Catalytic Asymmetric Fluorination of Alkyl 1-indanone-2-carboxylates Ruled by Pybox-Eu(III) Combination


Abstract
:
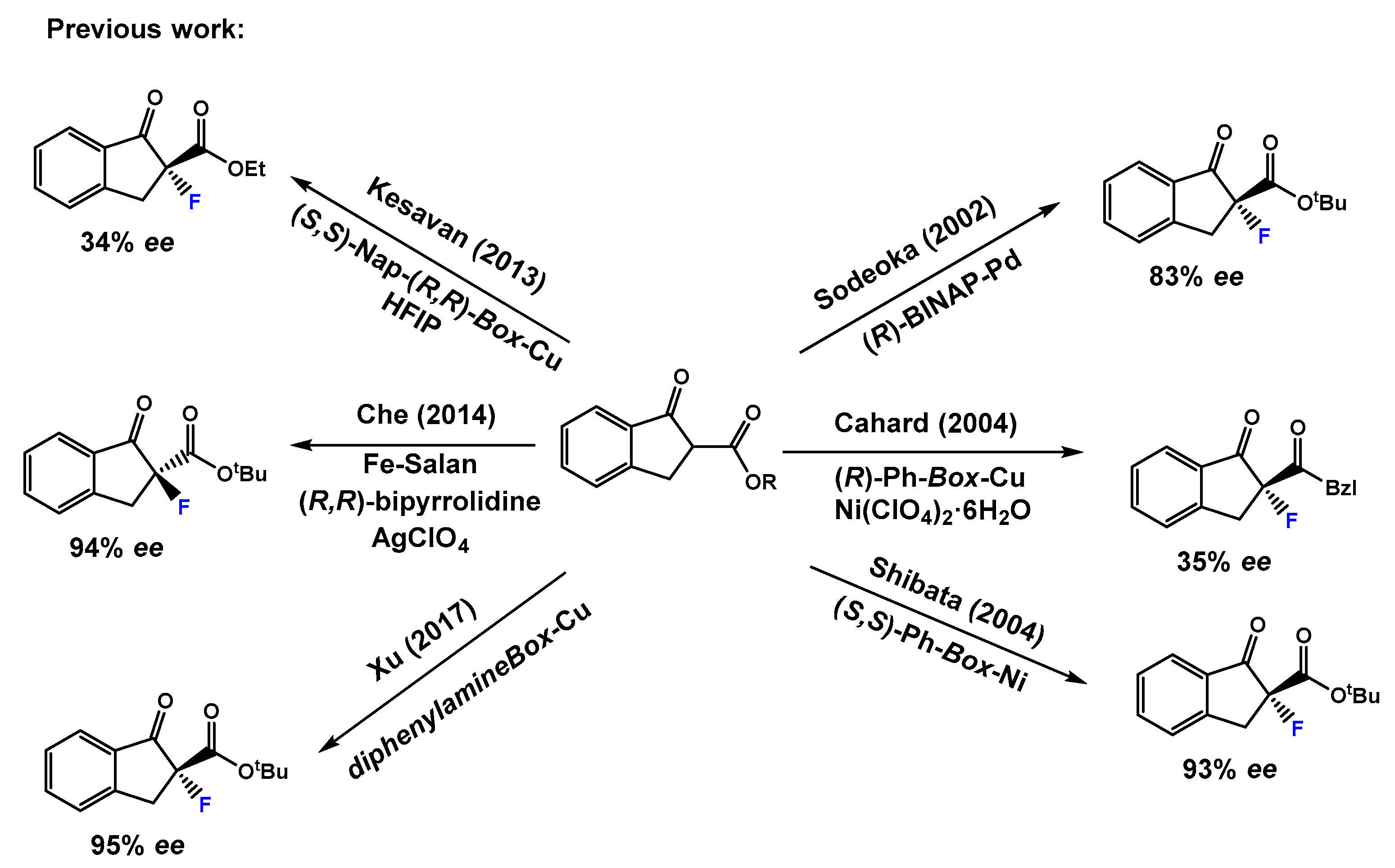
1. Introduction
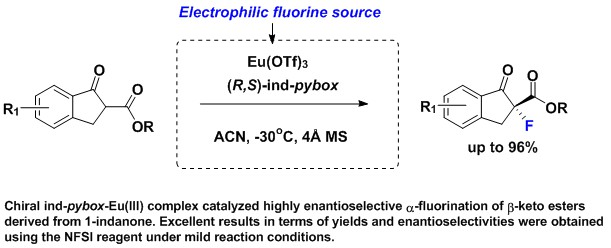
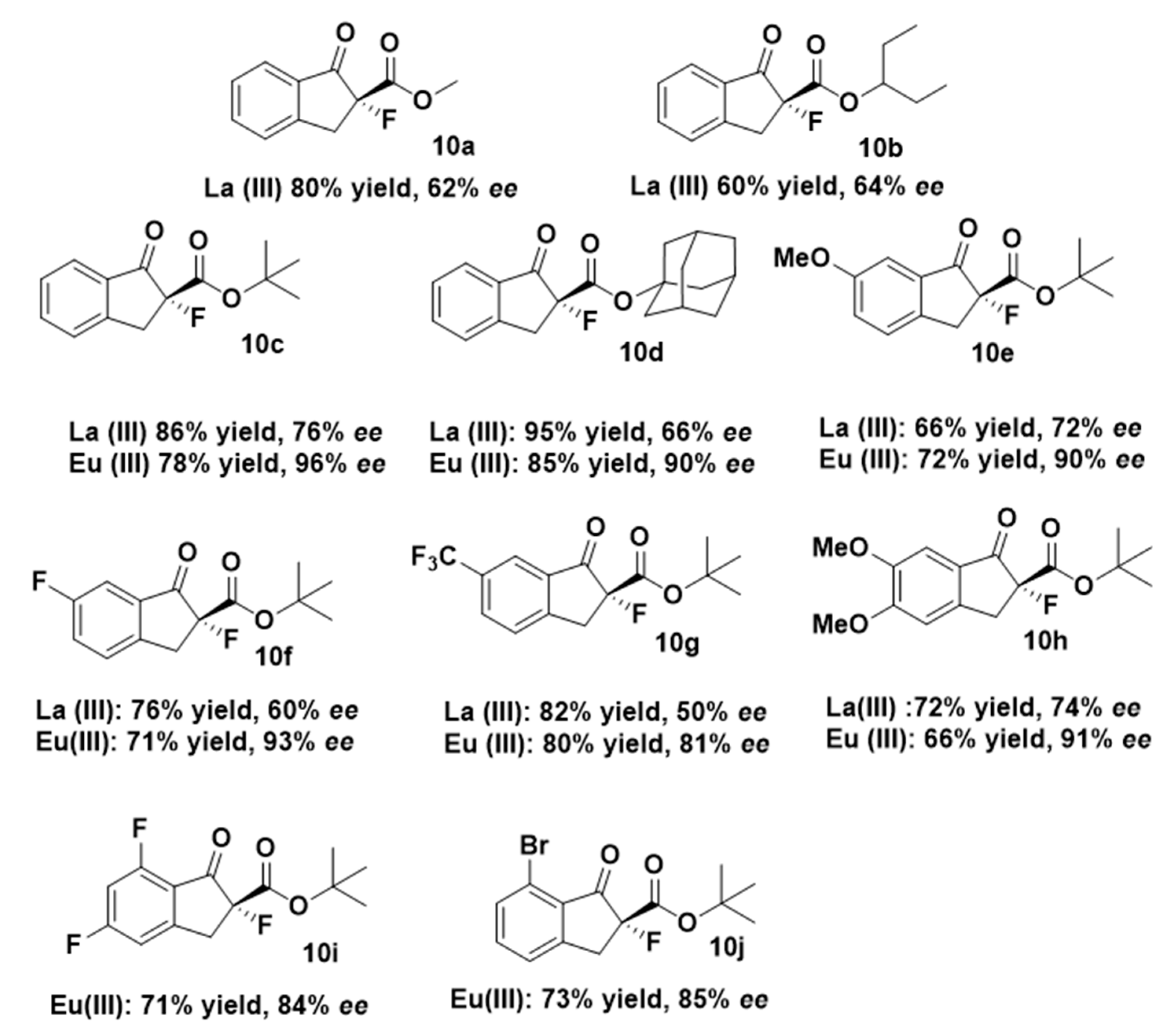
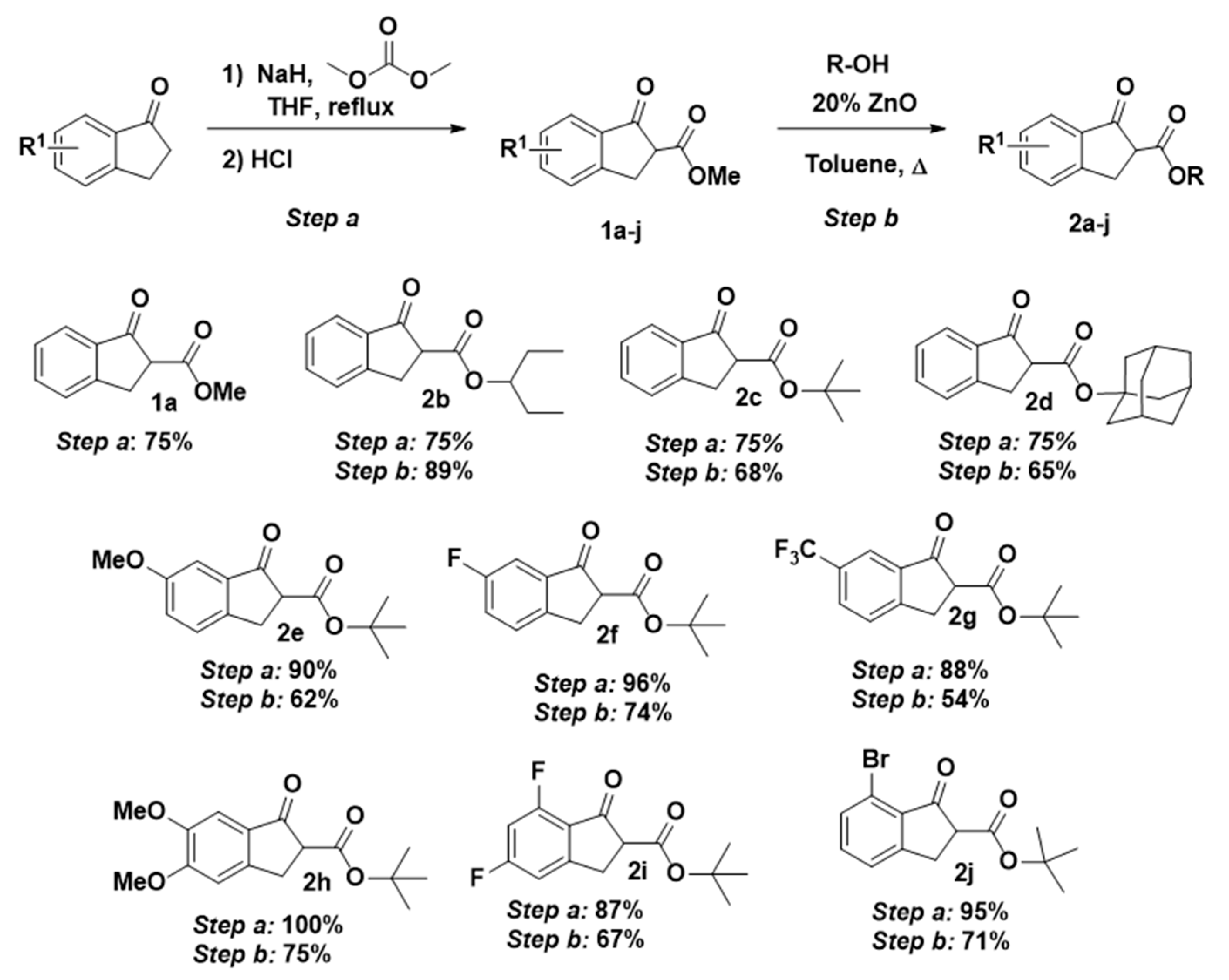
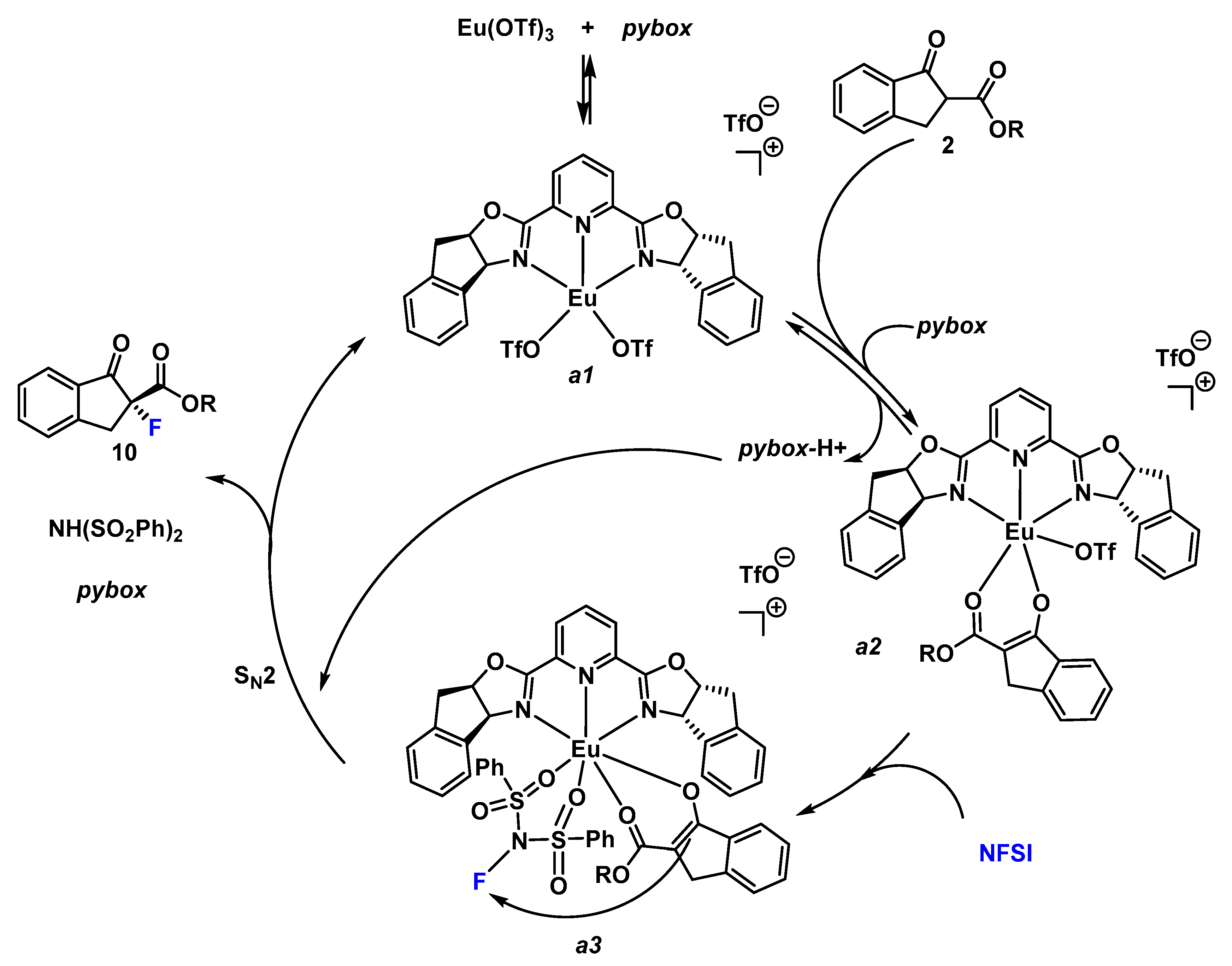
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Gladysz, J.A.; Curran, D.E.; Horváth, I.T. Handbook of Fluorous Chemistry; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smart, B.E. Fluorine Substituent Effects (on Bioactivity). J. Fluor. Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- O’Hagan, D. Understanding Organofluorine Chemistry. An Introduction to the C–F Bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Isanbor, C.; O’Hagan, D. Fluorine in Medicinal Chemistry: A Review of Anti-cancer Agents. J. Fluor. Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Hobpken, NY, USA, 2009. [Google Scholar]
- O’Hagan, D. Fluorine in Health Care: Organofluorine Containing Blockbuster Drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Ilardi, E.A.; Vitaku, E.; Njardarson, J.T. Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals To Reveal Opportunities for Drug Design and Discovery. J. Med. Chem. 2014, 57, 2832–2842. [Google Scholar] [CrossRef]
- Wang, J.; Sanchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Ghosh, A.; Nakanishi, T. Frontiers of Solvent-free Functional Molecular Liquids. Chem. Comm. 2017, 53, 10344–10357. [Google Scholar] [CrossRef]
- Pihko, P. Enantioselective α-Fluorination of Carbonyl Compounds: Organocatalysis or Metal Catalysis? Angew. Chem. Int. Ed. 2006, 45, 544–547. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.S.; Toste, F.D. Modern Approaches for Asymmetric Construction of Carbon–Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, W.; Zhang, L.; Luo, T.; Wan, X.; Gu, Y.; Hu, J. Enantioselective Synthesis of Cyclopropanes that Contain Fluorinated Tertiary Stereogenic Carbon Centers: A Chiral α-Fluoro Carbanion Strategy. Angew. Chem. Int. Ed. 2012, 51, 6966–6970. [Google Scholar] [CrossRef]
- Differding, E.; Ofner, H. N-Fluorobenzenesulfonimide: A Practical Reagent for Electrophilic Fluorinations. Synlett 1991, 187–189. [Google Scholar] [CrossRef]
- Nyffeler, P.T.; Gonzalez, S.; Burkart, M.D.; Vicent, S.P.; Wong, C.-H. Selectfluor: Mechanistic Insight and Applications. Angew. Chem. Int. Ed. 2005, 44, 192–202. [Google Scholar] [CrossRef]
- Hintermann, L.; Togni, A. Catalytic Enantioselective Fluorination of β-Ketoesters. Angew. Chem. Int. Ed. 2000, 39, 4359–4362. [Google Scholar] [CrossRef]
- Hamashima, Y.; Yagi, K.; Takano, H.; Tamás, L.; Sodeoka, M. An Efficient Enantioselective Fluorination of Various β-Ketoesters Catalyzed by Chiral Palladium Complexes. J. Am. Chem. Soc. 2002, 124, 14530–14531. [Google Scholar] [CrossRef]
- Hamashima, Y.; Takano, H.; Hotta, D.; Sodeoka, M. Immobilization and Reuse of Pd Complexes in Ionic Liquid: Efficient Catalytic Asymmetric Fluorination and Michael Reactions with β-Ketoesters. Org. Lett. 2003, 5, 3225–3228. [Google Scholar] [CrossRef]
- Suzuki, T.; Goto, T.; Hamashima, Y.; Sodeoka, M. Enantioselective Fluorination of tert-Butoxycarbonyl Lactones and Lactams Catalyzed by Chiral Pd(II)-Bisphosphine Complexes. J. Org. Chem. 2007, 72, 246–250. [Google Scholar] [CrossRef]
- Ibrahim, H.; Togni, A. Enantioselective Halogenation Reactions. Chem. Commun. 2004, 0, 1147–1155. [Google Scholar] [CrossRef]
- Huber, D.P.; Stanek, K.; Togni, A. Consecutive Catalytic Electrophilic Fluorination/Amination of β-Keto esters: Toward α-Fluoro-α-amino Acids? Tetrahedron-Asymmetr 2006, 17, 658–664. [Google Scholar] [CrossRef]
- Ma, J.-A.; Cahard, D. Copper(II) Triflate-bis(oxazoline)-catalysed Enantioselective Electrophilic Fluorination of β-Ketoesters. Tetrahedron-Asymmetr 2004, 15, 1007. [Google Scholar] [CrossRef]
- Shibata, N.; Ishimaru, T.; Nagai, T.; Kohno, J.; Toru, T. First Enantio-Flexible Fluorination Reaction Using Metal-Bis(oxazoline) Complexes. Synlett 2004, 10, 1703–1706. [Google Scholar] [CrossRef]
- Balaraman, K.; Vasanthan, R.; Kesavan, V. Enantioselective Fluorination of β-Ketoesters using Tartrate Derived Bidentate Bisoxazoline-Cu(II) Complexes. Tetrahedron-Asymmetr 2013, 24, 919–924. [Google Scholar] [CrossRef]
- Gu, X.; Zhang, Y.; Xu, Z.-J.; Che, C.-M. Iron(III)–salan complexes catalysed highly enantioselective fluorination and hydroxylation of β-keto esters and N-Boc oxindoles. Chem. Commun. 2014, 50, 7870–7873. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Jiang, Y.; Zhang, C.; Shao, J.; Xu, D. Fast, solvent-free and highly enantioselective fluorination of β-keto esters catalyzed by chiral copper complexes in a ball mill. Green Chem. 2017, 19, 1674–1677. [Google Scholar] [CrossRef]
- Comelles, J.; Pericas, A.; Moreno-Mañas, M.; Vallribera, A.; Drudis-Solé, G.; Lledós, A.; Parella, T.; Roglans, A.; García-Granda, S.; Roces-Fernández, L. Highly Enantioselective Electrophilic Amination and Michael Addition of Cyclic β-Ketoesters Induced by Lanthanides and (S,S)-ip-pybox: The Mechanism. J. Org. Chem. 2007, 72, 2077–2087. [Google Scholar] [CrossRef]
- Pericas, A.; Shafir, A.; Vallribera, A. Asymmetric Synthesis of l-Carbidopa Based on a Highly Enantioselective α-Amination. Org. Lett. 2013, 15, 1448–1451. [Google Scholar] [CrossRef]
- Pericas, A.; Jiménez, R.; Granados, A.; Shafir, A.; Vallribera, A.; Roglans, A.; Molins, E. Lanthanides–pybox: An Excellent Combination for Highly Enantioselective Electrophilic α-Amination of Acyclic β-Keto Esters. Isolation of Ternary Pybox/Ln/β-Keto Ester Complexes. ChemistrySelect. 2016, 1, 4305–4312. [Google Scholar] [CrossRef]
- Granados, A.; del Olmo, A.; Peccati, F.; Billard, T.; Sodupe, M.; Vallribera, A. Fluorous l-Carbidopa Precursors: Highly Enantioselective Synthesis and Computational Prediction of Bioactivity. J. Org. Chem. 2018, 83, 303–313. [Google Scholar] [CrossRef]
- Pericas, A.; Shafir, A.; Vallribera, A. ZnO-Catalyzed Transesterification of.β-Keto Esters. Tetrahedron 2008, 64, 9258–9263. [Google Scholar] [CrossRef]
- Meng, J.-C.; Fokin, V.V.; Finn, M.G. Kinetic resolution by copper-catalyzed azide-alkyne cycloaddition. Tetrahedron Lett. 2005, 46, 4543–4546. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Qiu, J.; Kong, D.; Gao, Y.; Lu, F.; Karmarker, P.G.; Chen, F.-X. The direct electrophilic cyanation of β-keto esters and amides with cyano benziodoxole. Org. Biomol. Chem. 2015, 13, 365–368. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | 2 | Metal | Ligand | Fluorinating Reagent | Yield (%) | ee (%)[a] |
|---|---|---|---|---|---|---|
| 1 | 2b | Eu+3 | 3 | NFSI | 76 | 2 |
| 2 | 2b | La+3 | 3 | NFSI | 74 | 5 |
| 3 | 2b | Yb+3 | 3 | NFSI | 68 | 2 |
| 4 | 2b | La+3 | 4 | NFSI | 81 | 56 |
| 5 | 2b | Eu+3 | 4 | NFSI | 45 | 30 |
| 6 | 2b | Yb+3 | 4 | NFSI | 78 | 54 |
| 7 | 2b | La+3 | 5 | NFSI | 85 | 50 |
| 8 | 2b | La+3 | 6 | NFSI | 87 | 60 |
| 9 | 2b | Eu+3 | 6 | NFSI | 72 | 78 |
| 10 | 2b | Yb+3 | 6 | NFSI | 70 | 60 |
| 11 | 2b | La+3 | 6 | 8 | 59 | 14 |
| 12 | 2b | La+3 | 6 | 9 | 0 | n.d. |
| 13[b] | 1b | La+3 | 6 | NFSI | 60 | 64 |
| 14[b] | 1a | La+3 | 6 | NFSI | 80 | 63 |
| 15[b] | 2c | La+3 | 6 | NFSI | 86 | 76 |
| 16[b] | 2c | Eu+3 | 6 | NFSI | 78 | 96 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granados, A.; Sarró, P.; Vallribera, A. Catalytic Asymmetric Fluorination of Alkyl 1-indanone-2-carboxylates Ruled by Pybox-Eu(III) Combination. Molecules 2019, 24, 1141. https://doi.org/10.3390/molecules24061141
Granados A, Sarró P, Vallribera A. Catalytic Asymmetric Fluorination of Alkyl 1-indanone-2-carboxylates Ruled by Pybox-Eu(III) Combination. Molecules. 2019; 24(6):1141. https://doi.org/10.3390/molecules24061141
Chicago/Turabian StyleGranados, Albert, Pau Sarró, and Adelina Vallribera. 2019. "Catalytic Asymmetric Fluorination of Alkyl 1-indanone-2-carboxylates Ruled by Pybox-Eu(III) Combination" Molecules 24, no. 6: 1141. https://doi.org/10.3390/molecules24061141
APA StyleGranados, A., Sarró, P., & Vallribera, A. (2019). Catalytic Asymmetric Fluorination of Alkyl 1-indanone-2-carboxylates Ruled by Pybox-Eu(III) Combination. Molecules, 24(6), 1141. https://doi.org/10.3390/molecules24061141