1. Introduction
Molecular computing is a fast-developing interdisciplinary field which uses molecules to perform computations rather than traditional silicon chips. DNA is one such biomolecule which has complementary base pairing properties that allow for both specificity in molecular recognition and self-assembly and for massively parallel reactions which can take place in minute volumes of DNA samples. Pioneering work by Adleman demonstrate solutions to combinatorial problems using molecular computing [
1].
Since then, research exploring and exploiting these DNA properties in DNA computing provide the core nanotechnologies required to build DNA devices that are capable of decision-making at a molecular level. Such examples are the implementation of logic gates [
2,
3,
4], storing and retrieving information [
5,
6], simple computations for differentiating biological information [
7], classifying analogue patterns [
8], training molecular automatons [
9] and even playing games [
10]. In particular, molecular computing based on enzymes has also attracted much attention as enzymes can respond to a range of small molecule inputs, have advantage in signal amplification, and are highly specific in recognition capabilities [
11].
As DNA computing has the advantage of biocompatibility with living tissue or organisms, implementing molecular computations has also led to promising biomedical applications. An example of this is the logic gates used to specifically target cells or tissue types, thereby minimizing side effects and releasing chemicals at a specific location [
12]. Nanostructures have also been used to efficiently deliver cytotoxic drugs to targeted cancerous cells as a therapeutic drug delivery system for various cancers [
13,
14].
However, these molecular computation approaches were generally preprogrammed, rule-based, or used logic gates for simple forms of computations which may not exceed the ability of reflex action from the perspective of intelligence. Such as in the work of [
15,
16] where a perceptron algorithm was designed with a weighted sum operation and [
17] where a feedforward and recurrent neural network was constructed with cascading nodes using DNA hybridization; although these studies realized pre-defined perceptrons, the idea of learning, where computational weight parameters were updated to train the model was lacking.
Another state-of-the-art molecular pattern recognition work using the winner-take-all model has been recently published, demonstrating molecular recognition using MNIST digit data and DNA-strand-displacement [
18]. This work recognizes patterns into defined categories of handwritten digits ‘1’ to ‘9’ using a simple neural network model called the winner-take-all model. Though similar to our study, a key difference is that this work focuses on ’remembering’ patterns during training for recognition and our study focuses on online learning of patterns for classification, where learning refers to the generalization of data following multiple iterations of update during molecular learning. Another key difference is the focus of our work to implement a complete in vitro molecular learning experiment, in wet lab conditions. This is further discussed in the results section as a comparative study with our work (
Section 3.4).
Another related area of research includes the implementation of dynamic reaction networks. in vitro biochemical systems, transcriptional circuits have been used to form complex networks by modifying the regulatory and coding sequence domains of DNA templates [
19]. A combination of switches with inhibitory and excitatory regulation are used as oscillators similar to that which are found as natural oscillators. Another study also use chemical reactions inspired from living organisms to demonstrate assembling of a de novo chemical oscillator, where the wiring of the corresponding network is encoded in a sequence of DNA templates [
20]. These studies use the synthetic systems to further understand the complex chemical reactions found in nature to deepen our understanding of the principle of biological dynamics. A key similarity to our work is the use of modular circuits to model more complex networks. However, it is important to note that these studies are all demonstrated in silico, although it illustrates the potential of in vitro transcriptional circuitry. Computational tools are also being developed, one example being the EXPonential Amplification Reaction (EXPAR), to facilitate the assay design of isothermal nucleic acid amplification [
21]. This method helps accelerate DNA assay design, identifying template performance links to specific sequence motifs.
These dynamic system programming paradigms could be valid approaches to implement machine learning algorithms, as programmable chemical synthesis and the instruction strands of DNA dictate which reaction sequence to perform. We ponder that this kind of powerful information-based DNA system technology could also be manipulated to perform defined reactions in specific orders similar to what our study strives to do, thus, implementation operations in vitro to demonstrate molecular learning with the hypernetwork or other machine learning algorithms [
22].
Recent work by [
23], implement mathematical functions using DNA strand displacement reactions. This study demonstrates considerably more complex mathematical functions to date, can be designed through chemical reaction networks in a systematic manner. It is similar to our work in that it strives to compute complex functions using DNA though a key difference is that the design and validation of this work were presented in silico whereas our work focuses on in vitro implementation of molecular learning. However, the mass-action simulations of the chemical kinetics of DNA strand displacement reactions may be key in developing in vitro learning implementations, as learning consists of target mathematical operations which need to be performed with DNA in a systematic manner to manipulate DNA datasets. Consequently, operations or computational constructs are crucial in implementing machine learning algorithms, from simple perceptrons to neural networks, and this is proposed by this system and thus shares our interests in building systemic molecular implementations of chemical reactions for molecular machine learning. Further examples include a study where an architecture of three autocatalytic amplifiers interacts together to perform computations [
24]. The square root, natural logarithm and exponential functions for x in tunable ranges are computed with DNA circuits.
Molecular learning algorithms for pattern recognition in vitro with DNA molecules may be a step towards more advanced systems with higher complexity, adaptability, robustness, and scalability. This could be useful for solving more advanced problems, and be more applicable to use with more intelligent molecular learning devices in order to function in dynamic in vitro and in vivo environments. Some in vitro and in silico studies which aim to create more complex molecular computing systems include associative recall and supervised learning frameworks using strand-displacement [
25,
26,
27,
28,
29].
There are many difficulties in implementing molecular learning in wet lab experimental settings. DNA may be a more stable biomolecule compared to others such as RNA, however it still requires storage in appropriate solutions and temperature and it is prone to contamination, manipulation techniques often result in heavily reduced yield, and performing and analyzing the molecular biology results can be tedious and time consuming. Furthermore, applying learning algorithms familiar in machine learning bears critical differences to the current demonstration of DNA computing, such as predefined or rule-based models and logic gates.
Our previous study displayed in vitro DNA classification results [
30] by retrieving information from a model that was trained with a pseudo-update like operation of increasing the concentration of the matched DNA strands. However, the adaptability and scalability of the model was limited, due in part to the restrictions in the representation of the model by creating single strand DNA (features) with a fixed variable length. Here, this refers to a fixed length of DNA sequence which encodes the variables. Additionally, from the machine learning perspective, updating only with the positive term has critical limitations not common in conventional machine learning methods [
31], such as in the log-linear models or neural networks, which require both the positive and the negative terms to perform classification. The accuracy was also somewhat guaranteed because the training and test set were not divided and the features (pool of DNA) were manually designed to have small errors.
In this paper, we introduce a self-improving molecular machine learning algorithm, the hypernetwork [
5,
27,
32], and a novel experimental scheme to implement in vitro molecular machine learning with enzymatic weight update. We present the preliminary in vitro experimental results of proof-of-concept experiments for constructing two types of DNA datasets, the training and test data, with the self-assembling processes of DNA with hybridization and ligation, and DNA cleavage with a nuclease enzyme for the weight update stage of the molecular learning. Our study provides a new method of implementing molecular machine learning with weight update including a negative term.
First, we consider a natural experimental situation typical to machine learning, where we separate the training and test data to evaluate the model. Secondly, by adopting a high order feature extraction method when creating single stranded DNA, a higher-order hypothesis space may be explored which allows for discovery of better solutions even with simple linear classifiers. Thirdly, unlike previous methods which only increased the weight of the model, our proposed method considers both the positive and negative terms of the weight update in the model for learning using an enzymatic weight update operation in vitro. This method is inspired by the energy-based models which use the energy-based objective function to solve problems, and is represented with exponentially proportional probabilities and consists of positive and negative terms to calculate the gradient [
33]. Lastly, by encompassing the concept of ensemble learning, the model uses its full feature space for the classification task and also guarantees best performance by voting the best classified labels between each ensemble model. These four aspects are the crucial differences that distinguish our study from previous demonstrations of molecular learning, where without these assumptions of machine learning based aspects, learning, adaptability, and scalability of the model is limited. We show in the results section that the performance of our model gradually increases with the continual addition of training data.
3. Results
To ensure that the experimental protocol is implemented to the highest degree of efficiency and accuracy, a series of preliminary experiments were undertaken to validate the experimental steps involved in demonstrating the molecular hypernetwork.
3.1. DNA Quantification of 3-Order Hyperedges
The formation of a random single-stranded library was critical in verifying the success of the full experimental scheme. The experimental steps and results are as follows:
Hybridization of upper and bottom strands of variable units.
Ligation of these variables in a random fashion, all in one tube to create a double-stranded DNA random library.
Purification of the sample from ligase.
Separation of the double-stranded library to a single-stranded random library using Streptavidin and Biotin.
Centrifugal filtering of DNA for concentration.
Verification of library formation with the use of complementary strands.
Each step listed above was carried out using wet DNA in a test tube, and at each step, the DNA concentration was measured using a NanoDrop Nucleic Acid Quantification machine, which is a common lab spectrophotometer (
Figure 6). Hybridization was performed on the PCR machine with a decrement of
from
to
100 pmol of each upper and lower strand was used.
Ligation was carried out using Thermo Fisher’s T4 ligase enzyme. This enzyme joins DNA fragments with staggered end and blunt ends and repairs nicks in double stranded DNA with 3’-OH and 5’-P ends. Three units of T4 ligase was used with 1 L of ligation buffer.
Annealing of the ligated DNA strands are then put into PCR conditions with a decrement of 1 degree from 30 to 4 degrees. Purification and extraction of DNA from the ligase inclusive sample was carried out using the QIAEX II protocol for desalting and concentrating DNA solutions. The standard procedures for this were used [
45,
46,
47]. This procedure is commonly used to remove impurities (phosphatases, endonucleases, dNTPs etc.) from DNA, and to concentrate DNA. The QIAEX desalting and concentration protocol gives quite a detailed description of the procedure.
While there was a significant loss of DNA content after ligation, a sufficient concentration was recovered from the centrifugal filtering step allowing identification of the nine complementary strands possible from the combinations of the three different variables initially used. Bands at the 70 bp marker were present for all nine types of sequences which confirm that all possible sequences were successfully constructed and retrieved during the experimental process. The results shown in
Figure 6 present DNA concentrations at various stages of the learning process, and the final confirmation of the success in making a random double-stranded library.
3.2. Creating Free-Order DNA Hypernetworks
For the creation of the free-order hyperedges or different lengths of hyperedges, the concentrations of DNA sequences according to its corresponding pixel greyscale value, was added to a tube and ligation performed as described above. The PAGE electrophoresis gel illustrates the free-order hyperedges which were produced from the ligation procedure (
Figure 4c and
Figure 7a). Against the 10 bp ladder, many bands of varying intensities can be seen, representing different lengths of DNA present in the sample. From the original tube of 15 bp single stranded DNA following the ligation protocol, the formation of random hyperedges from 30 bp to 300 bp are visible (
Figure 7a). This correlates to 0-order to 15-order hyperedges. The same procedure was carried out to produce free-order hyperedges for every dataset: Training data and test data.
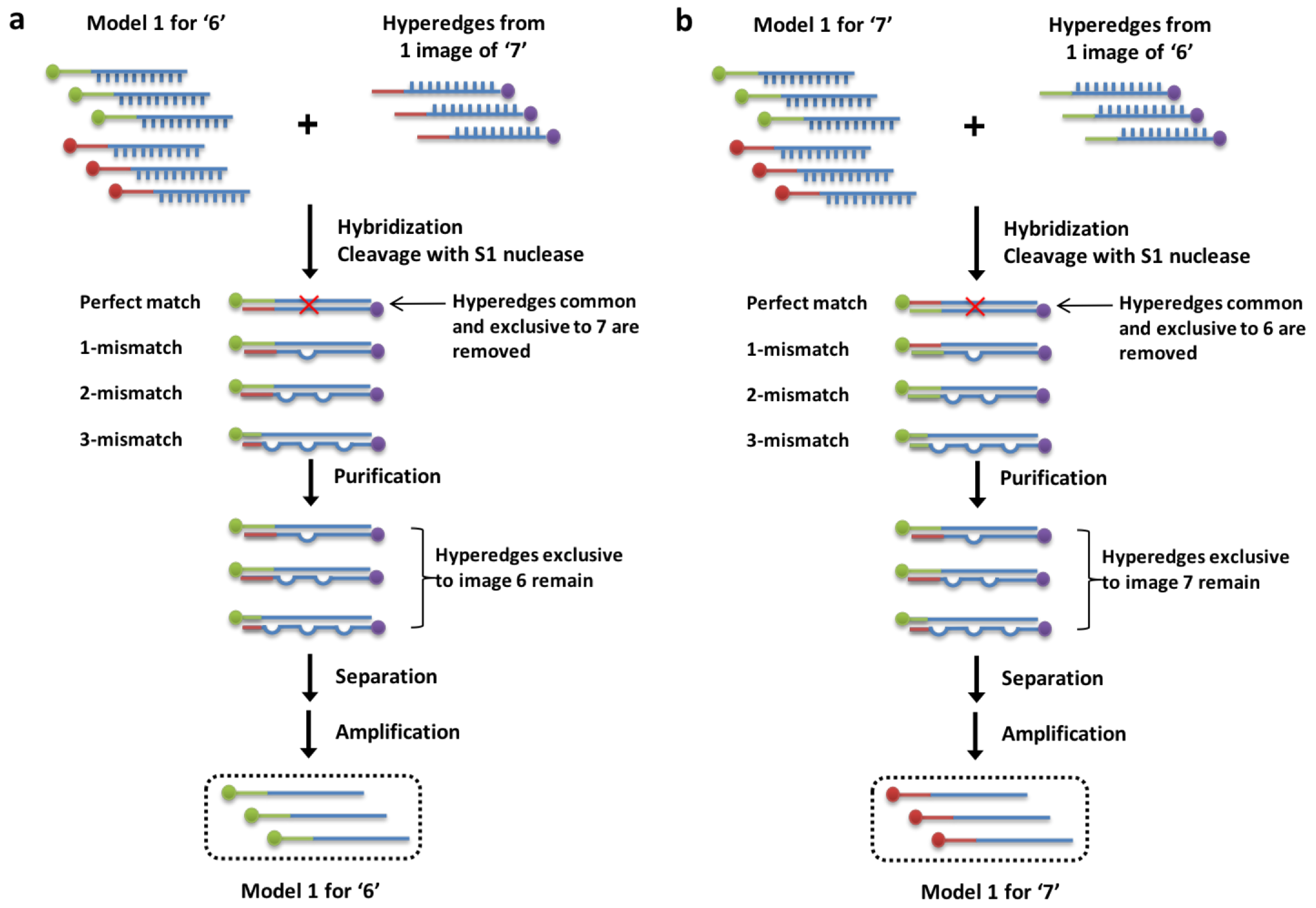
3.3. Weight Update Feedback Loop for DNA Hypernetwork
Figure 7b shows the result of enzyme treatment for S1 nuclease. DNA was incubated with the enzyme for 30 min at room temperature than enzyme inactivated with 0.2 M of EDTA at heating at
for 10 min. The control lane shows 4 bands, the perfectly matched strand, 1-mismatched, 2-mismatched, and 3-mismatched strands from the bottom to the top of the gel. The function of the S1 nuclease is investigated for decreasing weights or in this case DNA concentration, where perfectly matched DNA sequences are cleaved and mismatched sequences remain. In the 0.5 S1 lane, it is evident that only the perfectly matched DNA strands are cleaved and the mismatched strands remain in the mixture. This represents the sequences only present exclusively in the data for digits ‘6’ or ‘7’ can be reproduced with the use of S1 nuclease in the weight update algorithm.
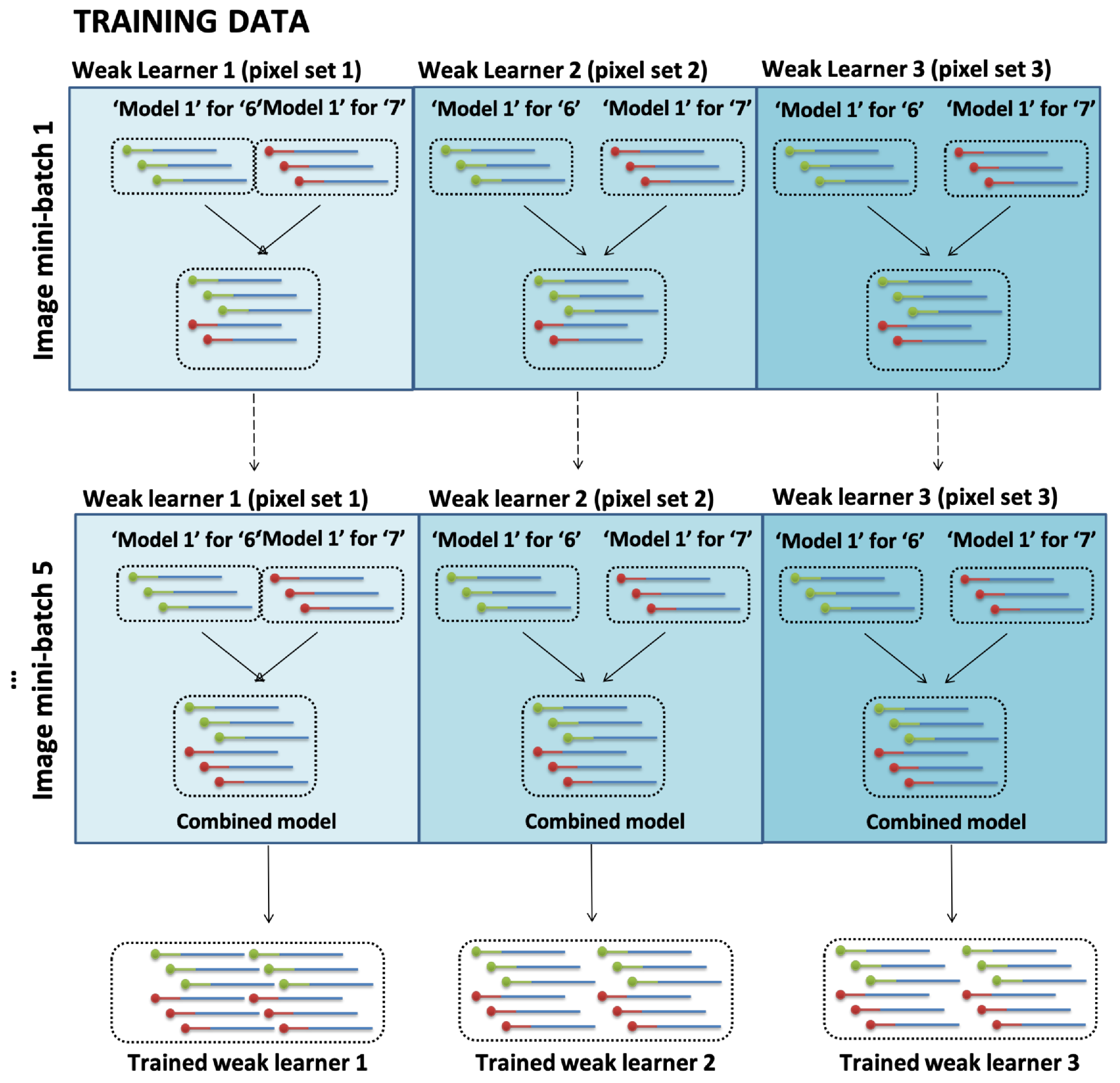
It is interesting to note that the issue of scalability may be addressed through our design which allows 10-class digit classification within the same number of experimental steps. More classes of training data could be added in the hybridization stage to all but the one class of training data for which the label is being learned. This provides a larger scale of digit classification without drastically increasing the workload, time or the need to order new sequences. This novel method of implementing digit classification and experimental results demonstrate the enzymatic reactions which is prerequisite to making this experimentally plausible.
3.4. Performance of In Silico Experiment of DNA Hypernetwork
As described in the Materials and Methods section, we used the MNIST dataset to measure the classifier accuracy [
48], which is defined as the estimation of number of correctly classified objects over the total number of objects.
To verify the learning capability of our proposed model, both incremental and online aspects, we compared our model to two existing models; the perceptron model described in [
18] and the conventional neural network [
49] as a representative example of non-linear classification.
In [
18], a basic perceptron model outputs the weighted sum for each class and selects the maximum value as their winning final output. 2-class classification between digits ‘6’ and ‘7’ is demonstrated and nine label 3 grouped class-classifier is described, where all methods first eliminate the outlier and the performance achieved by providing probabilistically calculated weights of the 10 most characteristic features to the designer as a prerequisite. However, in our study, we do not eliminate outliers or give prior weights and use the MNIST dataset as it is for our performance. We exploit the learning ability of a DNA computing model without the need for the designer to previously define weights. Not only do we reduce the labor required by the designer to define weights for selected features but we exploit the massively parallel processing capability of DNA computing whilst demonstrating molecular learning which improves performance with experience as our model is designed for implementation in vitro through molecular biology technique with wet DNA.
Two types of initialization of weights are introduced in our simulation results, 0 weight initialization which is easily implemented in DNA experiments, and random weight initialization which is harder to be conducted in vitro but is more conventionally used in perceptron and neural network models. The perceptron and neural networks convergence of performance are dependent on their initialized weights [
50]. We conducted these two methods of initializing the weights, first starting with 0 weight, and second providing random values to the weights.
For the 2-class classification, 1127 and 11,184 images were used for the test and training data respectively. For the 10-class classification 10,000 and 60,000 images were used for the test and training data respectively. As the MNIST dataset is balanced over all classes and not skewed to any class, the accuracy measurement is sufficient to evaluate the classifier’s performance [
51]. For all cases of learning, we randomly sampled five images. We did this to demonstrate our model’s capacity to implement online learning in only a few iterations and more significantly for our work, for the correlation to our wet lab molecular learning protocol, where only five iterations of molecular learning experiments need to be carried out for learning to produce classification performance.
For both the perceptron and neural network model, the learning rate was set to 0.1. For the perceptron model, of the 10 output values from the given input, the output with the biggest value was selected in a winner-take-all method. As the hyperedges produced from the hypernetwork in the 10-class classification (all with weights) was 284, we chose to use 300 hidden nodes for the neural network. All the source codes and relevant results can be found on github repository (
https://github.com/drafity/dnaHN).
Figure 8 shows the results of our in silico classification results. As the number of epochs increased, the test accuracy of the hypernetwork also increased. The accuracy of the hypernetwork was also higher than the comparative models of the perceptron and neural network. We note here the significance of having used an accuracy measure to evaluate our classifier. The DNA learning models implemented in vitro in the mentioned related works often lacked appropriate measures to evaluate the classifier’s performance. Furthermore, though recognition abilities have been reported through in vitro molecular learning, classification of data through learning and testing this, to consequently group or label the unknown test data to a category by a molecular learning model is to the authors’ knowledge a novelty in itself.
A key feature of our model in comparison to the perceptron or neural network models is the minimum number of iterations required to observe significant performance. Our proposed model only needs five iterations of learning to achieve significant classification performance. However, as the results show in
Figure 8 and
Figure 9, initializing the weight to 0 or giving random weights to the compared models still resulted in low accuracy in small epoch sizes. The perceptron and the neural network require a much larger epoch size for significant classification performance to be achieved.
This is crucial as in vitro experiments to perform molecular learning not only require time-consuming laborious work, but issues with contamination and denaturation can affect the quality of the experimental results. It is only more suitable for molecular learning experiments performed in wet lab conditions to be efficient, exploiting the massively parallel computing possible with DNA but also minimizing the protocol required to perform molecular learning. Our model is designed to do this by autonomously constructing higher order representations, using massively parallel DNA processes to create and update weights in minimal iterations. Furthermore, compared to state-of-the-art studies in molecular recognition, we were able to achieve over 90% accuracy and 60% accuracy in 2-class and 10-class classification respectively, through a molecular learning algorithm in five iterations. Thus, this result present that our model is a novel molecular learning model which learns in an online manner through minimal iterations of learning, suitable for wet lab implementations using DNA.
The hypernetwork, inspired by DNA chemical reactions, when computed in silico, clearly showed the disadvantage of sequential computing in silico and the massively parallel processing advantage of DNA computing in vitro. In an instant, DNA molecules hybridize when complementary strands are added together in an appropriate buffer and thus almost immediately the computing in that tube comes to an end. However, implementation of the hypernetwork in silico is iterative, sequential. For each training and test data, the number of matches and mismatches need to be calculated sequentially, and as the order of hyperedges increases, computational time complexity increase exponentially. As a result, with our computing power, empirically, 1000 iterations require min, a total of approximately 10 days to complete. Therefore it is important to note that there is a sheer advantage in DNA implementation of the hypernetwork compared to in silico. For the same reason, the neural network requires around 1000 iterations to converge and in the case of non-linearly separable data when using the perceptron model, it fails to converge. Thus, the proposed hypernetwork may also be introducing the possibility of a new computing method.
We also discuss the reasons why the perceptron does not perform as well. Due to the nature of perceptron models, as a representative linear classifier, it is difficult for it to solve linearly inseparable problems (XOR problems) without any preprocessing or adding layers to the perceptron to deal with non-linearity problems [
52,
53]. As illustrated in
Figure 8, the perceptron model shows performances close to what would be achieved from random picking for both small and large number of iterations. Depending on how the data is fed, 2-class classification performance levels show major fluctuations, where up to 80% performance is achieved at times and others much lower performance. This phenomenon is typically representative of unsuccessful generalization of the data also called overfitting. For example, in the case of the perceptron, as described in the reference, the performance is achieved only for the data that can be fitted linearly. To learn linearly inseparable data, the model needs a feature reduction or extraction preprocessing methods [
54] or a nonlinear kernel to model (e.g., Support Vector Machine [
55], Neural Network [
53] the high dimension input dataset. As this paper focuses on the implementation of a learning model in vitro only using easy, basic and fundamental learning processes, we believe this is out of the scope of our paper and omit further discussion.
As a support to such arguments, as shown in
Figure 8 and
Figure 9, both in our results and in Cherry and Qian’s work, there are cases where a variety of elimination conditions and previously providing the optimal weights of batch data by the designer can achieve significant performance in 2-class classification i.e., overfitted results (the maximum value of the error bars). However, as in the case of 10-class classification tasks, where the data is not linearly separable where it exceeds the model’s capacity, the range of performance levels are smaller and, as acknowledged by Cherry and Qian in their paper, it is difficult for the designer to find the optimal weights for the model.
4. Discussion
We have proposed a novel molecular machine learning algorithm with a validated experimental scheme for in vitro demonstration of molecular learning with enzymatic weight update. The experiments are designed for plausible pattern recognition with DNA through iterative processes of self-organization, hybridization, and enzymatic weight update through the hypernetwork algorithm. Natural DNA processes act in unison with the proposed molecular learning algorithm using appropriate enzymes which allowed updating of weights to be realized in vitro. Unlike in previous studies, a molecular learning algorithm with enzymatic weight update is proposed, where the positive and negative terms of weight update are considered in the model for learning. Using the validated experimental steps, the model can be used for repeated learning iterations for the selection of relevant DNA to cause the DNA pool to continuously change and optimize, allowing large instance spaces to reveal a mixture of molecules most optimized to function as a DNA pattern recognition classifier.
Our experiments showed a higher order feature extraction method was possible in vitro using higher-order DNA hyperedges which was demonstrated by constructing longer DNA sequence datasets. This method of DNA data construction dramatically reduced the number of unique DNA sequences required to cover the large search space of image feature sets. Finally, DNA ensemble learning is introduced for use of the full feature space in the classification tasks.
Although the complete iterations of learning are yet to be carried out, the aim of this paper was to provide a framework, with a synergistic approach between theoretical and experimental designs of molecular learning algorithm. In future experiments we will carry out the iterative molecular learning scheme wet laboratory conditions.
By harnessing the strength of using biomolecules as building blocks for basic computations, new and exciting concepts of information processing have the potential to be discovered through more molecular computing methods. In turn, the implementation of machine learning algorithms through DNA could also act as a starting point for emerging technologies of computational molecular devices, implicated in a diverse range of fields such as intelligent medical diagnostics or therapeutics, drug delivery, tissue engineering, and assembly of nanodevices. As more advanced applications are explored, more intelligent molecular computing systems, with suitable intelligence to navigate and function in dynamic in vivo environments, may bridge gaps in current molecular computing technologies, so that DNA systems can function in uncontrolled, natural environments containing countless unforeseeable variables.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








