS (TOF-MS): m/z calcd for C19H37O2Si2 [M + H]+ 353.2327. Found 353.2327.
3.3. (3,4-Bis((tert-butyldimethylsilyl)oxy)benzyl)triphenylphosphonium Bromide (4)
Under a nitrogen atmosphere, N-bromosuccinimide (7.28 g, 40.83 mmol) and AIBN (1.1 g, 6.81 mmol) were added to a solution of compound 8 (12.0 g, 34.03 mmol) in CCl4 (70 mL) and the resulting mixture heated at reflux for 1 h. The reaction mixture was filtered and the residue was washed with petroleum ether and concentrated in vacuo to afford the crude product, which was immediately used in the next step without any purification. Triphenylphosphine (13.38 g, 51.04 mmol) was then added to the crude product solution in dry MeCN (70 mL) and the resulting mixture was heated under reflux for 3 h. The reaction mixture was cooled to room temperature and concentrated in vacuo. Toluene was added to the crude product and the resulting suspension filtered to give phosphonium salt 4 (17.94 g, 76% over two steps) as a pale yellow solid. 1H-NMR (CDCl3): δ 7.79–7.62 (m, 15H), 6.61 (s, 2H), 6.47 (s, 1H), 5.16 (d, J = 13.6 Hz, 2H), 0.93 (s, 9H), 0.84 (s, 9H), 0.15 (s, 6H), −0.05 (s, 6H); HRMS (TOF-MS): m/z calcd for C37H50O2Si2P [M-Br-]+ 613.3081. Found 613.3082.
3.4. 2-Formyl-6-methoxyphenyl Acetate (10)
A catalytic quantity of 4-dimethylaminopyridine (0.96 g, 7.89 mmol) and acetic anhydride (8.07 mL, 85.44 mmol) were added to an o-vanillin (9, 10.01 g, 65.72 mmol) solution in N,N-diisopropylethylamine (33 mL) at 0 °C under a nitrogen atmosphere. The solution was allowed to warm to room temperature and stirred overnight. After compound 9 was completely consumed, hydrochloric acid (2 M, 100 mL) was added. The resulting mixture was stirred for another 10 min and then extracted with dichloromethane (100 mL × 3). The combined organic layers were concentrated in vacuo to afford a crude yellow solid, which was recrystallized from ethanol to give compound 10 (10.85 g, 85%) as a yellow solid. 1H-NMR (CDCl3): δ 10.12 (s, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.31 (t, J = 8.4 Hz, 1H), 7.20 (d, J = 8.4 Hz, 1H), 3.85 (s, 3H), 2.39 (s, 3H).; 13C-NMR (100 MHz, CDCl3): δ 188.7, 168.7, 151.8, 141.5, 129.2, 126.8, 121.2, 117.9, 56.3, 20.4.
3.5. 3-Bromo-2-formyl-6-methoxyphenyl Acetate (11)
Bromine (3.73 mL, 72.64 mmol) was added to a potassium bromide (21.47 g, 180.48 mmol) solution in H2O (133 mL). Aldehyde 10 (10.85 g, 55.88 mmol) was added to the resulting dark red solution and stirred overnight. The reaction was monitored by TLC until 10 was completely consumed. The reaction mixture was extracted with ethyl acetate, the layers separated and the organic layer concentrated in vacuo. The resulting residue was recrystallized from ethyl acetate–hexane to give compound 11 (11.44 g, 75%) as yellow crystals. 1H-NMR (CDCl3): δ 10.35 (s, 1H), 7.59 (d, J = 8.8 Hz, 1H), 7.14 (d, J = 8.8 Hz, 1H), 3.94 (s, 3H), 2.47 (s, 3H); 13C-NMR (CDCl3): δ 190.3, 168.7, 151.9, 140.4, 131.5, 126.6, 117.8, 116.3, 56.4, 20.5.
3.6. 6-Bromo-2-hydroxy-3-methoxybenzaldehyde (12)
Sodium bicarbonate (4.93 g, 58.64 mmol) was added to a solution of aldehyde 11 (11.44 g, 41.89 mmol) solution in a mixture of methanol and water (v/v = 2:1, 84 mL). The resulting turbid yellow solution was stirred for 2 h at room temperature. Hydrochloric acid (1 M) was then added to adjust the pH to 6–7 and the mixture extracted with ethyl acetate (100 mL × 3). The combined organic layers were concentrated in vacuo and the crude yellow solid recrystallized from ethyl acetate/hexane to give compound 12 (8.23 g, 85%) as yellow acicular crystals. 1H-NMR (CDCl3): δ 12.26 (s, 1H), 10.27 (s, 1H), 7.07 (d, J = 8.4 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 3.88 (s, 3H); 13C-NMR (CDCl3): δ 198.3, 154.4, 148.3, 123.3, 118.2, 117.1, 116.3, 56.3.
3.7. 6-Bromo-2,3-bis((tert-butyldimethylsilyl)oxy)benzaldehyde (5)
Boron tribromide (13.47 mL, 142.48 mmol) in CH2Cl2 (60 mL) was added dropwise to a solution of compound 12 (8.23 g, 35.62 mmol) in dry CH2Cl2 (120 mL) at 0 °C under a nitrogen atmosphere. The resulting mixture was then stirred for 2 h. Ice–water (100 mL) was added slowly to quench the reaction, the layers separated and the organic layer concentrated in vacuo. The residue was washed with water, filtered and the resulting yellow filter cake dried at 40 °C to give the crude product as a yellow solid, which was used in the next step without any further purification. Et3N (16.50 mL, 118.37 mmol), DMAP (0.826 g, 6.76 mmol) and tert-butyldimethylchlorosilane (15.29 g, 101.46 mmol) were added to the above crude solid (7.34 g, 33.82 mmol) in dry DMF (307 mL) at 0 °C under a nitrogen atmosphere. The resulting solution was stirred for 1 h at 0 °C. The reaction was quenched with brine, extracted with ethyl acetate (200 mL × 3) and the combined organic layers concentrated in vacuo. The resulting residue was purified by silica gel chromatography to give intermediate 5 (12.85 g, 81% over two steps). 1H-NMR (CDCl3): δ 10.29 (s, 1H), 7.11 (d, J = 8.4 Hz, 1H), 6.87 (d, J = 8.8 Hz, 1H), 1.00 (s, 9H), 0.96 (s, 9H), 0.23 (s, 6H), 0.12 (s, 6H); 13C-NMR (CDCl3): δ 189.4, 149.3, 146.9, 127.0, 126.0, 124.6, 112.8, 25.1, 25.0, 17.7, 17.5, −4.7, −4.8; HRMS (TOF-MS): m/z calcd for C19H34BrO3Si2 [M + H]+ 445.1224. Found 445.1230.
3.8. (E)-6-Bromo-2,3,3’,4’-tetrakis(tert-butyldimethylsilyloxy)stilbene (13)
n-BuLi (10.3 mL, 2.5 M in hexane, 25.81 mmol) was slowly added to a suspension of phosphonium salt 4 (19.47 g, 28.06 mmol) in dry THF (100 mL) at –78 °C under a nitrogen atmosphere and the resulting mixture was stirred for 30 min at –78 °C. Aldehyde 5 (5.0 g, 11.22 mmol) dissolved in dry THF (40 mL) was added dropwise to the reaction mixture and the resulting solution stirred for another 1 h at –78 °C. The reaction was quenched with a saturated aqueous solution of ammonium chloride. The mixture was extracted with ethyl acetate (100 mL × 3) and the combined organic layers washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue was purified by silica gel chromatography to give compound 13 (17.08 g, 78%, E:Z = 84:16) as a colorless oil. 1H-NMR (CDCl3): δ 7.11 (d, J = 8.4 Hz, 1H), 7.02 (d, J = 8.8 Hz, 1H), 7.00 (d, J = 17.2 Hz, 1H), 6.94 (s, 1H), 6.85 (d, J = 16.8 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 6.66 (d, J = 8.4 Hz, 1H), 1.04 (s, 18H), 1.02 (s, 9H), 0.95 (s, 9H), 0.28 (s, 6H), 0.26 (s, 12H), 0.09 (s, 6H).; 13C-NMR (CDCl3): δ 147.3, 146.9, 146.8, 145.6, 135.2, 131.8, 131.2, 125.4, 123.5, 121.0, 120.0, 119.1, 115.5, 26.2, 26.2, 26.0, 25.9, 18.8, 18.5, 18.5, 18.3, −3.5, −3.6, −4.0 −4.1; HRMS (TOF-MS): m/z calcd for C36H68BrO4Si4 [M + H]+ 779.3373. Found 779.3378.
3.9. (E)-2-(3,4-Bis((tert-butyldimethylsilyl)oxy)styryl)-3,4-bis((tert-butyldimethyl-silyl)oxy)benzaldehyde (2)
n-BuLi (6.7 mL, 2.5 M in hexane, 16.82 mmol) was added to a solution of compound 13 (6.56 g, 8.41 mmol) in dry THF (40 mL) at −78 °C under a nitrogen atmosphere. After the reaction mixture was stirred for 30 min at −78 °C, dry N,N-dimethylformamide (6.50 mL, 84.08 mmol) in dry THF (10 mL) was slowly added to the mixture and stirred for another 30 min at −78 °C until 13 was completely consumed. The reaction was quenched with a saturated aqueous solution of ammonium chloride and hydrochloric acid (1 M) added to adjust the pH to 1. The mixture was extracted with ethyl acetate (50 mL × 3) and the combined organic layers dried over anhydrous Na2SO4, filtered and concentrated in vacuo. The resulting residue was purified by silica gel chromatography to give compound 2 (4.66 g, 76%) as a yellow oil. 1H-NMR (CDCl3): δ 10.08 (s, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.18 (d, J = 16.4 Hz, 1H), 7.02 (s, 1H), 6.99 (d, J = 8.4 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 6.85 (d, J = 8.0 Hz, 1H), 6.46 (d, J = 16.4 Hz, 1H), 1.03 (s, 9H), 1.03 (s, 9H), 1.02 (s, 9H), 0.99 (s, 9H), 0.31 (s, 6H), 0.26 (s, 6H), 0.25 (s, 6H), 0.13 (s, 6H).; 13C-NMR (CDCl3): δ 192.6, 152.9, 148.4, 147.9, 145.5, 139.4, 137.3, 131.4, 130.2, 124.0, 122.0, 121.4, 120.5, 120.4, 120.2, 27.1, 27.0, 26.9, 19.8, 19.4, 19.4, 19.3, −2.4, −2.6, −3.1, −3.1; HRMS (TOF-MS): m/z calcd for C39H69O5Si4 [M + H]+ 729.4217. Found 729.4218.
3.10. (E)-methyl 3-(3,4-dihydroxyphenyl)acrylate (14)
Concentrated sulfuric acid (4 mL) was slowly added to a solution of caffeic acid (6, 2.0 g, 11.10 mmol) in methanol (140 mL). The resulting mixture was heated at reflux for 1 h. Aqueous NaOH (1 M) was added to quench the reaction and adjust the pH to 6–7. The aqueous phase was extracted with CH2Cl2 (100 mL × 3). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting crude product (2.15 g) was used in the next step without any further purification. 1H-NMR (CD3OD): δ 7.51 (d, J = 16.0 Hz, 1H), 7.03 (s, 1H), 6.90 (d, J = 8.0 Hz, 1H), 6.77 (d, J = 8.4 Hz, 1H), 6.22 (d, J = 16.0 Hz, 1H), 4.98 (br s, 2H), 3.71 (s, 3H).; 13C-NMR (CD3OD): δ 167.5, 147.1, 144.6, 144.4, 125.5, 120.7, 114.2, 113.0, 112.6, 49.8; HRMS (TOF-MS): m/z calcd for C10H11O4 [M + H]+ 195.0652. Found 195.0658.
3.11. (E)-methyl 3-(3,4-bis((tert-butyldimethylsilyl)oxy)phenyl)acrylate (15)
Imidazole (5.28 g, 77.51 mmol) and tert-butyldimethylchlorosilane (5.0 g, 33.22 mmol) were added to the crude compound 14 (2.15 g, 11.07 mmol) dissolved in dry DMF (60 mL) at 0 °C. The reaction mixture was allowed to warm up to room temperature and stirred overnight. The reaction was quenched with brine, extracted with ethyl acetate (100 mL × 3) and the combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue was purified by silica gel chromatography to afford compound 15 (4.74 g, 98%) as a white solid. 1H-NMR (CDCl3): δ 7.56 (d, J = 16.0 Hz, 1H), 7.01 (s, 1H), 6.99 (d, J = 5.6 Hz, 1H), 6.81 (d, J = 8.4 Hz, 1H), 6.23 (d, J = 16.0 Hz, 1H), 3.77 (s, 3H), 0.98 (s, 9H), 0.97 (s, 9H), 0.20 (s, 6H), 0.20 (s, 6H).; 13C-NMR (CDCl3): δ 167.7, 149.4, 147.2, 144.8, 128.0, 122.2, 121.1, 120.4, 115.4, 51.5, 25.9, 25.9, 18.5, 18.4, −4.1, −4.1; HRMS (TOF-MS): m/z calcd for C22H39O4Si2 [M + H]+ 423.2381. Found 423.2376.
3.12. Methyl 3-(3,4-bis((tert-butyldimethylsilyl)oxy)phenyl)-2,3-dihydroxypropanoate (16)
Methyl caffeate (15, 2.30 g, 5.43 mmol) dissolved in tert-butyl alcohol (20 mL) was added to a suspension of K2OsO4 (2.80 g, 7.60 mmol), K3[Fe(CN)6] (2.50 g, 7.60 mmol), K2CO3 (1.05 g, 7.60 mmol) and methanesulfonamide (10 mmol) in tert-butyl alcohol (40 mL) under a nitrogen atmosphere at 0 °C. The mixture was stirred for 28 h until 15 was completely consumed. The reaction was quenched with a saturated solution of sodium sulfite. The mixture was allowed to warm up to room temperature and stirred for 1 h. The reaction mixture was extracted with CH2Cl2 (100 mL × 3) and the combined organic layers washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue was purified by silica gel chromatography to give dihydroxyl ester 16 (2.10 g, 85%) as a white solid. 1H-NMR (CDCl3): δ 6.91 (s, 1H), 6.81 (s, 2H), 4.85 (s, 1H), 4.30 (s, 1H), 3.77 (s, 3H), 3.12 (s, 1H), 2.70 (s, 1H), 0.99 (s, 9H), 0.98 (s, 9H), 0.19 (s, 6H), 0.19 (s, 6H); 13C-NMR (CDCl3): δ 173.2, 146.8, 146.8, 133.0, 120.8, 119.5, 119.4, 74.9, 74.3, 52.6, 25.9, 18.4, 18.4, −4.1, −4.1.
3.13. Methyl 3-(3,4-bis((tert-butyldimethylsilyl)oxy)phenyl)-2-hydroxypropanoate (17)
Et3SiH (7.78 mL, 48.83 mmol) and trifluoroacetic acid (7.26 mL, 97.7 mmol) were added to a solution of dihydroxyl ester 16 (4.46 g, 9.77 mmol) dissolved in dry CH2Cl2 (40 mL) at 0 °C. The reaction mixture was allowed to warm up to room temperature and stirred for 10 h. The reaction was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with CH2Cl2 (80 mL × 3). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue was purified by silica gel chromatography to give the monohydroxy product 17 (3.70 g, 86%) as a white semi-solid. 1H-NMR (CDCl3): δ 6.76 (d, J = 8.0 Hz, 1H), 6.75 (s, 1H), 6.66 (d, J = 8.0 Hz, 1H), 4.41 (br s, 1H), 3.74 (s, 3H), 3.00 (dd, J = 4.4 and 14.0 Hz, 1H), 2.89 (br s, 1H), 2.88 (dd, J = 6.4 and 14.0 Hz, 1H), 1.01 (s, 18H), 0.21 (s, 12H).; 13C-NMR (CDCl3): δ 174.5, 146.6, 145.8, 129.3, 122.5, 122.4, 120.9, 71.4, 52.2, 39.9, 26.0, 18.4, −4.1.
3.14. Methyl 3-(3,4-bis((tert-butyldimethylsilyl)oxy)phenyl)-2-(2-(dimethoxyphosphoryl)acetoxy)-propanoate (3)
Dicyclohexylcarbodiimide (DCC, 2.58 g, 12.49 mmol) and a catalytic amount of 4-dimethylaminopyridine (0.19 g, 1.56 mmol) were added to a solution of the monohydroxy product 17 (3.44 g, 7.81 mmol) and 2-(dimethoxyphosphoryl)acetic acid 18 (2.10 g, 12.49 mmol) in dry CH2Cl2 (40 mL) at 0 °C under a nitrogen atmosphere, and the resulting mixture was then stirred for 1 h. The reaction was quenched with brine, extracted with CH2Cl2 (50 mL × 3) and the combined organic layers dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue was purified by silica gel chromatography to give compound 3 (4.15 g, 90%) as a white semi-solid. 1H-NMR (CDCl3): δ 6.69 (d, J = 8.0 Hz, 1H), 6.64 (s, 1H), 6.60 (d, J = 8.4 Hz, 1H), 5.15 (dd, J = 4.8 and 7.2 Hz, 1H), 3.72 (d, J = 11.2 Hz, 3H), 3.70 (d, J = 11.2 Hz, 3H), 3.64 (s, 3H), 2.97 (d, J = 21.6 Hz, 2H), 2.94–3.00 (m, 2H), 0.93 (s, 9H), 0.92 (s, 9H), 0.14 (s, 6H), 0.13 (s, 6H).; 13C-NMR (CDCl3) : δ 169.4, 165.1 (d, J = 4.7 Hz), 146.7, 146.0, 128.4, 122.2, 122.2, 120.9, 74.1, 53.2, 52.2, 36.6, 32.9 (d, J = 135.0 Hz), 25.9, 18.4, 18.4, −4.1; HRMS (TOF-MS): m/z calcd for C26H47O9PSi2Na [M + Na]+ 613.2388. Found 613.2386.
3.15. 2-(3,4-Bis(tert-butyldimethylsilyloxy)phenyl-1-methoxycarbonyl)ethyl(E)-3-((E)-2,3,3′,4′-tetrakis(tert -butyldimethylsilyloxy)stylben-6-yl)acrylate (19)
NaH (30.9 mg, 0.77 mmol, 60% in mineral oil) was added to a solution of 3 (0.453 g, 0.77 mmol) in dry THF (4 mL) at 0 °C under a nitrogen atmosphere and the reaction mixture stirred for 30 min. Aldehyde 2 (0.427 g, 0.59 mmol) dissolved in dry THF (1.0 mL) was added slowly to the mixture and stirred for another 3 h at 0 °C. The reaction was quenched with a saturated aqueous solution of ammonium chloride, extracted with ethyl acetate (10 mL × 3) and the combined organic layers dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue purified by silica gel chromatography to give compound 19 (0.60 g, 65%, E:Z = 77:23) as a yellow oil. 1H- NMR (CDCl3): δ 8.04 (d, J = 15.6 Hz, 1H), 7.18 (d, J = 8.0 Hz, 1H), 7.01 (d, J = 16.0 Hz, 1H), 6.99 (s, 1H), 6.96 (d, J = 8.8 Hz, 1H), 6.81 (d, J = 8.4 Hz, 1H), 6.73 (s, 1H), 6.72 (d, J = 8.8 Hz, 1H), 6.63 (d, J = 7.2 Hz, 1H), 6.45 (d, J = 16.8 Hz, 1H), 6.32 (d, J = 15.6 Hz, 1H), 5.25 (br s, 1H), 3.69 (s, 3H), 2.97–3.08 (m, 2H), 1.06 (s, 18H), 0.98 (s, 18H), 0.96 (s, 18H), 0.28 (s, 6H), 0.23 (s, 12H), 0.19 (s, 12H), 0.11 (s, 6H).; 13C-NMR (CDCl3): δ 169.9, 165.9, 148.3, 146.6, 146.4, 146.1, 145.5, 145.3, 144.2, 136.7, 133.0, 130.7, 128.6, 126.5, 121.9, 121.7, 121.1, 120.6, 120.4, 119.9, 119.1, 118.7, 115.0, 72.6, 51.6, 36.4, 25.7, 25.5, 25.5, 18.3, 18.0, 17.9, −3.8, −4.0, −4.0, −4.5; HRMS (TOF-MS): m/z calcd for C63H108O10Si6Na [M + Na]+ 1215.6450. Found 1215.6457.
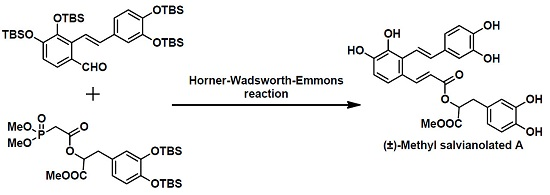
3.16. 3-(3,4-Dihydroxyphenyl)-1-methoxy-1-oxopropan-2-y(E)-3-(2-((E)-3,4-dihydroxystyryl)-3,4-dihydroxyphenyl)acrylate (1)
Et3N•3HF (0.81 g, 5.02 mmol) was added to a solution of 19 (0.50 g, 0.42 mmol) in dry pyridine (4 mL) at 0 °C under a nitrogen atmosphere. The reaction mixture was stirred for 1 h and then diluted with methanol. The mixture was extracted with CH2Cl2 (10 mL × 3) and the combined organic layers washed with brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The resulting residue was purified by silica gel chromatography to give (±)-methyl salvianolate A (1, 0.196 g, 92%) as a yellow solid. 1H-NMR (CD3OD): δ 8.00 (d, J = 16.0 Hz, 1H), 7.07 (d, J = 16.4 Hz, 1H), 7.05 (d, J = 8.0 Hz, 1H), 6.98( s, 1H), 6.81 (d, J = 7.2 Hz, 1H), 6.69 (d, J = 8.0 Hz, 1H), 6.68 (d, J = 8.4 Hz, 1H), 6.58 (s, 1H), 6.57 (d, J = 7.6 Hz, 1H), 6.56 (d, J = 16.0 Hz, 1H), 6.44 (d, J = 8.0 Hz, 1H), 6.21 (d, J = 16.0 Hz, 1H), 5.11 (dd, J = 5.2 and 7.2 Hz, 1H), 3.61 (s, 3H), 2.89-2.94 (m, 1H).; 13C-NMR (CD3OD): δ 170.8, 167.1, 146.9, 146.2, 145.3, 145.1, 144.7, 143.9, 143.0, 136.5, 130.0, 127.4, 127.1, 124.7, 120.6, 119.3, 119.0, 118.7, 116.0, 115.1, 115.0, 113.9, 113.5, 112.7, 73.3, 51.3, 36.5; HRMS (TOF-MS): m/z calcd for C27H24O10Na [M + Na]+ 531.1262. Found 531.1262.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
