Design and Synthesis of New Quinoxaline Derivatives as Anticancer Agents and Apoptotic Inducers


Abstract
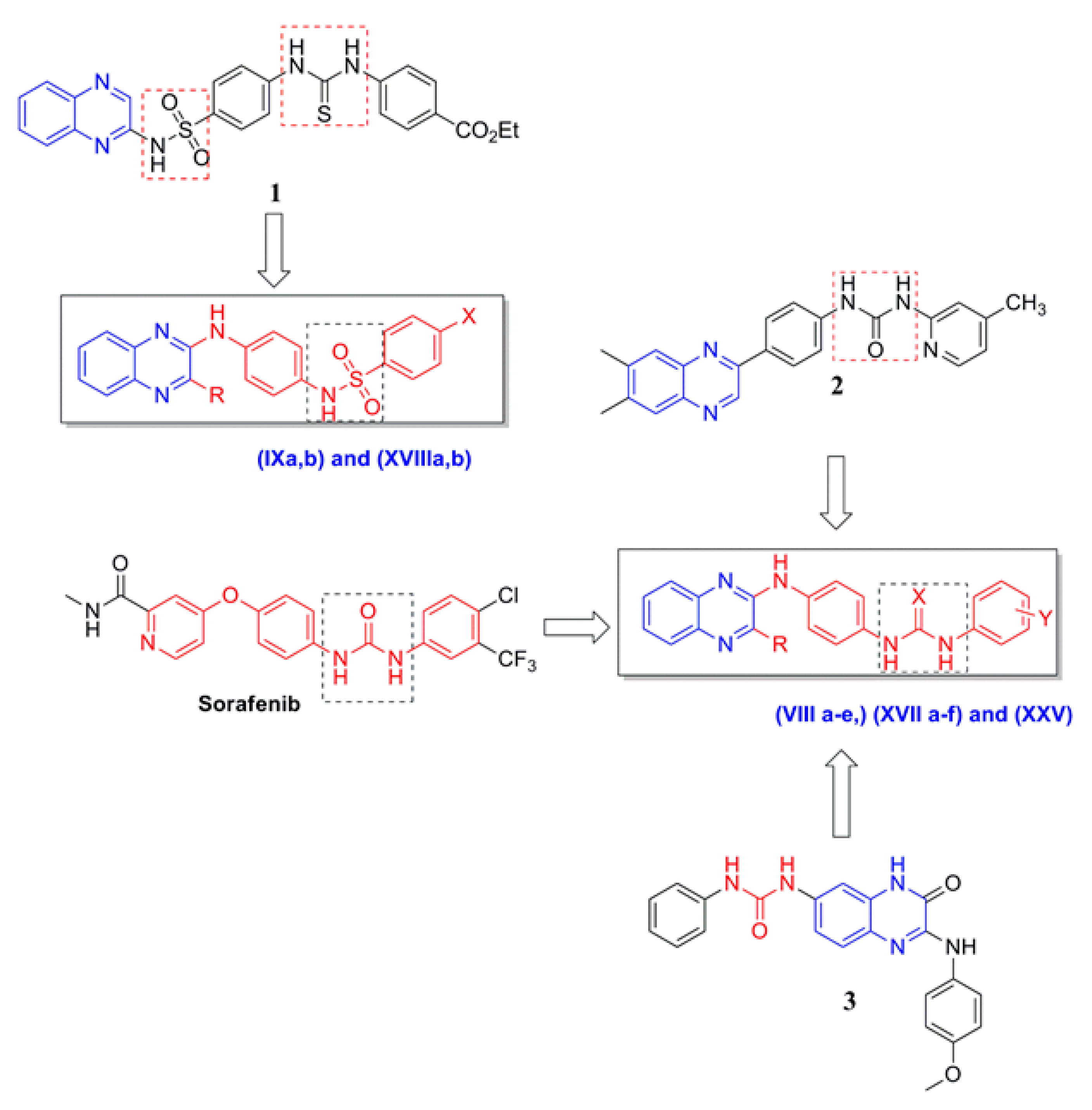
:1. Introduction
2. Results and Discussion
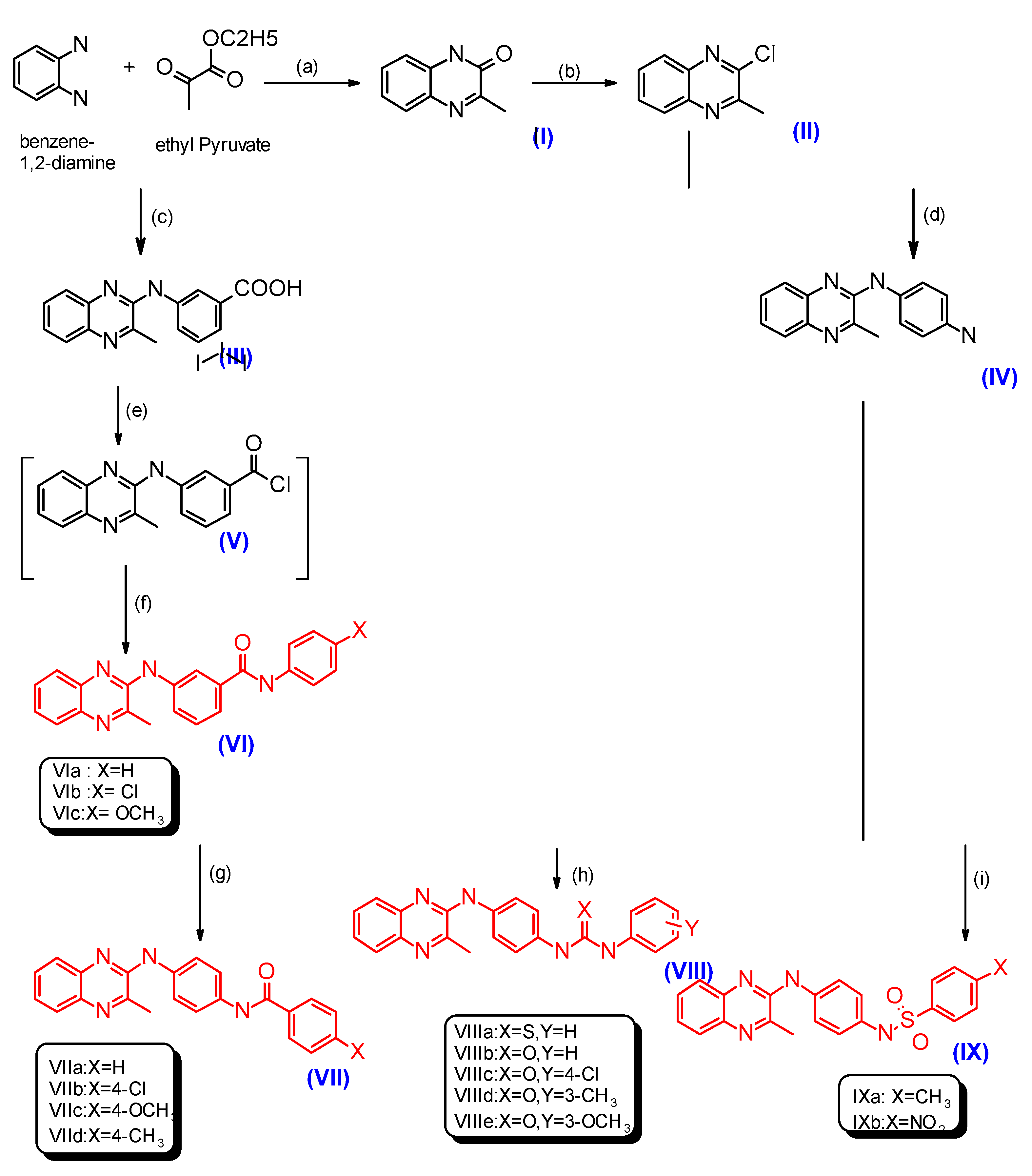
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In Vitro Cell Proliferation Assay
2.2.2. In Vitro VEGFR-2 Inhibition Assay
2.2.3. Cell Cycle Analysis
2.2.4. Apoptosis Determination
2.2.5. In Vitro Cytotoxic Assay
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.2.1. General Procedures for the Synthesis of Compounds VIa–c, XVa–c and XXIVa,b
3.2.2. 3-((3-Methylquinoxalin-2-yl)amino)-N-phenylbenzamide (VIa)
3.2.3. N-(4-Chlorophenyl)-3-((3-methylquinoxalin-2-yl)amino)benzamide (VIb)
3.2.4. N-(4-Methoxyphenyl)-3-((3-methylquinoxalin-2-yl)amino)benzamide (VIc)
3.2.5. 3-((3-Chloroquinoxalin-2-yl)amino)-N-phenylbenzamide (XVa)
3.2.6. N-(4-Chlorophenyl)-3-((3-chloroquinoxalin-2-yl)amino)benzamide (XVb)
3.2.7. 3-((3-Chloroquinoxalin-2-yl)amino)-N-(4-methoxyphenyl)benzamide (XVc)
3.2.8. N-(4-chlorophenyl)-3-(quinoxalin-2-ylamino)benzamide (XXIVa)
3.2.9. N-(4-Methoxyphenyl)-3-(quinoxalin-2-ylamino)benzamide (XXIVb)
3.2.10. General Procedures for the Synthesis of Compounds VIIa–d and XVIa–d
3.2.11. N-(4-((3-Methylquinoxalin-2-yl)amino)phenyl)benzamide (VIIa)
3.2.12. 4-Chloro-N-(4-((3-methylquinoxalin-2-yl)amino)phenyl)benzamide (VIIb)
3.2.13. 4-Methoxy-N-(4-((3-methylquinoxalin-2-yl)amino)phenyl)benzamide (VIIc)
3.2.14. 4-Methyl-N-(4-((3-methylquinoxalin-2-yl)amino)phenyl)benzamide (VIId)
3.2.15. N-(4-((3-Chloroquinoxalin-2-yl)amino)phenyl)benzamide (XVIa)
3.2.16. 4-Chloro-N-(4-((3-chloroquinoxalin-2-yl)amino)phenyl) benzamide (XVIb)
3.2.17. N-(4-((3-Chloroquinoxalin-2-yl)amino)phenyl)-4-methoxybenzamide (XVIc)
3.2.18. N-(4-((3-Chloroquinoxalin-2-yl)amino)phenyl)-4-methylbenzamide (XVId)
3.2.19. General Procedures for the Synthesis of Compounds VIIIa–e, XVIIa–f and XXV
3.2.20. 1-(4-((3-Methylquinoxalin-2-yl)amino)phenyl)-3-phenylthiourea (VIIIa)
3.2.21. 1-(4-((3-Methylquinoxalin-2-yl)amino)phenyl)-3-phenylurea (VIIIb)
3.2.22. 1-(4-Chlorophenyl)-3-(4-((3-methylquinoxalin-2-yl)amino)phenyl)urea (VIIIc)
3.2.23. 1-(4-((3-Methylquinoxalin-2-yl)amino)phenyl)-3-(m-tolyl)urea (VIIId)
3.2.24. 1-(3-Methoxyphenyl)-3-(4-((3-methylquinoxalin-2-yl)amino)phenyl)urea (VIIIe)
3.2.25. 1-(4-((3-Chloroquinoxalin-2-yl)amino)phenyl)-3-phenylthiourea (XVIIa)
3.2.26. 1-(4-((3-Chloroquinoxalin-2-yl)amino)phenyl)-3-phenylurea (XVIIb)
3.2.27. 1-(4-Chlorophenyl)-3-(4-((3-chloroquinoxalin-2-yl)amino)phenyl)urea (XVIIc)
3.2.28. 1-(4-((3-Chloroquinoxalin-2-yl)amino)phenyl)-3-(m-tolyl) urea (XVIId)
3.2.29. 1-(4-((3-Chloroquinoxalin-2-yl)amino)phenyl)-3-(3-methoxyphenyl) urea (XVIIe)
3.2.30. 1-(3-Methoxyphenyl)-3-(4-(quinoxalin-2-ylamino)phenyl)urea (XXV)
3.2.31. 1-(4-((3-Chloroquinoxalin-2-yl)amino)phenyl)-3-(4-methoxyphenyl)urea (XVIIf)
3.2.32. General Procedures for the Synthesis of Compounds IXa,b and XVIIIa,b
3.2.33. 4-Methyl-N-(4-((3-methylquinoxalin-2-yl)amino)phenyl) benzenesulphonamide (IXa)
3.2.34. N-(4-((3-Methylquinoxalin-2-yl)amino)phenyl)-4-nitrobenzenesulfonamide (IXb)
3.2.35. N-(4-((3-Chloroquinoxalin-2-yl)amino)phenyl)-4-methylbenzenesulfonamide (XVIIIa)
3.2.36. N-(4-((3-Chloroquinoxalin-2-yl)amino)phenyl)-4-nitrobenzenesulfonamide (XVIIIb)
3.3. Biological Studies
3.3.1. In-Vitro Anti-Cancer Activity
3.3.2. In-Vitro VEGFR-2 Inhibition Assay
3.3.3. Cell Cycle Analysis
3.3.4. Apoptosis Determination
3.3.5. In Vitro Cytotoxic Assay
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zong, X.; Chen, J.; Li, L.; Cai, J.; Sun, C.; Ji, M. Discovery of 3,3a,4,5-tetrahydro-2H-benzo[g]indazole containing quinoxaline derivatives as novel EGFR/HER-2 dual inhibitors. RSC Adv. 2015, 5, 24814–24823. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Madhusudan, S.; Ganesan, T.S. Tyrosine kinase inhibitors in cancer therapy. Clin. Biochem. 2004, 37, 618–635. [Google Scholar] [CrossRef] [PubMed]
- Hoefnagel, A.J.; Van Koningsveld, H.; Van Meurs, F.; Peters, J.A.; Sinnema, A.; Van Bekkum, H. Reactions of hydroxyglycines. New synthetic routes to 4-phenylquinazoline derivatives. Tetrahedron 1993, 49, 6899–6912. [Google Scholar] [CrossRef]
- Noolvi, M.N.; Patel, H.M.; Bhardwaj, V.; Chauhan, A. Synthesis and in vitro antitumor activity of substituted quinazoline and quinoxaline derivatives: Search for anticancer agent. Eur. J. Med. Chem. 2011, 46, 2327–2346. [Google Scholar] [CrossRef]
- El Newahie, A.M.S.; Ismail, N.S.M.; El Ella, D.A.A.; Abouzid, K.A.M. Quinoxaline-Based Scaffolds Targeting Tyrosine Kinases and Their Potential Anticancer Activity. Arch. Pharm. Chem. Life Sci. 2016, 349, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Zghaib, Z.; Guichou, J.-F.; Vappiani, J.; Bec, N.; Hadj-Kaddour, K.; Vincent, L.-A.; Paniagua-Gayraud, S.; Larroque, C.; Moarbess, G.; Cuq, P.; et al. New imidazoquinoxaline derivatives: Synthesis, biological evaluation on melanoma, effect on tubulin polymerization and structure–activity relationships. Bioorg. Med. Chem. 2016, 24, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Balderas-Renteria, I.; González-Barranco, P.; García, A.; Banik, B.K.; Rivera, G. Anticancer Drug Design Using Scaffolds of β-Lactams, Sulfonamides, Quinoline, Quinoxaline and Natural Products. Drugs Advances in Clinical Trials. CMC 2012, 19, 4377–4398. [Google Scholar] [CrossRef]
- Ghorab, M.; Ragab, F.; Heiba, H.; El-Gazzar, M.; El-Gazzar, M. Synthesis, in vitro anticancer screening and radiosensitizing evaluation of some new 4-[3-(substituted)thioureido]-N-(quinoxalin-2-yl)-benzenesulfonamide derivatives. Acta Pharmaceutica 2011, 61, 415–425. [Google Scholar] [CrossRef]
- Göring, S.; Bensinger, D.; Naumann, E.C.; Schmidt, B. Computer-Guided Design, Synthesis, and Biological Evaluation of Quinoxalinebisarylureas as FLT3 Inhibitors. ChemMedChem 2015, 10, 511–522. [Google Scholar] [CrossRef]
- Gali-Muhtasib, H.U.; Diab-Assaf, M.; Haddadin, M.J. Retraction Note to: Quinoxaline 1,4-dioxides induce G2/M cell cycle arrest and apoptosis in human colon cancer cells. Cancer Chemother. Pharmacol. 2018, 81, 627. [Google Scholar] [CrossRef]
- Weng, Q.; Wang, D.; Guo, P.; Fang, L.; Hu, Y.; He, Q.; Yang, B. Q39, a novel synthetic Quinoxaline 1,4-Di-N-oxide compound with anti-cancer activity in hypoxia. Eur. J. Pharmacol. 2008, 581, 262–269. [Google Scholar] [CrossRef]
- Shahin, M.I.; El Ella, D.A.A.; Ismail, N.S.; Abouzid, K.A. Design, synthesis and biological evaluation of type-II VEGFR-2 inhibitors based on quinoxaline scaffold. Bioorg. Chem. 2014, 56, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Ramurthy, S.; Costales, A.; Jansen, J.M.; Levine, B.; Renhowe, P.A.; Shafer, C.M.; Subramanian, S. Design and synthesis of 6,6-fused heterocyclic amides as raf kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 1678–1681. [Google Scholar] [CrossRef] [PubMed]
- Ghanbarimasir, Z.; Bekhradnia, A.; Morteza-Semnani, K.; Rafiei, A.; Razzaghi-Asl, N.; Kardan, M. Design, synthesis, biological assessment and molecular docking studies of new 2-aminoimidazole-quinoxaline hybrids as potential anticancer agents. Spectrochim. Acta A 2018, 194, 21–35. [Google Scholar] [CrossRef]
- Lu, C.; Tang, K.; Li, Y.; Li, P.; Lin, Z.; Yin, D.; Chen, X.; Huang, H. Design, synthesis and evaluation of novel diaryl urea derivatives as potential antitumor agents. Eur. J. Med. Chem. 2014, 77, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Yao, Q.; Yu, S.; Gong, P.; Qin, M. Synthesis and Antitumor Activity of Triazole-Containing Sorafenib Analogs. Molecules 2017, 22, 1759. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib Blocks the RAF/MEK/ERK Pathway, Inhibits Tumor Angiogenesis, and Induces Tumor Cell Apoptosis in Hepatocellular Carcinoma Model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [Green Version]
- Gris, J.; Glisoni, R.; Fabian, L.; Fernández, B.; Moglioni, A.G. Synthesis of potential chemotherapic quinoxalinone derivatives by biocatalysis or microwave-assisted Hinsberg reaction. Tetrahedron Lett. 2008, 49, 1053–1056. [Google Scholar] [CrossRef]
- Westphal, G.; Wasicki, H.; Zielinski, U.; Weber, F.G.; Tonew, M.; Tonew, E. Potential virostatics. 1. Quinoxalines. Die Pharmazie 1977, 32, 570–571. [Google Scholar]
- Singh, D.P.; Deivedi, S.K.; Hashim, S.R.; Singhal, R.G. Synthesis and Antimicrobial Activity of Some New Quinoxaline Derivatives. Pharmaceuticals 2010, 3, 2416–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.S.; Kim, H.M.; Park, Y.T.; Kim, H.S. Heterocyclic Compounds with Sulfone Functional Groups (II): Synthesis of 1-Arenesulfonyl-2-Quinoxalinones. Bull. Korean Chem. Soc. 2000, 21, 133–136. [Google Scholar]
- Romer, D.R. Synthesis of 2,3-dichloroquinoxalines via Vilsmeier-reagent chlorination. J. Heterocycl. Chem. 2009, 46, 317–319. [Google Scholar] [CrossRef]
- Galal, S.A.; Abdelsamie, A.S.; Tokuda, H.; Suzuki, N.; Lida, A.; Elhefnawi, M.M.; Ramadan, R.A.; Atta, M.H.; El Diwani, H.I.; Ayoub, S.A.G. Part I: Synthesis, cancer chemopreventive activity and molecular docking study of novel quinoxaline derivatives. Eur. J. Med. Chem. 2011, 46, 327–340. [Google Scholar] [CrossRef]
- Sastry, C.R.; Krishnan, V.; Narayan, G.; Vemana, K.; Vairamani, M. Reaction of 2, 3-Dichloroquinoxaline with Acid Hydrazides: A Convenient Synthesis of 1, 6-Disubstituted (1, 2, 4) Ditriazolo (4, 3-a: 3′, 4′-c)-and 3-Aryl/Heteroaryl (1, 3, 4) oxadiazino (5, 6-b) quinoxalines. Indian J. Chem. 1991, 30, 936–940. [Google Scholar] [CrossRef]
- Deng, J.; Feng, E.; Ma, S.; Zhang, Y.; Liu, X.; Li, H.; Huang, H.; Zhu, J.; Zhu, W.; Shen, X.; et al. Design and Synthesis of Small Molecule RhoA Inhibitors: A New Promising Therapy for Cardiovascular Diseases? J. Med. Chem. 2011, 54, 4508–4522. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, C.; Rao, V.S.; Deuther-Conrad, W.; Sridhar, D.; Nagesh, H.N.; Kumar, V.S.; Brust, P.; Kumar, M.M.K. Design, synthesis, and preliminary in vitro and in vivo pharmacological evaluation of 4-{4-[2-(4-(2-substitutedquinoxalin-3-yl) piperazin-1-yl) ethyl]phenyl} thiazoles as atypical antipsychotic agents. Med. Chem. Res. 2013, 22, 1660–1673. [Google Scholar] [CrossRef]
- Wang, S.; Yan, J.; Wang, X.; Yang, Z.; Lin, F.; Zhang, T. Synthesis and evaluation of the α-glucosidase inhibitory activity of 3-[4-(phenylsulfonamido)benzoyl]-2H-1-benzopyran-2-one derivatives. Eur. J. Med. Chem. 2010, 45, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Kakuta, H.; Zheng, X.; Oda, H.; Harada, S.; Sugimoto, Y.; Sasaki, K.; Tai, A. Cyclooxygenase-1-Selective Inhibitors Are Attractive Candidates for Analgesics That Do Not Cause Gastric Damage. Design and in Vitro/in Vivo Evaluation of a Benzamide-Type Cyclooxygenase-1 Selective Inhibitor. J. Med. Chem. 2008, 51, 2400–2411. [Google Scholar] [CrossRef]
- Cee, V.J.; Deak, H.L.; Du, B.; Geuns-Meyer, S.D.; Hodous, B.L.; Nguyen, H.N.; Olivieri, P.R.; Patel, V.F.; Romero, K.; Schenkel, L. Preparation of Substituted Phthalazinamines as Aurora Kinase Modulators. Patent WO2007087276, 2 August 2007. [Google Scholar]
- Dai, Y.; Hartandi, K.; Ji, Z.; Ahmed, A.A.; Albert, D.H.; Bauch, J.L.; Bouska, J.J.; Bousquet, P.F.; Cunha, G.A.; Glaser, K.B.; et al. Discovery of N-(4-(3-Amino-1H-indazol-4-yl)phenyl)-N‘-(2-fluoro-5-methylphenyl)urea (ABT-869), a 3-Aminoindazole-Based Orally Active Multitargeted Receptor Tyrosine Kinase Inhibitor. J. Med. Chem. 2007, 50, 1584–1597. [Google Scholar] [CrossRef]
- Brown, E.; Moudachirou, M. Agents de dédoublement. 2. Synthèse d’aryluréthanes de l’acide (S)-lactique et leur utilisation dans le dédoublement de bases racémiques. Tetrahedron 1994, 50, 10309–10320. [Google Scholar] [CrossRef]
- González-Álvarez, M.; Alzuet, G.; Borrás, J.; Agudo, L.D.C.; Garcĺa-Granda, S.; Bernardo, J.M.M. Strong protective action of Copper(II) N-substituted sulfonamide complexes against reactive oxygen species. J. Inorg. Biochem. 2004, 98, 189–198. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- A Scudiero, D.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M.R. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar] [PubMed]
Sample Availability: Some samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | Ring A (R) | Ring B (X) | HCT116 Cell Line IC50 (µM) | Hep G2 Cell Line IC50 (µM) | MCF-7 Cell Line IC50 (µM) |
|---|---|---|---|---|---|
| VIa | CH3 | H | 602.5 | >1000 | 692 |
| VIb | CH3 | Cl | 537 | 955 | 692 |
| VIc | CH3 | OCH3 | 275 | 479 | 288 |
| XVa | Cl | H | 4.4 | 10 | 5.3 |
| XVb | Cl | Cl | 15.5 | 27.5 | 21.4 |
| XVc | Cl | OCH3 | 182 | 912 | 380 |
| XXIVa | H | Cl | 219 | 436.5 | 182 |
| XXIVb | H | OCH3 | 302 | 490 | 219 |
| Doxorubicin | 0.62 | 1.2 | 0.9 |

| Compound | Ring A (R) | Ring B (X) | HCT116 Cell Line IC50 (µM) | Hep G2 Cell Line IC50 (µM) | MCF-7 Cell Line IC50 (µM) |
|---|---|---|---|---|---|
| VIIa | CH3 | H | 21.9 | 27.5 | 25.7 |
| VIIb | CH3 | Cl | 27.5 | 114.8 | 138 |
| VIIc | CH3 | OCH3 | 33.9 | 19 | 24 |
| VIId | CH3 | CH3 | 7.8 | 25.7 | 60.3 |
| XVIa | Cl | H | 346.7 | 512.9 | 323.6 |
| XVIb | Cl | Cl | >1000 | >1000 | >1000 |
| XVIc | Cl | 4-OCH3 | >1000 | >1000 | >1000 |
| Doxorubicin | 0.62 | 1.2 | 0.9 |

| Compound | Ring A R | X | Ring B Y | HCT116 Cell Line IC50 (µM) | Hep G2 Cell Line IC50 (µM) | MCF-7 Cell Line IC50 (µM) |
|---|---|---|---|---|---|---|
| VIIIa | CH3 | S | H | 12.3 | 9.8 | 15.5 |
| VIIIb | CH3 | O | H | 38 | 98 | 13.5 |
| VIIIc | CH3 | O | 4-Cl | 2.5 | 22 | 9 |
| VIIId | CH3 | O | 3-CH3 | 11 | 36 | 30 |
| VIIIe | CH3 | O | 3-OCH3 | 8.4 | 21.4 | 24.5 |
| XVIIa | Cl | S | H | 309 | 575 | 170 |
| XVIIb | Cl | O | H | 170 | 513 | 148 |
| XVIIc | Cl | O | 4-Cl | 257 | 616.5 | 692 |
| XVIId | Cl | O | 3-CH3 | 190.5 | 513 | 64.5 |
| XVIIe | Cl | O | 3-OCH3 | 40.7 | 55 | 47.9 |
| XVIIf | Cl | O | 4-OCH3 | 457 | 724 | 436.5 |
| XXV | H | O | 3-OCH3 | 31 | 64.5 | 32 |
| Doxorubicin | 0.62 | 1.2 | 0.9 |

| Compound | R | X | HCT116 Cell Line IC50 (µM) | Hep G2 Cell Line IC50 (µM) | MCF-7 Cell Line IC50 (µM) |
|---|---|---|---|---|---|
| IXa | CH3 | CH3 | 21.9 | 22.9 | 22.9 |
| IXb | CH3 | NO2 | 12 | 24.5 | 10.23 |
| XVIIIa | Cl | CH3 | 302 | 489.8 | 537 |
| XVIIIb | Cl | NO2 | >1000 | >1000 | 55 |
| Doxorubicin | 0.62 | 1.2 | 0.9 |
| Compounds | % of VEGFR-2 Inhibition | Compounds | % of VEGFR-2 Inhibition | Compounds | % of VEGFR-2 Inhibition |
|---|---|---|---|---|---|
| VIIa | 2 | VIIIa | 7 | VIa | 7 |
| XVIa | 8 | XVIIa | 10 | XVb | 7 |
| VIIb | 4 | VIIIb | 4 | XVc | 5 |
| XVIb | 4 | XVIIb | 10 | VIc | 5 |
| VIIc | 6 | XVIIf | 6 | XXIVb | 9 |
| XVIc | 4 | VIIIc | 7 | XXIVa | 3 |
| VIId | 3 | XVIIc | 6 | XXV | 21 |
| XVId | 3 | VIIId | 5 | Staurosporine | 100 |
| IXa | 3 | XVIId | 5 | ||
| XVIIIa | 4 | VIIIe | 6 | ||
| IXb | 3 | XVIIe | 8 | ||
| XVIIIb | 9 | XVa | 6 |
| Compound | IC50 (µM) |
|---|---|
| XVa | 163.6 ± 9.2 |
| VIIIa | 126.19 ± 6.81 |
| VIIIc | 97.2 ± 5.66 |
| VIId | 40.13 ± 3.42 |
| VIIIe | 96.4 ± 5.72 |
| staurosporine | 101.86 ± 6.34 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Newahie, A.M.S.; Nissan, Y.M.; Ismail, N.S.M.; Abou El Ella, D.A.; Khojah, S.M.; Abouzid, K.A.M. Design and Synthesis of New Quinoxaline Derivatives as Anticancer Agents and Apoptotic Inducers. Molecules 2019, 24, 1175. https://doi.org/10.3390/molecules24061175
El Newahie AMS, Nissan YM, Ismail NSM, Abou El Ella DA, Khojah SM, Abouzid KAM. Design and Synthesis of New Quinoxaline Derivatives as Anticancer Agents and Apoptotic Inducers. Molecules. 2019; 24(6):1175. https://doi.org/10.3390/molecules24061175
Chicago/Turabian StyleEl Newahie, Aliya M. S., Yassin M. Nissan, Nasser S. M. Ismail, Dalal A. Abou El Ella, Sohair M. Khojah, and Khaled A.M. Abouzid. 2019. "Design and Synthesis of New Quinoxaline Derivatives as Anticancer Agents and Apoptotic Inducers" Molecules 24, no. 6: 1175. https://doi.org/10.3390/molecules24061175
APA StyleEl Newahie, A. M. S., Nissan, Y. M., Ismail, N. S. M., Abou El Ella, D. A., Khojah, S. M., & Abouzid, K. A. M. (2019). Design and Synthesis of New Quinoxaline Derivatives as Anticancer Agents and Apoptotic Inducers. Molecules, 24(6), 1175. https://doi.org/10.3390/molecules24061175