
Oligonucleotide–Palladacycle Conjugates as Splice-Correcting Agents


 , , ,
, , , 
Abstract
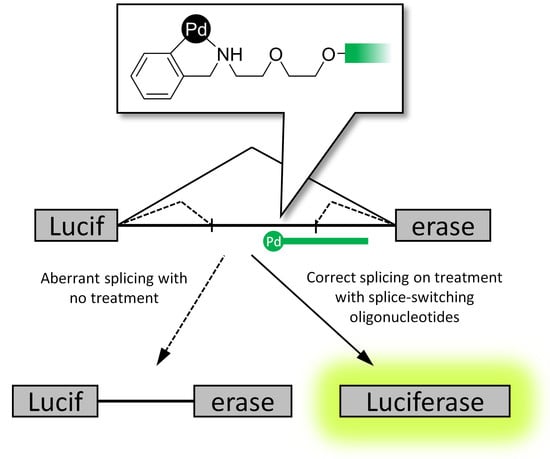
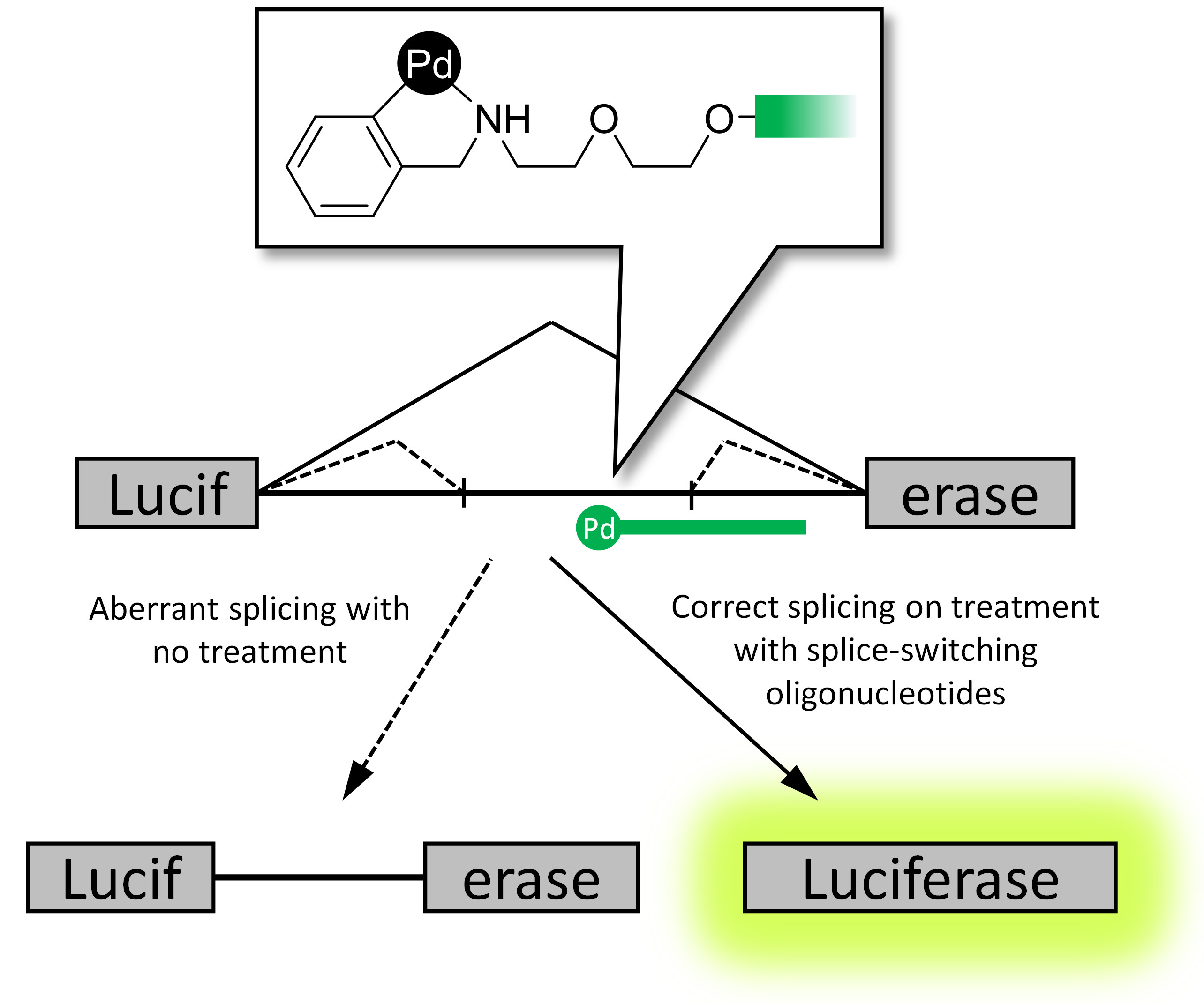
:
1. Introduction
2. Results
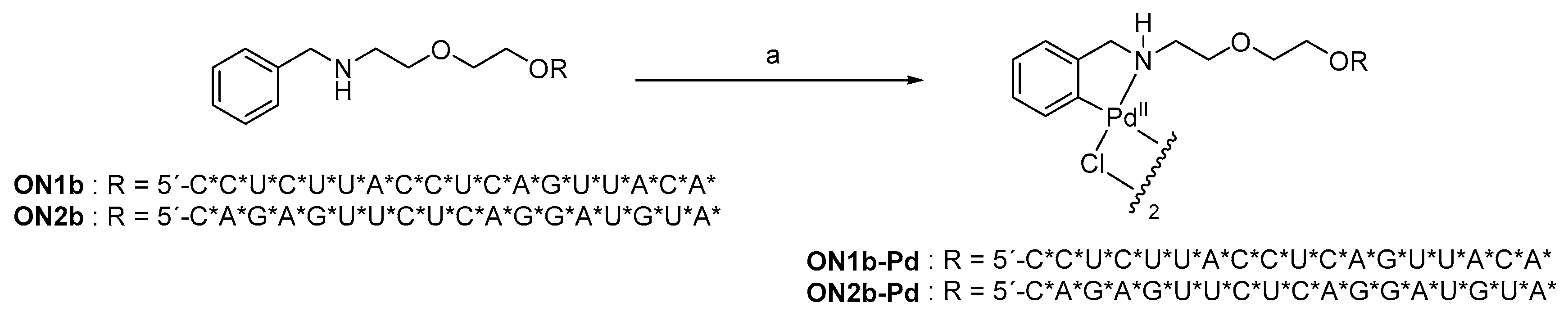
2.1. Oligonucleotide Synthesis
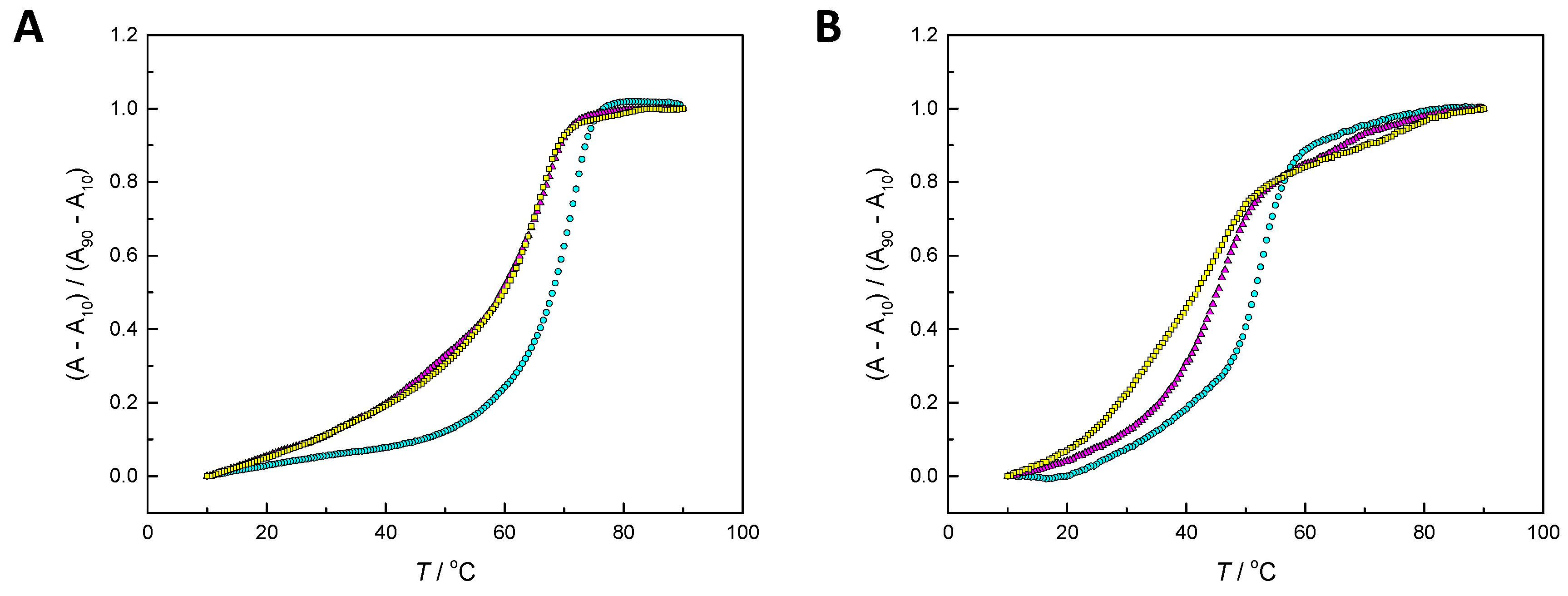
2.2. Hybridization Studies
2.3. Reporter Cell Lines
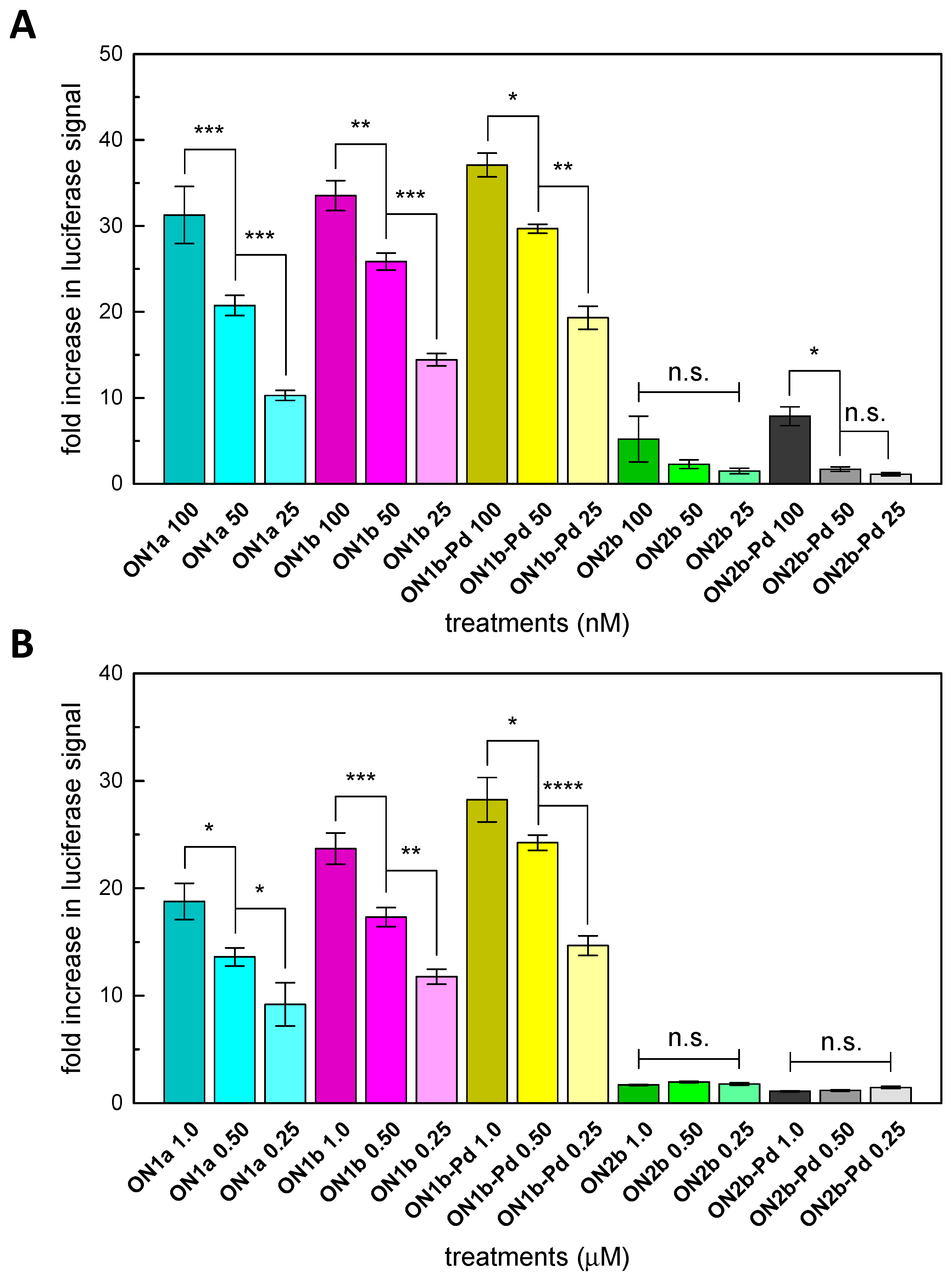
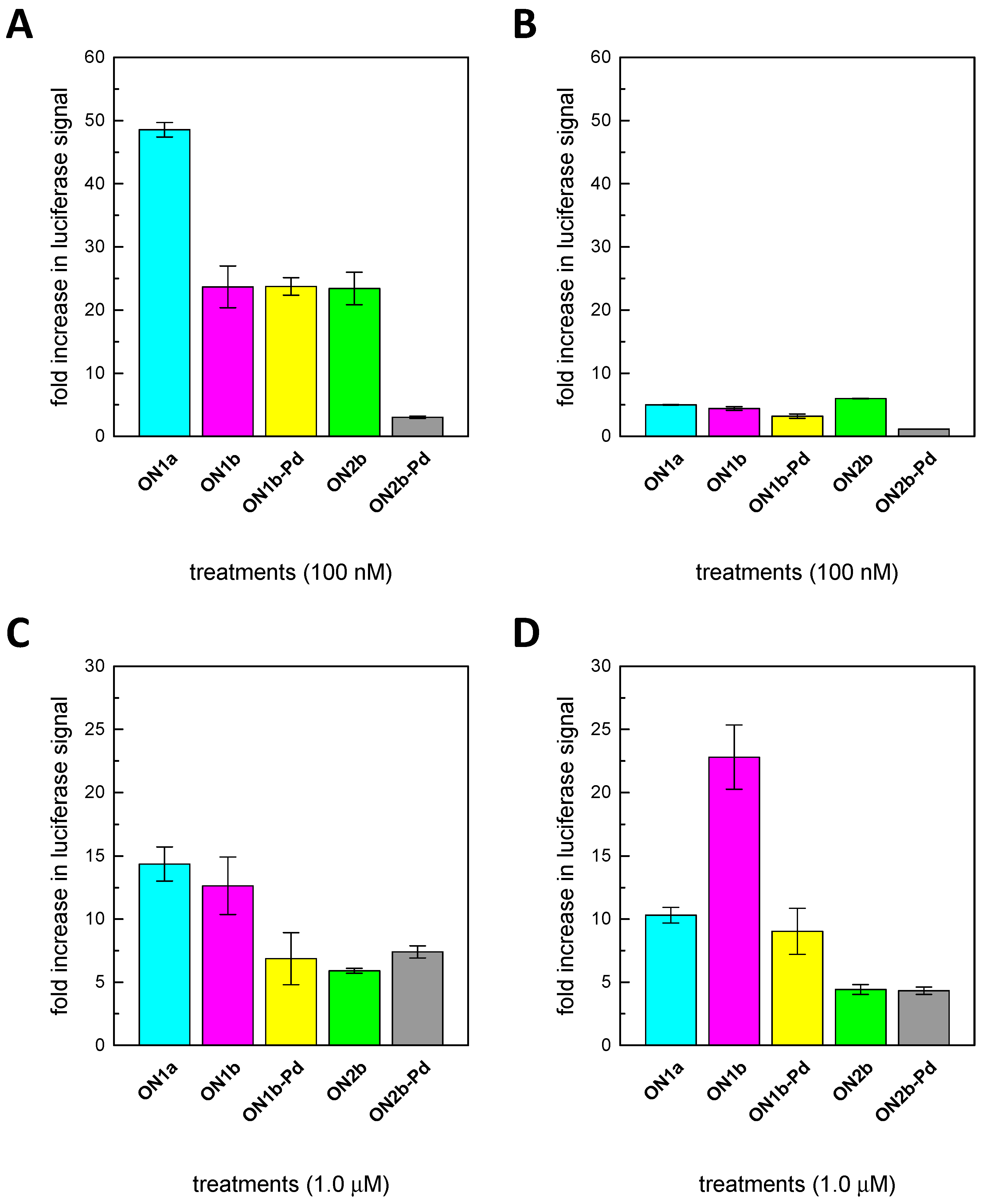
2.4. Splice-Correction Mediated by Lipofectamine 2000 Transfection of ONs
2.5. Splice-Correction Mediated by Gymnosis
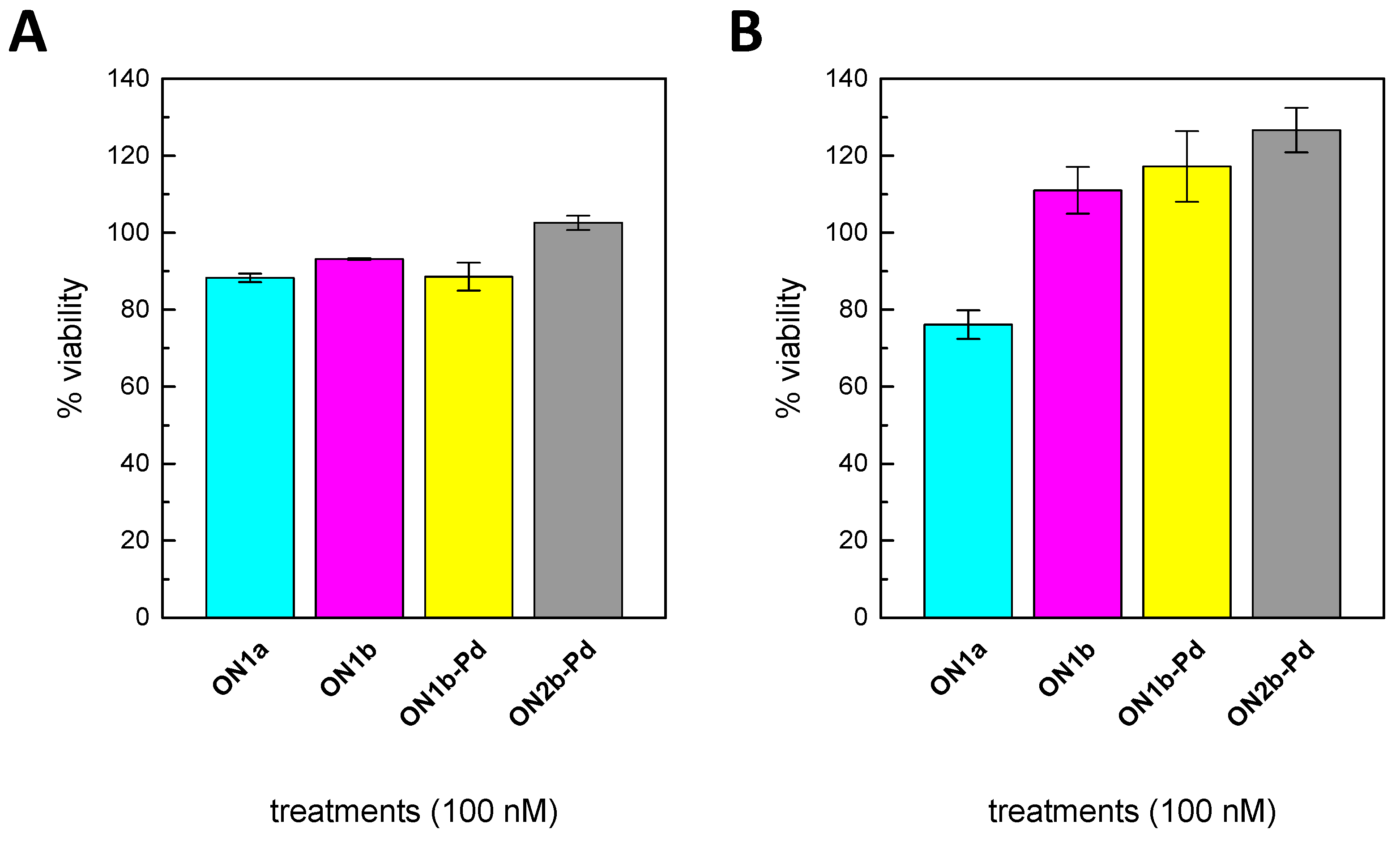
2.6. Cell Viability
3. Discussion
3.1. Hybridization Affinity of the Modified Oligonucleotides
3.2. Splice-Correction by the Modified Oligonucleotides
4. Materials and Methods
4.1. General Methods
4.2. Oligonucleotide Synthesis
4.3. UV Melting Temperature Measurements
4.4. Cell Lines and Culture Conditions
4.5. Transfection with Lipofectamine 2000
4.6. Gymnosis Experiments
4.7. Luciferase Assay
4.8. RNA Expression Analysis
4.9. Cell Viability Assay
4.10. Data analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lundin, K.E.; Gissberg, O.; Smith, C.I.E. Oligonucleotide Therapies: The Past and the Present. Hum. Gene Ther. 2015, 26, 475–485. [Google Scholar] [CrossRef]
- Smith, C.I.E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 605–630. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Stein, C.A. Antisense Oligonucleotides: Basic Concepts and Mechanisms. Mol. Cancer Ther. 2002, 1, 347. [Google Scholar]
- Gurav, B.; Srinivasan, G. Antisense oligonucleotides as therapeutics and their delivery. Curr. Sci. 2017, 112, 490–498. [Google Scholar] [CrossRef]
- Malik, R.; Roy, I. Design and development of antisense drugs. Expert Opin. Drug Discov. 2008, 3, 1189–1207. [Google Scholar] [CrossRef]
- Mansoor, M.; Melendez, A.J. Advances in antisense oligonucleotide development for target identification, validation, and as novel therapeutics. Gene Regul. Syst. Bio. 2008, 2008, 275–295. [Google Scholar] [CrossRef]
- Sahu, N.K.; Shilakari, G.; Nayak, A.; Kohli, D.V. Antisense technology: A selective tool for gene expression regulation and gene targeting. Curr. Pharm. Biotechnol. 2007, 8, 291–304. [Google Scholar] [CrossRef]
- Aboul-Fadl, T. Antisense oligonucleotides: The state of the art. Curr. Med. Chem. 2005, 12, 2193–2214. [Google Scholar] [CrossRef]
- Scherer, L.J.; Rossi, J.J. Approaches for the sequence-specific knockdown of mRNA. Nat. Biotechnol. 2003, 21, 1457. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.C. Inhibition of microRNA with antisense oligonucleotides. Methods 2008, 44, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Parsons, C.; Walker, L.; Zhang, W.C.; Slack, F.J. Targeting noncoding RNAs in disease. J. Clin. Invest. 2017, 127, 761–771. [Google Scholar] [CrossRef] [Green Version]
- van Rooij, E.; Kauppinen, S. Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef] [Green Version]
- Esau, C.C.; Monia, B.P. Therapeutic potential for microRNAs. Adv. Drug Deliv. Rev. 2007, 59, 101–114. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.-S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [Green Version]
- Havens, M.A.; Duelli, D.M.; Hastings, M.L. Targeting RNA splicing for disease therapy. Wiley Interdiscip. Rev. RNA 2013, 4, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.; Seth, P.P.; Bennett, C.F. Antisense Oligonucleotide-Based Therapies for Diseases Caused by pre-mRNA Processing Defects. In Systems Biology of RNA Binding Proteins; Yeo, G.W., Ed.; Springer: New York, NY, USA, 2014; pp. 303–352. [Google Scholar]
- Disterer, P.; Kryczka, A.; Liu, Y.; Badi, Y.E.; Wong, J.J.; Owen, J.S.; Khoo, B. Development of Therapeutic Splice-Switching Oligonucleotides. Hum. Gene Ther. 2014, 25, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Marlin, F.; Simon, P.; Saison-Behmoaras, T.; Giovannangeli, C. Delivery of Oligonucleotides and Analogues: The Oligonucleotide Conjugate-Based Approach. ChemBioChem 2010, 11, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Gooding, M.; Malhotra, M.; Evans, J.C.; Darcy, R.; O’Driscoll, C.M. Oligonucleotide conjugates—Candidates for gene silencing therapeutics. Eur. J. Pharm. Biopharm. 2016, 107, 321–340. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennox, K.A.; Behlke, M.A. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011, 18, 1111. [Google Scholar] [CrossRef]
- Saleh, A.F.; Arzumanov, A.A.; Gait, M.J. Overview of Alternative Oligonucleotide Chemistries for Exon Skipping. In Exon Skipping: Methods and Protocols; Aartsma-Rus, A., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 365–378. [Google Scholar]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef]
- Takezawa, Y.; Shionoya, M. Metal-Mediated DNA Base Pairing: Alternatives to Hydrogen-Bonded Watson–Crick Base Pairs. Acc. Chem. Res. 2012, 45, 2066–2076. [Google Scholar] [CrossRef]
- Takezawa, Y.; Müller, J.; Shionoya, M. Artificial DNA Base Pairing Mediated by Diverse Metal Ions. Chem. Lett. 2017, 46, 622–633. [Google Scholar] [CrossRef] [Green Version]
- Taherpour, S.; Golubev, O.; Lönnberg, T. On the feasibility of recognition of nucleic acid sequences by metal-ion-carrying oligonucleotides. Inorg. Chim. Acta. 2016, 452, 43–49. [Google Scholar] [CrossRef]
- Jash, B.; Müller, J. Metal-Mediated Base Pairs: From Characterization to Application. Chem. Eur. J. 2017, 23, 17166–17178. [Google Scholar] [CrossRef]
- Marchán, V.; Grandas, A. Platinated Oligonucleotides: Synthesis and Applications for the Control of Gene Expression. In Metal Complex–DNA Interactions; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2009; pp. 273–300. [Google Scholar]
- Colombier, C.; Lippert, B.; Leng, M. Interstrand Cross-linking Reaction in Triplexes Containing a Monofunctional Transplatin-Adduct. Nucleic Acids Res. 1996, 24, 4519–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brabec, V.; Reedijk, J.; Leng, M. Sequence-dependent distortions induced in DNA by monofunctional platinum(II) binding. Biochemistry 1992, 31, 12397–12402. [Google Scholar] [CrossRef] [PubMed]
- Algueró, B.; Pedroso, E.; Marchán, V.; Grandas, A. Incorporation of two modified nucleosides allows selective platination of an oligonucleotide making it suitable for duplex cross-linking. J. Biol. Inorg. Chem. 2007, 12, 901–911. [Google Scholar] [CrossRef]
- Algueró, B.; López de la Osa, J.; González, C.; Pedroso, E.; Marchán, V.; Grandas, A. Selective Platination of Modified Oligonucleotides and Duplex Cross-Links. Angew. Chem. Int. Ed. 2006, 45, 8194–8197. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.S.; Boudvillain, M.; Schwartz, A.; van der Marel, G.A.; van Boom, J.H.; Reedijk, J.; Lippert, B. Monofunctionally trans-Diammine Platinum(II)-Modified Peptide Nucleic Acid Oligomers: A New Generation of Potential Antisense Drugs. Chem. Eur. J. 2002, 8, 5566–5570. [Google Scholar] [CrossRef]
- Hibino, M.; Aiba, Y.; Watanabe, Y.; Shoji, O. Peptide Nucleic Acid Conjugated with Ruthenium-Complex Stabilizing Double-Duplex Invasion Complex Even under Physiological Conditions. ChemBioChem 2018, 19, 1601–1604. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.K.; Lönnberg, T. Oligonucleotides Incorporating Palladacyclic Nucleobase Surrogates. Chem. Eur. J. 2018, 24, 1274–1277. [Google Scholar] [CrossRef] [PubMed]
- Hande, M.; Maity, S.; Lönnberg, T. Palladacyclic Conjugate Group Promotes Hybridization of Short Oligonucleotides. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Kang, S.-H.; Cho, M.-J.; Kole, R. Up-Regulation of Luciferase Gene Expression with Antisense Oligonucleotides: Implications and Applications in Functional Assay Development. Biochemistry 1998, 37, 6235–6239. [Google Scholar] [CrossRef]
- Rocha, C.S.J.; Lundin, K.E.; Behlke, M.A.; Zain, R.; El Andaloussi, S.; Smith, C.I.E. Four Novel Splice-Switch Reporter Cell Lines: Distinct Impact of Oligonucleotide Chemistry and Delivery Vector on Biological Activity. Nucleic Acid Ther. 2016, 26, 381–391. [Google Scholar] [CrossRef]
- Dominski, Z.; Kole, R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1993, 90, 8673. [Google Scholar] [CrossRef]
- Zaghloul, E.M.; Viola, J.R.; Zuber, G.; Smith, C.I.; Lundin, K.E. Formulation and Delivery of Splice-Correction Antisense Oligonucleotides by Amino Acid Modified Polyethylenimine. Mol. Pharm. 2010, 7, 652–663. [Google Scholar] [CrossRef]
- Saher, O.; Rocha, C.S.J.; Zaghloul, E.M.; Wiklander, O.P.B.; Zamolo, S.; Heitz, M.; Ezzat, K.; Gupta, D.; Reymond, J.-L.; Zain, R.; et al. Novel peptide-dendrimer/lipid/oligonucleotide ternary complexes for efficient cellular uptake and improved splice-switching activity. Eur. J. Pharm. Biopharm. 2018, 132, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.-I.; Yamamoto, T.; Waki, R.; Wada, S.; Wada, F.; Noda, M.; Obika, S. Ca2+ enrichment in culture medium potentiates effect of oligonucleotides. Nucleic Acids Res. 2015, 43, e128. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds (ON1b and ON2b) are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligonucleotide | Sequence 1 |
|---|---|
| ON1a | 5′-C*C*U* C*U*U* A*C*C* U*C*A* G*U*U* A*C*A*-3′ |
| ON1b | 5′-B- C*C*U* C*U*U* A*C*C* U*C*A* G*U*U* A*C*A*-3′ |
| ON1b-Pd | 5′-BPd- C*C*U* C*U*U* A*C*C* U*C*A* G*U*U* A*C*A*-3′ |
| ON2a | 5´-C*A*G* A*G*U* U*C*U* C*A*G* G*A*U* G*U*A*-3′ |
| ON2b | 5′-B- C*A*G* A*G*U* U*C*U* C*A*G* G*A*U* G*U*A*-3′ |
| ON2b-Pd | 5′-BPd- C*A*G* A*G*U* U*C*U* C*A*G* G*A*U* G*U*A*-3′ |
| ON3 | 5′-AUU GUA ACU GAG GUA AGA GGU U-3′ |
| ON4 | 5′-att gta act gag gta aga ggt t-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hande, M.; Saher, O.; Lundin, K.E.; Smith, C.I.E.; Zain, R.; Lönnberg, T. Oligonucleotide–Palladacycle Conjugates as Splice-Correcting Agents. Molecules 2019, 24, 1180. https://doi.org/10.3390/molecules24061180
Hande M, Saher O, Lundin KE, Smith CIE, Zain R, Lönnberg T. Oligonucleotide–Palladacycle Conjugates as Splice-Correcting Agents. Molecules. 2019; 24(6):1180. https://doi.org/10.3390/molecules24061180
Chicago/Turabian StyleHande, Madhuri, Osama Saher, Karin E. Lundin, C. I. Edvard Smith, Rula Zain, and Tuomas Lönnberg. 2019. "Oligonucleotide–Palladacycle Conjugates as Splice-Correcting Agents" Molecules 24, no. 6: 1180. https://doi.org/10.3390/molecules24061180
APA StyleHande, M., Saher, O., Lundin, K. E., Smith, C. I. E., Zain, R., & Lönnberg, T. (2019). Oligonucleotide–Palladacycle Conjugates as Splice-Correcting Agents. Molecules, 24(6), 1180. https://doi.org/10.3390/molecules24061180