
Acid Catalyzed Formation of C–C and C–S Bonds via Excited State Proton Transfer


Abstract
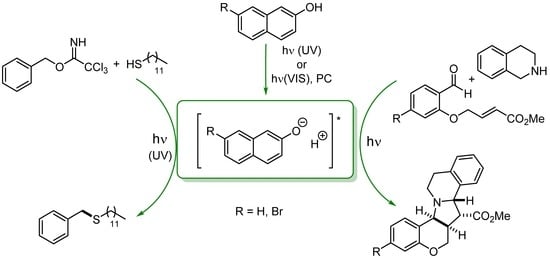
:
1. Introduction
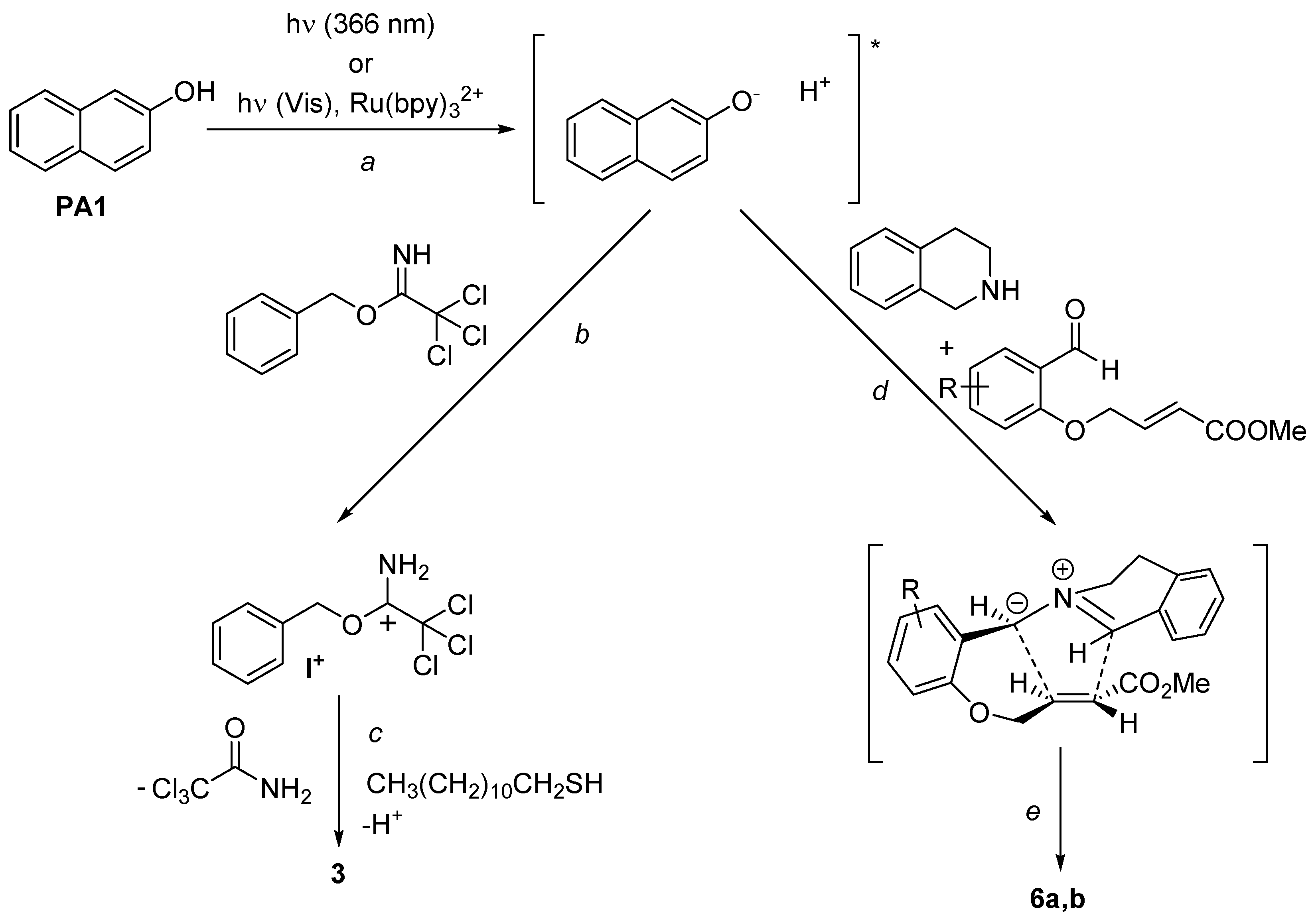
2. Results and Discussion
3. Material and Methods
3.1. General
3.2. Analytical Data for Compounds 3–6
3.2.1. Synthesis of Benzyl(dodecyl)sulfide (3)
3.2.2. Photochemical synthesis of 3
3.2.3. Synthesis of (E)-methyl 4-(2-formylphenoxy)but-2-enoate (4a)
3.2.4. Synthesis of Methyl-6a,7,7a,12,13,14a-hexahydro-6H chromeno[3′,4′:4,5]pyrrolo[2,1-a]isoquinoline-7-carboxylate (6a)
3.2.5. Synthesis of (E)-methyl 4-(2-formyl-5-methoxyphenoxy)but-2-enoate 4b
3.2.6. Synthesis of Methyl-methoxy-6a,7,7a,12,13,14a-hexahydro-6H-chromeno[3′,4′:4,5]pyrrolo[2,1-a]isoquinoline-7-carboxylate 6b
3.2.7. Photochemical synthesis of 6a,b
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Oelgemöller, M. Solar Photochemical Synthesis: From the Beginnings of Organic Photochemistry to the Solar Manufacturing of Commodity Chemicals. Chem. Rev. 2016, 116, 9664–9682. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, D.; Protti, S.; Fagnoni, M. Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates. Chem. Rev. 2016, 116, 9850–9913. [Google Scholar] [CrossRef]
- Marzo, L.; Pagire, S.K.; Reiser, O.; König, B. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Ed. 2018, 57, 10034–10072. [Google Scholar] [CrossRef]
- Yoon, T.P.; Ischay, M.A.; Du, J. Visible Light Photocatalysis As A Greener Approach To Photochemical Synthesis. Nat. Chem. 2010, 2, 527–532. [Google Scholar] [CrossRef]
- Albini, A.; Protti, S. Photochemistry; The Royal Society of Chemistry: London, UK, 2019. [Google Scholar]
- Hoffmann, N. Photochemical reactions of aromatic compounds and the concept of the photon as a traceless reagent. Photochem. Photobiol. Sci. 2012, 11, 1613–1641. [Google Scholar] [CrossRef]
- Albini, A.; Fagnoni, M. Green chemistry and photochemistry were born at the same time. Green Chem. 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Nagib, D.A.; MacMillan, D.W.C. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 2011, 480, 224–228. [Google Scholar] [CrossRef]
- Pham, P.V.; Nagib, D.A.; MacMillan, D.W.C. Photoredox Catalysis: A Mild, Operationally Simple Approach to the Synthesis of α-Trifluoromethyl Carbonyl Compounds. Angew. Chem. Int. Ed. 2011, 50, 6119–6122. [Google Scholar] [CrossRef]
- Ischay, M.A.; Anzovino, M.E.; Du, J.; Yoon, T.P. Efficient Visible Light Photocatalysis of 2 + 2 Enone Cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. [Google Scholar] [CrossRef]
- Protti, S.; Fagnoni, M.; Ravelli, D.D. Photocatalytic C-H Activation by Hydrogen-Atom Transfer in Synthesis. ChemCatChem 2015, 7, 1516–1523. [Google Scholar] [CrossRef]
- Yavorskyy, A.; Shvydkiv, O.; Hoffmann, N.; Nolan, K.; Oelgemoeller, M. Parallel Microflow Photochemistry: Process Optimization, Scale-up, and Library Synthesis. Org. Lett. 2012, 14, 4342–4345. [Google Scholar] [CrossRef]
- Ravelli, D.; Protti, S.; Fagnoni, M. Decatungstate Anion for Photocatalyzed “Window Ledge” Reactions. Acc. Chem. Res. 2016, 49, 2232–2242. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Shaikh, R.S.; König, B. Sensitization-Initiated Electron Transfer for Photoredox Catalysis. Angew. Chem. Int. Ed. 2017, 56, 8544–8549. [Google Scholar] [CrossRef] [PubMed]
- Marchini, M.; Bergamini, G.; Cozzi, P.G.; Ceroni, P.; Balzani, V. Photoredox Catalysis: The Need to Elucidate the Photochemical Mechanism. Angew. Chem. Int. Ed. 2017, 56, 12820–12821. [Google Scholar] [CrossRef]
- Tolbert, L.M.; Solntsev, K.M. Excited-State Proton Transfer: From Constrained Systems to “Super” Photoacids to Superfast Proton Transfer. Acc. Chem. Res. 2002, 35, 19–27. [Google Scholar] [CrossRef]
- Solntsev, K.M.; Huppert, D.; Tolbert, L.M.; Agmon, N. Solvatochromic Shifts of “Super” Photoacids. J. Am. Chem. Soc. 1998, 120, 7981–7982. [Google Scholar] [CrossRef]
- Solntsev, K.M.; Huppert, D.; Agmon, N.; Tolbert, L.M. Photochemistry of “Super” Photoacids. 2. Excited-State Proton Transfer in Methanol/Water Mixtures. J. Phys. Chem. A 2000, 104, 4658–4669. [Google Scholar] [CrossRef]
- Claya, A.; Krishnan, R.; Sibi, M.; Websterd, D.; Jockusch, S.; Sivaguru, J. Photoacidity of vanillin derivatives. J. Photochem. Photobiol. A Chem. 2018, 355, 38–41. [Google Scholar] [CrossRef]
- Nishikubo, Y.; Kanzaki, S.; Matsumura, S.; Toshima, K. The use of organophotoacids for deprotection reactions in organic synthesis. Tetrahedron Lett. 2006, 47, 8125–8128. [Google Scholar] [CrossRef]
- Oates, R.P.; Jones, P.B. Photosensitized Tetrahydropyran Transfer. J. Org. Chem. 2008, 73, 4743–4745. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Niu, L.; Wang, S.; Liu, T.; Singh, A.K.; Lei, A. Visible-Light-Induced Acetalization of Aldehydes with Alcohols. Org. Lett. 2017, 19, 122–125. [Google Scholar] [CrossRef]
- Yan, D.-M.; Chen, J.-R.; Xiao, W.-J. New Roles for Photoexcited Eosin Y in Photochemical Reactions. Angew. Chem. Int. Ed. 2019, 58, 378–380. [Google Scholar] [CrossRef]
- Zhao, G.; Wang, T. Stereoselective Synthesis of 2-Deoxyglycosides from Glycals by Visible-Light-Induced Photoacid Catalysis. Angew. Chem. Int. Ed. 2018, 57, 6120–6124. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Eto, T.; Takahashi, D.; Toshima, K. Stereocontrolled Photoinduced Glycosylation Using an Aryl Thiourea as an Organo Photoacid. Org. Lett. 2016, 18, 3190–3193. [Google Scholar] [CrossRef]
- Das, A.; Banerjee, T.; Hanson, K. Protonation of silylenol ether via excited state proton transfer catalysis. Chem. Commun. 2016, 52, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Ayad, S.; Hanson, K. Enantioselective Protonation of Silyl Enol Ether Using Excited State Proton Transfer Dyes. Org. Lett. 2016, 18, 5416–5419. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.Y.; Roush, W.R. Concerning the Origin of the High β-Selectivity of Glycosidation Reactions of 2-Deoxy-2-iodo-glucopyranosyl Trichloroacetimidates. Org. Lett. 2002, 4, 4523–4526. [Google Scholar] [CrossRef]
- Iwata, R.; Uda, K.; Takahashi, D.; Toshima, K. Photo-induced glycosylation using reusable organophotoacids. Chem. Commun. 2014, 50, 10695–10698. [Google Scholar] [CrossRef] [PubMed]
- Mantelingu, K.; Lin, Y.; Seidel, D. Intramolecular [3 + 2]-Cycloadditions of Azomethine Ylides Derived from Secondary Amines via Redox-Neutral C–H Functionalization. Org. Lett. 2014, 16, 5910–5913. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Cheng, Y.-C. Molecular electrostatic potential on the proton-donating atom as a theoretical descriptor of excited state acidity. Phys. Chem. Chem. Phys. 2018, 20, 4351–4359. [Google Scholar] [CrossRef]
- Acharya, A.; Chaudhuri, S.; Batista, V.S. Can TDDFT Describe Excited Electronic States of Naphthol Photoacids? A Closer Look with EOM-CCSD. J. Chem. Theory Comput. 2018, 14, 867–876. [Google Scholar] [CrossRef]
- Pines, E. UV-Visible Spectra and Photoacidity of Phenols, Naphthols and Pyrenols. In The Chemistry of Phenols-PATAI’S Chemistry of Functional Groups; Rappoport, Z., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Tolbert, L.M.; Haubrich, J.E. Photoexcited Proton Transfer from Enhanced Photoacids. J. Am. Chem. Soc. 1994, 116, 10593–10600. [Google Scholar] [CrossRef]
- Mcclure, D.S.; Blake, N.W.; Hanst, P.L. Singlet-Triplet Absorption Bands in Some Halogen Substituted Aromatic Compounds. J. Chem. Phys. 1954, 22, 255–258. [Google Scholar] [CrossRef]
- Eckenberg, P.; Groth, U.; Huhn, T.; Richter, N.; Schmeck, C. A Useful Application of Benzyl Trichloroacetimidate for the Benzylation of Alcohols. Tetrahedron 1993, 49, 1619–1624. [Google Scholar] [CrossRef]
- Yin, J.; Pidgeon, C. A simple and efficient method for preparation of unsymmetrical sulfides. Tetrahedron Lett. 1997, 38, 5953–5954. [Google Scholar] [CrossRef]
- Tanaka, K.; Ajiki, K. Rhodium-Catalyzed Reaction of Thiols with Polychloroalkanes in the Presence of Triethylamine. Org. Lett. 2005, 7, 1537–1539. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |



{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Conditions a | Light Source (nm) | Yield b (%) |
| 1 | DMSO, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | - |
| 2 | Hexane, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 13 |
| 3 | PhMe, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 15 |
| 4 | DCE, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 21 |
| 5 | Et2O, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 8 |
| 6 | MeCN, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 4 |
| 7 | DCM, 1 (0.1 M), 2 (0.5 M), PA1 (30 mol%) | 366 | 28 |
| 8 | DCM, 1 (0.1 M), 2 (1.0 M), PA1 (30 mol%) | 366 | 40 |
| 9 | DCM, 1 (0.1 M), 2 (0.2 M), PA1 (30 mol%) | 366 | 12 |
| 10 | DCM, 1 (0.1 M), 2 (0.5 M), PA1 (50 mol%) | 366 | 40 |
| 11 | DCM, 1 (0.1 M), 2 (0.5 M), PA1 (50 mol%) | 310 | 21 |
| 12 | DCM, 1 (0.1 M), 2 (0.5 M), PA2 (30 mol%) | 366 | 32 |
| 13 | DCM, 1 (0.1 M), 2 (0.5 M), PA2 (50 mol%) | 366 | 47 |
| 14 | MeCN, 1 (0.1 M), 2 (0.5 M), PA2 (50 mol%) c | 366 | 9 |
| 15 | DCM, 1 (0.1 M), 2 (0.5 M) | 366 | 12 |
| 16 | DCM, 1 (0.1 M), 2 (0.5 M), PA2 (50 mol%) d | - d | - |
 | |||
|---|---|---|---|
| Entry | Conditions a | Light Source (nm) | Product (% Yield) b |
| 1 | 4a (0.1 M) 5 (0.12 M), PA1 or PA2 (50 mol%) | 366 | 6a, < 5% |
| dry DCM | |||
| 2 | 4a (0.1 M), 5 (0.12 M), PA1 (50 mol%), Ru[(bpy)3]2+ | 410 | 6a, 19% |
| (5 mol%), dry MeCN | |||
| 3 | 4a (0.1 M), 5 (0.1 M), PA1 (50 mol%), Ru[(bpy)3]2+ | 410 | 6a, 43% |
| (5 mol%), dry MeCN | |||
| 4 | 4a (0.15 M), 5 (0.10 M), PA1 (50 mol%), Ru[(bpy)3]2+ | 410 | 6a, 65% |
| (5 mol%), dry MeCN | |||
| 5 | 4a (0.2 M), 5 (0.10 M), PA1 (50 mol%), Ru[(bpy)3]2+ | 410 | 6a, 63% |
| (5 mol%), dry MeCN c | |||
| 6 | 4a (0.15 M), 5 (0.10 M), Ru[(bpy)3]2+ | 410 | 6a, 20% |
| (5 mol%), dry MeCN | |||
| 7 | 4a (0.15 M), 5 (0.10 M), PA2 (30 mol%), Ru[(bpy)3]2+ | 410 | 6a, 62% |
| (5 mol%), dry MeCN | |||
| 8 | 4b (0.15 M), 5 (0.10 M), PA1 (50 mol%), Ru[(bpy)3]2+ | 410 | 6b, 25% |
| (5 mol%), dry MeCN | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strada, A.; Fredditori, M.; Zanoni, G.; Protti, S. Acid Catalyzed Formation of C–C and C–S Bonds via Excited State Proton Transfer. Molecules 2019, 24, 1318. https://doi.org/10.3390/molecules24071318
Strada A, Fredditori M, Zanoni G, Protti S. Acid Catalyzed Formation of C–C and C–S Bonds via Excited State Proton Transfer. Molecules. 2019; 24(7):1318. https://doi.org/10.3390/molecules24071318
Chicago/Turabian StyleStrada, Alessandro, Mattia Fredditori, Giuseppe Zanoni, and Stefano Protti. 2019. "Acid Catalyzed Formation of C–C and C–S Bonds via Excited State Proton Transfer" Molecules 24, no. 7: 1318. https://doi.org/10.3390/molecules24071318
APA StyleStrada, A., Fredditori, M., Zanoni, G., & Protti, S. (2019). Acid Catalyzed Formation of C–C and C–S Bonds via Excited State Proton Transfer. Molecules, 24(7), 1318. https://doi.org/10.3390/molecules24071318