
Silicon Isotope Geochemistry: Fractionation Linked to Silicon Complexations and Its Geological Applications

 , ,
, ,
Abstract
:1. Introduction
2. Analytical Techniques
2.1. Gas-Source Mass Spectrometry (GS-MS)
2.2. Second Ion Mass Spectrometry (SIMS)
2.3. Multicollector Inductively Plasma Mass Spectrometry (MC-ICP-MS)
2.4. Laser Ablation Multicollector Inductively Coupled Plasma Mass Spectrometry (fsLA-MC-ICP-MS)
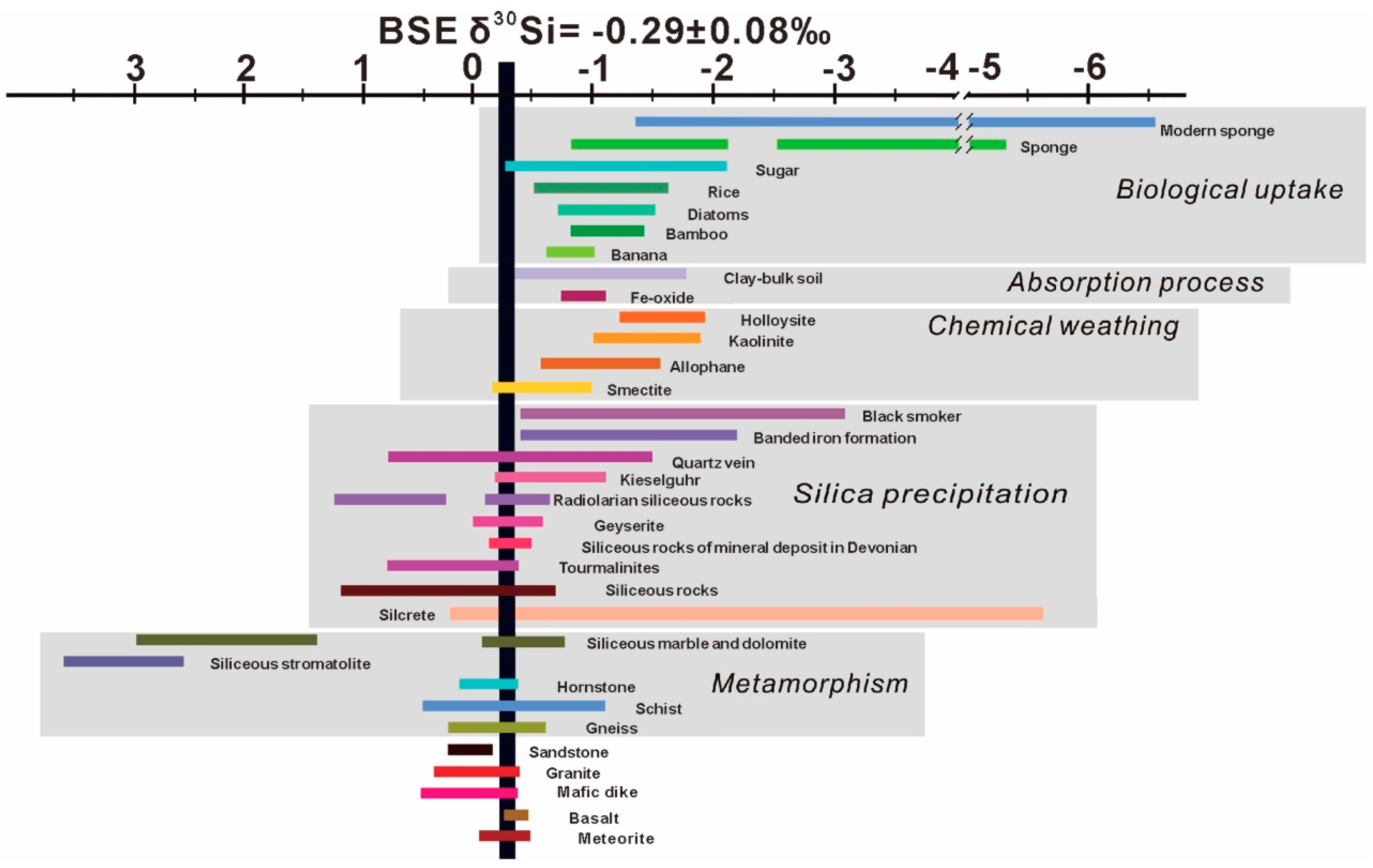
3. Silicon Isotope Variations in Major Reservoirs and Geological Processes
4. Silicon Isotope Fractionations Linked to Silicon Coordination/Complexation
4.1. Silicate Minerals with Variable Structures and Chemical Compositions
4.2. Silica Precipitation and Diagenesis
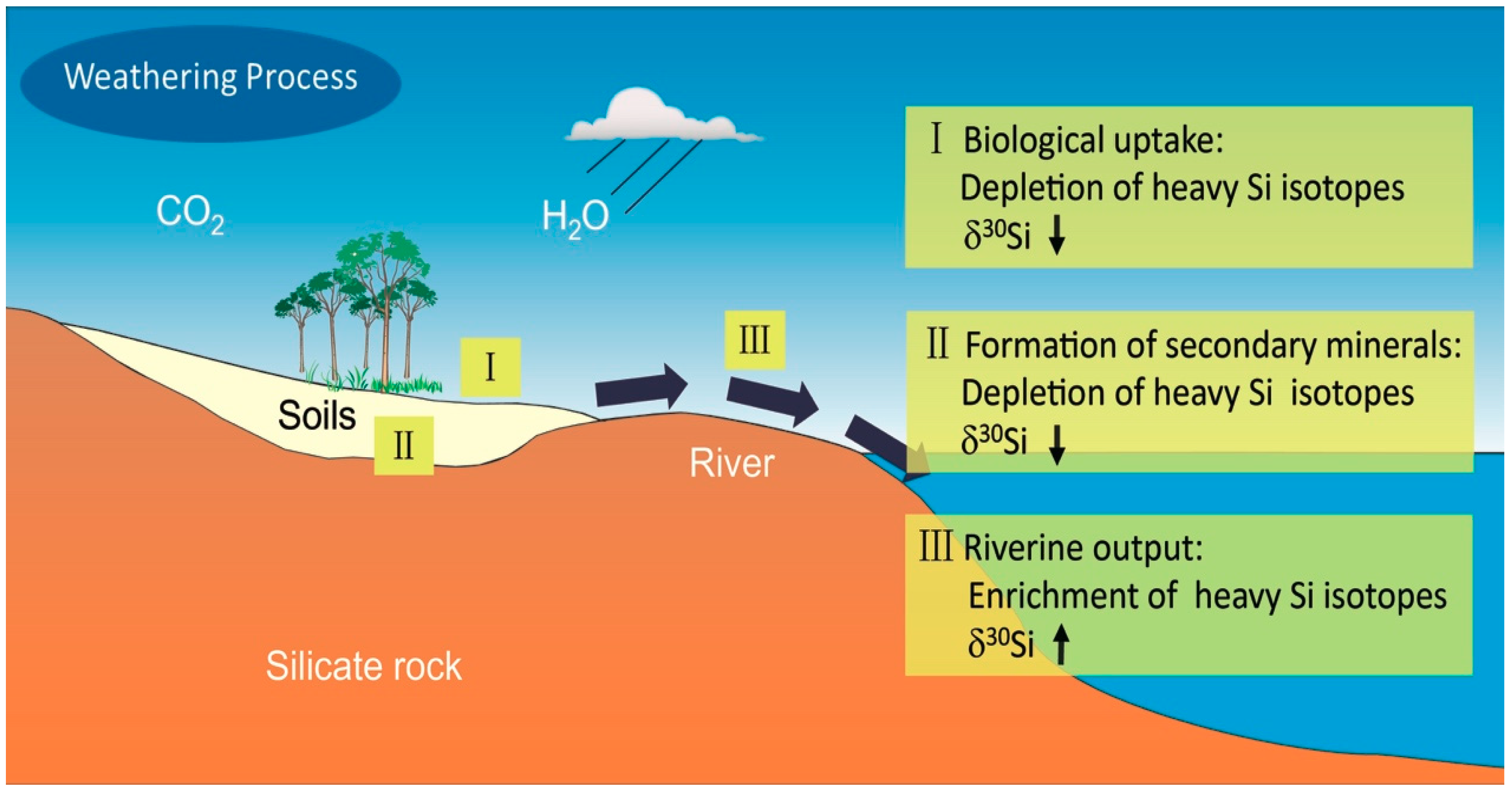
4.3. Chemical Weathering of Crustal Surface Silicate Rocks
4.4. Biological Uptake
4.5. Silicon Cycling in Hydrosphere
4.5.1. Global Spatial and Temporal Variability/Heterogeneity in Oceanic Si Cycle and δ30Si
4.5.2. δ30Si Variation in Terrestrial Hydrosphere
5. Geological Applications of Silicon Isotopes
5.1. Implication for Meteorites and Planetary Core Formation
5.1.1. Origin of the Lunar Planetary Materials
5.1.2. Formation of the Earth Core
5.2. Implication for Core Deposits Formation and Hydrothermal Fluids Activities
5.2.1. BIF Deposits
5.2.2. Hydrothermal Polymetallic Core Deposits
5.2.3. Clay Minerals Deposits
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McDonough, W.F. Compositional Model for the Earth’s Core; Carlson, R.W., Ed.; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Wedepohl, K.H. The composition of the continental–Crust. Geochim. Cosmochim. Acta 1995, 59, 1217–1232. [Google Scholar] [CrossRef]
- Savage, P.S.; Georg, R.B.; Armytage, R.M.G.; Williams, H.M.; Halliday, A.N. Silicon isotope homogeneity in the mantle. Earth Planet. Sci. Lett. 2010, 295, 139–146. [Google Scholar] [CrossRef]
- De Laeter, J.R.; Bohlke, J.K.; De Biever, P.; Hidaka, H.; Peiser, H.S.; Rosman, K.J.R.; Taylor, P.D.P. Atomic weight of the elements: Review 2000 IUPAC technical report. Pure Appl. Chem. 2003, 75, 683–800. [Google Scholar] [CrossRef]
- Molinivelsko, C.; Mayeda, T.K.; Clayton, R.N. Isotopic composition of silicon in meteorites. Geochim. Cosmochim. Acta 1986, 50, 2719–2726. [Google Scholar] [CrossRef]
- Fitoussi, C.; Bourdon, B.; Kleine, T.; Oberli, F.; Reynolds, B. Silicon isotope composition of meteorites and peridotitis: Si in the earth’s core, influence of nebular processes and terrestrial magmatic processes. Meteorit. Planet. Sci. 2009, 44, A70. [Google Scholar]
- Armytage, R.M.G.; Georg, R.B.; Savage, P.S.; Williams, H.M.; Halliday, A.N. Silicon isotopes in meteorites and planetary core formation. Earth Planet. Sci. Lett. 2011, 75, 3662–3676. [Google Scholar] [CrossRef]
- Zambardi, T.; Poitrasson, F.; Corgne, A.; Méheut, M.; Quitté, G.; Anand, M. Silicon isotope variations in the inner solar system: Implications for planetary formation, differentiation and composition. Geochim. Cosmochim. Acta 2013, 127, 67–83. [Google Scholar] [CrossRef]
- Douthitt, C.B. The geochemistry of the stable isotopes of silicon. Geochim. Cosmochim. Acta 1982, 46, 1449–1458. [Google Scholar] [CrossRef]
- Ding, T.P.; Jiang, S.Y.; Wan, D.F.; Li, Y.H.; Li, J.; Song, H.; Liu, Z.; Yao, X. Silicon Isotope Geochemistry; Geological Publishing House: Beijing, China, 1996. [Google Scholar]
- Oelze, M.; von Blanckenburg, F.; Hoellen, D.; Dietzel, M.; Bouchez, J. Si stable isotope fractionation during adsorption and the competition between kinetic and equilibrium isotope fractionation: Implications for weathering systems. Chem. Geol. 2014, 380, 161–171. [Google Scholar] [CrossRef]
- Opfergelt, S.; Georg, R.B.; Delvaux, B.; Cabidoche, Y.M.; Burton, K.W.; Halliday, A.N. Silicon isotopes and the tracing of desilication in volcanic soil weathering sequences. Chem. Geol. 2012, 326, 113–122. [Google Scholar] [CrossRef]
- Opfergelt, S.; Delmelle, P. Silicon isotopes and continental weathering processes: Assessing controls on Si transfer to the ocean. Comptes Rendus Geosci. 2012, 344, 723–738. [Google Scholar] [CrossRef]
- Basile–Doelsch, I.; Meunier, J.D.; Parron, C. Another continental pool in the terrestrial silicon cycle. Nature 2005, 433, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.P.; Ma, G.R.; Shui, M.X.; Wan, D.F.; Li, R.H. Silicon isotope study on rice plants from the Zhejiang province, China. Chem. Geol. 2005, 218, 41–50. [Google Scholar] [CrossRef]
- De La Rocha, C.L.; Brzezinski, M.A.; DeNiro, M.J. Fractionation of silicon isotopes by marine diatoms during biogenic silica formation. Geochim. Cosmochim. Acta 1997, 61, 5051–5056. [Google Scholar] [CrossRef]
- Engström, E.; Rodushkin, I.; Öhlander, B.; Ingri, J.; Baxter, D.C. Silicon isotopic composition of boreal forest vegetation in Northern Sweden. Chem. Geol. 2008, 257, 247–256. [Google Scholar] [CrossRef]
- Engström, E.; Rodushkin, I.; Ingri, J.; Baxter, D.C.; Ecke, F.; Osterlund, H.; Ohlander, B. Temporal isotopic variations of dissolved silicon in a pristine boreal river. Chem. Geol. 2010, 271, 142–152. [Google Scholar] [CrossRef]
- Cardinal, D.; Alleman, L.Y.; Dehairs, F.; Savoye, N.; Trull, T.W.; Andre, L. Relevance of silicon isotopes to Si–nutrient utilization and Si–source assessment in Antarctic. Global Biogeochem. Cycles 2005, 19, GB2007. [Google Scholar] [CrossRef]
- Cardinal, D.; Savoye, N.; Trull, T.; Dehairs, F.; Kopczynska, E.E.; Fripiat, F.; Tison, J.L.; Andre, L. Silicon isotopes in spring Southern Ocean diatoms: Large zonal changes despite homogeneity among size fractions. Mar. Chem. 2007, 106, 46–62. [Google Scholar] [CrossRef]
- Ding, T.P.; Tian, S.H.; Sun, L.; Wu, L.H.; Zhou, J.X.; Chen, Z.Y. Silicon isotope fractionation between rice plants and nutrient solution and its significance to the study of the silicon cycle. Geochim. Cosmochim. Acta 2008, 72, 5600–5615. [Google Scholar] [CrossRef]
- Ding, T.P.; Zhou, J.X.; Wan, D.F.; Chen, Z.Y.; Wang, C.Y.; Zhang, F. Silicon isotope fractionation in bamboo and its significance to the biogeochemical cycle of silicon. Geochim. Cosmochim. Acta 2009, 72, 1381–1395. [Google Scholar] [CrossRef]
- Opfergelt, S.; Cardinal, D.; Henriet, C.; Draye, X.; Andre, L.; Delvaux, B. Silicon isotopic fractionation by banana (Musa spp.) grown in a continuous nutrient flow device. Plant Soil. 2006, 285, 333–345. [Google Scholar] [CrossRef]
- Hendry, K.R.; Georg, R.B.; Rickaby, R.E.M.; Robinson, L.F.; Halliday, A.N. Deep ocean nutrients during the Last Glacial Maximum deduced from sponge silicon isotopic compositions. Earth Planet. Sci. Lett. 2010, 292, 290–300. [Google Scholar] [CrossRef]
- Hendry, K.R.; Robinson, L.F. The relationship between silicon isotope fractionation in sponges and silicic acid concentration: Modern and core–top studies of biogenic opal. Geochim. Cosmochim. Acta 2012, 81, 1–12. [Google Scholar] [CrossRef]
- Opfergelt, S.; Cardinal, D.; André, L.; Delvigne, C.; Bremond, L.; Delvaux, B. Variations of δ30Si and Ge/Si with weathering and biogenic input in tropical basalticash soils under monoculture. Geochim. Cosmochim. Acta 2010, 74, 225–240. [Google Scholar] [CrossRef]
- Cao, Z.M.; Frank, M.; Dai, M.H.; Grasse, P.; Ehlert, C. Silicon isotope constraints on sources and utilization of silicic acid in the northern South China Sea. Geochim. Cosmochim. Acta 2012, 97, 876–889. [Google Scholar] [CrossRef]
- Cornelis, J.T.; Delvaux, B.; Georg, R.B.; Lucas, Y.; Ranger, J.; Opfergelt, S. Tracing the origin of dissolved silicon transferred from various soil–plant systems towards rivers: a review. Biogeosciences 2011, 8, 89–112. [Google Scholar] [CrossRef]
- Geilert, S.; Vroon, P.Z.; Keller, N.S.; Gudbrandsson, S.; Stefansson, A.; van Bergen, M.J. Silicon isotope fractionation during silica precipitation from hot–spring waters: Evidence from the Geysir geothermal field, Iceland. Geochim. Cosmochim. Acta 2015, 164, 403–427. [Google Scholar] [CrossRef]
- Tatzel, M.; von Blanckenburg, F.; Oelze, M.; Schuessler, J.A.; Bohrmann, G. The silicon isotope record of early silica diagenesis. Geochim. Cosmochim. Acta 2015, 428, 293–303. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Ding, T.P.; Wan, D.F. A study on silicon and oxygen isotope geochemistry of some lead–zinc ore deposits in China. Chin. Sci. Bull. 1992, 37, 1022–1027. (in Chinese). [Google Scholar]
- Jiang, S.Y.; Ding, T.P.; Wan, D.F.; Li, Y.H. Silicon isotopic compositions of archean banded Si–Fe formation (BIF) in the Gongchangling ore deposit, Liaoning province, China. Sci. China Ser. B 1993, 36, 482–489. [Google Scholar]
- Jiang, S.Y.; Palmer, M.R.; Ding, T.P. Silicon isotope geochemistry of the Sullivan Pb–Zn deposit, Canada: A preliminary study. Econ. Geol. 1994, 89, 1623–1629. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Han, F.; Shen, J.Z.; Palmer, M.R. Chemical and Rb–Sr, Sm–Nd isotopic systematics of tourmaline from the Dachang Sn–polymetallic ore deposit, Guangxi Province, P.R. China. Chem. Geol. 1999, 157, 49–67. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Palmer, M.R.; Slack, J.F.; Shaw, D.R. Boron isotope systematics of tourmaline formation in the Sullivan Pb–Zn–Ag deposit, British Columbia, Canada. Chem. Geol. 1999, 158, 131–144. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Palmer, M.R.; Slack, J.F.; Ding, T.P.; Wan, D.F. Silicon isotope compositions of tourmalinites and selected silicate minerals from the Sullivan deposit and vicinity, and their significance to mineral exploration and ore genesis. In The Geological Environment of the Sullivan Deposit British Columbia; Lydon, J.W., Hoy, T., Slack, J.F., Knapp, M.E., Eds.; Geological Association of Canada Special Publication: St. John’s, NL, Canada, 2000; pp. 782–790. [Google Scholar]
- Roerdink, D.L.; van den Boorn, S.H.J.M.; Geilert, S.; Vroon, P.Z.; van Bergen, M.J. Experimental constraints on kinetic and equilibrium silicon isotope fractionation during the formation of non–biogenic chert deposits. Chem. Geol. 2015, 402, 40–51. [Google Scholar] [CrossRef]
- Jaeger, F.M.; Dijkstra, D.W. The mass ratio of isotopes in chemical elements. Phlebol–ANN. Vas. 1924, 27, 393–406. [Google Scholar]
- Reynolds, J.H.; Verhoogen, J. Natural variations in the isotopic constitution of silicon. Geochim. Cosmochim. Acta 1953, 3, 224–234. [Google Scholar] [CrossRef]
- Allenby, R.J. Determination of the isotopic ratios of silicon in rocks. Geochim. Cosmochim. Acta 1954, 5, 540–548. [Google Scholar] [CrossRef]
- Grant, F.S. The geological significance of variations in the abandances of the isotopes of silicon in rocks. Geochim. Cosmochim. Acta 1954, 5, 225–242. [Google Scholar] [CrossRef]
- Esptein, S.; Taylor, H.P., Jr. 18O/16O, 30Si/28Si, D/H and 13C/12C studies of lunar rocks and minerals. Science 1970, 167, 533–535. [Google Scholar]
- Clayton, R.N.; Mayeda, T.K.; Epstein, S. Isotopic fractionation of silicon in Allende inclusions. In Proceedings of the 9th Lunar and Planetary Science Conference, Huston, TX, USA, 13–17 March 1978; pp. 1267–1278. [Google Scholar]
- Ding, T.P.; Wan, D.; Wang, C.; Zhang, F. Silicon isotope compositions of dissolved silicon and suspended matter in the Yangtze River, China. Geochim. Cosmochim. Acta 2004, 68, 205–216. [Google Scholar] [CrossRef]
- De La Rocha, C.L.; Brzezinski, M.A.; DeNiro, M.J.; Shemesh, A. Silicon–isotope composition of diatoms as an indicator of past oceanic change. Nature 1998, 395, 680–683. [Google Scholar] [CrossRef]
- De La Rocha, C.L.; Brzezinski, M.A.; DeNiro, M.J. A first look at the distribution of the stable isotopes of silicon in natural waters. Geochim. Cosmochim. Acta 2000, 64, 2467–2477. [Google Scholar] [CrossRef]
- Varela, D.E.; Pride, C.J.; Brzezinski, M.A. Biological fractionation of silicon isotopes in Southern Ocean surface waters. Global Biogeochem. Cycles 2004, 18. [Google Scholar] [CrossRef]
- Georg, R.B.; Reynolds, B.C.; Frank, M.; Halliday, A.N. New sample preparation techniques for the determination of Si isotopic compositions using MC–ICPMS. Chem. Geol. 2006, 235, 95–104. [Google Scholar] [CrossRef]
- Georg, R.B.; Reynolds, B.C.; Frank, M.; Halliday, A.N. Mechanisms controlling the silicon isotopic compositions of river waters. Earth Planet. Sci. Lett. 2006, 249, 290–306. [Google Scholar] [CrossRef]
- Georg, R.B.; Reynolds, B.C.; West, A.J.; Burton, K.W.; Halliday, A.N. Silicon isotope variations accompanying basalt weathering in Iceland. Earth Planet. Sci. Lett. 2007, 261, 476–490. [Google Scholar] [CrossRef]
- Georg, R.B.; West, A.J.; Basu, A.R.; Halliday, A.N. Silicon fluxes and isotope composition of direct groundwater discharge into the Bay of Bengal and the effect on the global ocean silicon isotope budget. Earth Planet. Sci. Lett. 2009, 283, 67–74. [Google Scholar] [CrossRef]
- Pokrovsky, O.S.; Reynolds, B.C.; Prokushkin, A.S.; Schott, J.; Viers, J. Silicon isotope variations in Central Siberian rivers during basalt weathering in permafrost–dominated larch forests. Chem. Geol. 2013, 355, 103–116. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Jacobsen, S.B. Silicon isotopes in the inner Solar System: Implications for core formation, solar nebular processes and partial melting. Geochim. Cosmochim. Acta 2010, 74, 6921–6933. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Jacobsen, S.B. The isotopic composition of magnesium in the inner Solar System. Earth Planet. Sci. Lett. 2010, 293, 349–358. [Google Scholar] [CrossRef]
- Fitoussi, C.; Bourdon, B. Silicon isotope evidence against an enstatite chondrite earth. Science 2012, 335, 1477–1480. [Google Scholar] [CrossRef]
- Ziegler, K.; Young, E.D.; Schauble, E.A.; Wasson, J.T. Metal–silicate silicon isotope fractionation in enstatite meteorites and constraints on Earth’s core formation. Earth Planet. Sci. Lett. 2010, 295, 487–496. [Google Scholar] [CrossRef]
- Savage, P.S.; Moynier, F. Silicon isotopic variation in enstatite meteorites: Clues to their origin and Earth–forming material. Earth Planet. Sci. Lett. 2013, 361, 487–496. [Google Scholar] [CrossRef]
- Savage, P.S.; Armytage, R.M.G.; Georg, R.B.; Halliday, A.N. High temperature silicon isotope geochemistry. Lithos 2014, 190, 500–519. [Google Scholar] [CrossRef]
- Poitrasson, F. Silicon isotope geochemistry. Rev. Mineral. Geochem. 2017, 82, 289–344. [Google Scholar] [CrossRef]
- Frings, F. Revisiting the dissolution of biogenic Si in marine sediments: A key term in the ocean Si budget. Acta Geochim. 2017, 36, 429–432. [Google Scholar] [CrossRef]
- Conley, D.J.; Frings, P.J.; Fontorbe, G.; Clymans, W.; Stadmark, J.; Hendry, K.R.; Marron, A.O.; De La Rocha, C.L. Biosilicification drives a decline of dissolved Si in the oceans through geologic time. Front. Mar. Sci. 2017, 4. [Google Scholar] [CrossRef]
- Sutton, J.N.; André, L.; Cardinal, D.; Conley, D.J.; de Souza, G.F.; Dean, J.; Dodd, J.; Ehlert, C.; Ellwood, M.J.; Frings, P.J.; et al. A review of the stable isotope bio-geochemistry of the global silicon cycle and its sssociated trace elements. Front. Earth Sci. 2018, 5. [Google Scholar] [CrossRef]
- Huneke, J.C.; Armstrong, J.T.; Wasserburg, G.J. Fun with panurge–High mass resolution ion microprobe measurements of Mg in allende inclusions. Geochim. Cosmochim. Acta 1983, 47, 1635–1650. [Google Scholar] [CrossRef]
- Zinner, E.; Tang, M.; Anders, E. Large isotopic anomalies of Si, C, N and noble–gases in interstellar silicon–carbide from the murray meteorite. Nature 1987, 330, 730–732. [Google Scholar] [CrossRef]
- Stone, J.; Hutcheou, I.D.; Epstein, S.; Wasserburg, G.J. Correlated 29Si and 30Si enrichements in a family of SiC grains from Orgueil and Murchison meteorites. Earth Planet. Sci. Lett. 1991, 107, 570–581. [Google Scholar] [CrossRef]
- Walder, A.J.; Freedman, P.A. Isotopic ration measurement using a double focusing magnetic–sector mass analyzer with in inductively coupled plasma as an ion–source. J. Anal. At. Spectrom. 1992, 7, 571–575. [Google Scholar] [CrossRef]
- Clayton, R.N.; Mayeda, T.K. The use of bromine pentafluoride in the extraction of oxygen from oxides and silicates for isotope analysis. Geochim. Cosmochim. Acta 1963, 27, 43–52. [Google Scholar] [CrossRef]
- Brzezinski, M.A.; Jones, J.L.; Beucher, C.P.; Demarest, M.S.; Berg, H.L. Automated determination of silicon isotope natural abundance by the acid decomposition of cesium hexafluosilicate. Anal. Chem. 2006, 78, 6109–6114. [Google Scholar] [CrossRef]
- Clayton, R.N. High temperature isotope effects in the early solar system. Rev. Mineral. Geochem. 1986, 16, 129–140. [Google Scholar]
- Robert, F.; Chaussidon, M. A palaeotemperature curve for the Precambrian oceans based on silicon isotopes in cherts. Nature 2006, 443, 969–972. [Google Scholar] [CrossRef]
- Heck, P.R.; Huberty, J.M.; Kita, N.T.; Ushikubo, T.; Kozdon, R.; Valley, J.W. SIMS analyses of silicon and oxygen isotope ratios for quartz from Archean and Paleoproterozoic banded iron formations. Geochim. Cosmochim. Acta 2011, 75, 5879–5891. [Google Scholar] [CrossRef]
- Reynolds, B.C.; Georg, R.B.; Oberli, F.; Wiechert, U.; Halliday, A.N. Re–assessment of silicon isotope reference materials using high–resolution multi–collector ICP–MS. J. Anal. At. Spectrom. 2006, 21, 266–269. [Google Scholar] [CrossRef]
- van den Boorn, S.H.J.M.; Vroon, P.Z.; van Bergen, M.J. Sulfur–induced offsets in MC–ICP–MS silicon–isotope measurements. J. Anal. At. Spectrom. 2009, 24, 1111–1114. [Google Scholar] [CrossRef]
- Hughes, H.J.; Delvigne, C.; Korntheuer, M.; de Jong, J.; Andre, L.; Cardinal, D. Controlling the mass bias introduced by anionic and organic matrices in silicon isotopic measurements by MC–ICP–MS. J. Anal. At. Spectrom. 2011, 26, 1892–1896. [Google Scholar] [CrossRef]
- Schuessler, J.A.; von Blanckenburg, F. Testing the limits of micro–scale analyses of Si stable isotopes by femtosecond laser ablation multicollector inductively coupled plasma mass spectrometry with application to rock weathering. Spectrochim. Acta Part B 2014, 98, 1–18. [Google Scholar] [CrossRef]
- Frick, D.A.; Schuessler, J.A.; von Blanckenburg, F. Development of routines for simultaneous in situ chemical composition and stable Si isotope ratio analysis by femtosecond laser ablation inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2016, 938, 33–43. [Google Scholar] [CrossRef]
- Yu, H.M.; Lia, Y.H.; Gao, Y.J.; Huang, J.; Huang, F. Silicon isotopic compositions of altered oceanic crust: Implications for Si isotope heterogeneity in the mantle. Chem. Geol. 2018, 479, 1–9. [Google Scholar] [CrossRef]
- Wang, W.; Wei, H.Z.; Jiang, S.Y.; Christopher, J.C.; Guo, Q.; Lin, Y.B. Adsorption behavior of metasilicate on N–Methyl D–Glucamine functional groups and associated silicon isotope fractionation. Langmuir 2016, 32, 8872–8881. [Google Scholar] [CrossRef]
- O’Neil, J.R. Theoretical and Experimental Aspects of Isotopic Fractionation. Rev. Mineral. Geochem. 1986, 16, 1–40. [Google Scholar]
- Huang, F.; Wu, Z.Q.; Huang, S.C.; Wu, F. First–principles calculations of equilibrium silicon isotope fractionation among mantle minerals. Geochim. Cosmochim. Acta 2014, 140, 509–520. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Huang, F.; Huang, S.C. Isotope fractionation induced by phase transformation: First–principles investigation for Mg2SiO4. Earth Planet. Sci. Lett. 2015, 409, 339–347. [Google Scholar] [CrossRef]
- Méheut, M.; Lazzeri, M.; Balan, E.; Mauri, F. Equilibrium isotopic fractionation in the kaolinite, quartz, water system: Prediction from first–principles density–functional theory. Geochim. Cosmochim. Acta 2007, 71, 3170–3181. [Google Scholar]
- Méheut, M.; Lazzeri, M.; Balan, E.; Mauri, F. Structural control over equilibrium silicon and oxygen isotopic fractionation: A first–principles density–functional theory study. Chem. Geol. 2009, 258, 28–37. [Google Scholar] [CrossRef]
- Méheut, M.; Schauble, E.A. Silicon isotope fractionation in silicate minerals: Insights from first–principles models of phyllosilicates, albite and pyrope. Geochim. Cosmochim. Acta 2014, 134, 137–154. [Google Scholar] [CrossRef]
- Gunnarsson, I.; Arnorsson, S. Amorphous silica solubility and the thermodynamic properties of H4SiO4 degrees in the range of 0 degrees to 350 degrees C at P–sat. Geochim. Cosmochim. Acta 2000, 64, 2295–2307. [Google Scholar] [CrossRef]
- Fujii, T.; Pringle, E.A.; Chaussidon, M.; Moynier, F. Isotope fractionation of Si in protonation/deprotonation reaction of silicic acid: A new pH proxy. Geochim. Cosmochim. Acta 2015, 168, 193–205. [Google Scholar] [CrossRef]
- Dupuis, R.; Benoit, M.; Nardin, E.; Méheut, M. Fractionation of silicon isotopes in liquids: The importance of configurational disorder. Chem. Geol. 2015, 396, 239–254. [Google Scholar] [CrossRef]
- Marshall, H.R.; Foster, G.L. Boron Isotopes in the Earth and Planetary Sciences—A Short History and Introduction. In Boron Isotopes; Marschall, H., Foster, G., Eds.; Springer Int. Publishing: Berlin, Germany, 2018; pp. 1–11. [Google Scholar]
- Wonisch, H.; Gérard, F.; Dietzel, M.; Jaffrain, J.; Nestroy, O.; Boudot, J.P. Occurrence of polymerized silicic acid and aluminum species in two forest soil solutions with different acidity. Geoderma 2008, 144, 435–445. [Google Scholar] [CrossRef]
- Nelson, D.M.; Brzezinski, M.A. Diatom growth and productivity in an oligotrophic midocean gyre: A 3–yr record from the Sargasso Sea near Bermuda. Linol. Oceanogr. 1997, 42, 473–486. [Google Scholar]
- Dixit, S.; Van, C.; Philippe, V.B.; Johan, A. Processes controlling solubility of biogenic silica and pore water build–up of silicic acid in marine sediments. Mar. Chem. 2001, 73, 333–352. [Google Scholar] [CrossRef]
- Fleming, B.A.; Crerar, D.A. Silicic acid ionization and calculation of silica solubility at elevated temperature and pH: Application to geothermal fluid processing and reinjection. Geothermics 1982, 11, 15–29. [Google Scholar] [CrossRef]
- Laudise, R.A. Kinetics of hydrothermal quartz crystallization 1. J. Am. Chem. Soc. 1959, 3, 81. [Google Scholar]
- Bohlmann, E.G.; Mesmer, R.E.; Berlinski, P. Kinetics of silica deposition from simulated geothermal brines. Soc. Pet. Eng. J. 1980, 20, 103–111. [Google Scholar]
- Hosaka, M.; Taki, S. Hydrothermal growth of quartz crystals in NaCl solution. J. Cryst. Growth 1981, 52, 837–842. [Google Scholar] [CrossRef]
- Hosaka, M.; Taki, S. Growth patterns on the rhombohedral faces of quartz crystals grown hydrothermally in NaCl and KCl solutions. J. Cryst. Growth 1981, 55, 363–368. [Google Scholar] [CrossRef]
- Weres, O.; Yee, A.; Tsao, L. Kinetics of silica polymerization. J. Colloid Interface Sci. 1981, 84, 379–402. [Google Scholar] [CrossRef]
- Fleming, B.A. Kinetics of reaction between silicic acid and amorphous silica surfaces in NaCl solutions. J. Colloid Interface Sci. 1986, 110, 40–64. [Google Scholar] [CrossRef]
- Carroll, S.; Mroczek, E.; Alai, M.; Ebert, M. Amorphous silica precipitation (60 to 120 °C) comparison of laboratory and field rates. Geochim. Cosmochim. Acta 1998, 62, 1379–1396. [Google Scholar] [CrossRef]
- Iler, R.K. The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Chemistry, and Biochemistry; John Wiley Sons: Hoboken, NJ, USA, 1979; pp. 2951–2956. [Google Scholar]
- Moulik, S.P.; Mullick, D.K. On kinetics of polymerisation of vanadic acid its complexes with ortho–phosphoric acid. Zeitschrift für Physikalische Chemie 1966, 48, 196–205. [Google Scholar] [CrossRef]
- Li, Y.H.; Ding, T.P.; Wan, D.F. Experimental study of silicon isotope dynamic fractionation and its application in geology. Chinese J. Geochem. 1995, 14, 212–219. [Google Scholar]
- Geilert, S.; Vroon, P.Z.; van Bergen, M.J. Silicon isotopes and trace elements in chert record early Archean basin evolution. Chem. Geol. 2014, 386, 133–142. [Google Scholar] [CrossRef]
- Skulan, J.L.; Beard, B.L.; Johnson, C.M. Kinetic and equilibrium Fe isotope fractionation between aqueous Fe(III) and hematite. Geochim. Cosmochim. Acta 2002, 66, 2995–3015. [Google Scholar] [CrossRef]
- DePaolo, D.J. Calcium isotopic variations produced by biological, kinetic, radiogenic and nucleosynthetic processes. Rev. Mineral. Geochem. 2004, 55, 255–288. [Google Scholar]
- Chen, X.Y.; Chafetz, H.S.; Andreasen, R.; Lapen, T.J. Silicon isotope compositions of euhedral authigenic quartz crystals: Implications for abiotic fractionation at surface temperatures. Chem. Geol. 2016, 423, 61–73. [Google Scholar] [CrossRef]
- Pollington, A.D.; Kozdon, R.; Anovitz, L.M.; Georg, R.B.; Spicuzza, M.J.; Valley, J.W. Experimental calibration of silicon and oxygen isotope fractionations between quartz and water at 250 °C by in situ microanalysis of experimental products and application to zoned low δ30Si quartz overgrowths. Chem. Geol. 2016, 421, 127–142. [Google Scholar] [CrossRef]
- Li, Y.H. Some applications of isotope–tracing in geology. Earth Sci. Front. 1998, 5, 106–112. [Google Scholar]
- van den Boorn, S.H.J.M.; van Bergen, M.J.; Vroon, P.Z.; de Vries, S.T.; Nijman, W. Silicon isotope and trace element constraints on the origin of 3.5 Ga cherts: Implications for Early Archaean marine environments. Geochim. Cosmochim. Acta 2010, 74, 1077–1103. [Google Scholar] [CrossRef]
- Abraham, K.; Hofmann, A.; Foley, S.F.; Cardinal, D.; Harris, C.; Barth, M.G.; Andre, L. Coupled silicon–oxygen isotope fractionation traces Archaean silicification. Earth Planet. Sci. Lett. 2011, 301, 222–230. [Google Scholar] [CrossRef]
- Oelze, M.; von Blanckenburg, F.; Bouchez, J.; Hoellen, D.; Dietzel, M. The effect of Al on Si isotope fractionation investigated by silica precipitation experiments. Chem. Geol. 2015, 397, 94–105. [Google Scholar] [CrossRef]
- Delstanche, S.; Opfergelt, S.; Cardinal, D.; Elsass, F.; André, L.; Delvaux, B. Silicon isotopic fractionation during adsorption of aqueous monosilicic acid onto iron oxide. Geochim. Cosmochim. Acta 2009, 73, 923–934. [Google Scholar] [CrossRef]
- Pokrovski, G.S.; Schott, J.; Garges, F.; Hazemann, J.L. Iron (III)–silica interactions in aqueous solution: Insights from X–ray absorption fine structure spectroscopy. Geochim. Cosmochim. Acta 2003, 67, 3559–3573. [Google Scholar] [CrossRef]
- Hiemstra, T.; van Riemsdijk, W.H. Adsorption and surface oxidation of Fe(II) on metal (hydr) oxides. Geochim. Cosmochim. Acta 2007, 71, 5913–5933. [Google Scholar] [CrossRef]
- Swedlund, P.J.; Webster, J.G. Adsorption and polymerization of silicic acid on ferrihydrite, and its effect on arsenic adsorption. Water Res. 1999, 33, 3413–3422. [Google Scholar] [CrossRef]
- Opfergelt, S.; de Bournonville, G.; Cardinal, D.; André, L.; Delstanche, S.; Delvaux, B. Impact of soil weathering degree on silicon isotopic fractionation during adsorption onto iron oxides in basaltic ash soils, Cameroon. Geochim. Cosmochim. Acta 2009, 73, 7226–7240. [Google Scholar] [CrossRef]
- Bern, C.R.; Brzezinski, M.A.; Beucher, C.; Ziegler, K.; Chadwick, O.A. Weathering, dust, and biocycling effects on soil silicon isotope ratios. Geochim. Cosmochim. Acta 2010, 74, 876–889. [Google Scholar] [CrossRef]
- Ziegler, K.; Chadwick, O.A.; Brzezinski, M.A.; Kelly, E.F. Natural variations of delta δ30Si rations during progressive basalt weathering, Hawalian Islands. Geochim. Cosmochim. Acta 2005, 69, 4597–4610. [Google Scholar] [CrossRef]
- Steinhoefel, G.; Breuer, J.; von Blanckenburg, F.; Horn, I.; Kaczorek, D.; Sommer, M. Micrometer silicon isotope diagnostics of soils by UV femtosecond laser ablation. Chem. Geol. 2011, 286, 280–289. [Google Scholar] [CrossRef]
- Savage, P.S.; Georg, R.B.; Williams, H.M.; Halliday, A.N. Silicon isotopes in granulite xenoliths: Insights into isotopic fractionation during igneous processes and the composition of the deep continental crust. Earth Planet. Sci. Lett. 2013, 365, 221–231. [Google Scholar] [CrossRef]
- Cornelis, J.T.; Weis, D.; Lavkulich, L.; Vermeire, M.L.; Delvaux, B.; Barling, J. Silicon isotopes record dissolution and re–precipitation of pedogenic clay minerals in a podzolic soil chronosequence. Geoderma 2014, 235, 19–29. [Google Scholar] [CrossRef]
- Barber, D.A.; Shone, M.G.T. Absorption of silica from aqueous sloution by plants. J. Exp. Bot. 1966, 17, 569. [Google Scholar] [CrossRef]
- Umemura, M.; Takenaka, C. Biological cycle of silicon in moso bamboo (Phyllostachys pubescens) forests in central Japan. Ecol. Res. 2014, 29, 501–510. [Google Scholar] [CrossRef]
- Alexandre, A.; Aunier, J.D.; Colin, F.; Koud, J.M. Plant impact on the biogeochemical cycle of silicon and related weathering processes. Geochim. Cosmochim. Acta 1997, 61, 677–682. [Google Scholar] [CrossRef]
- Sangster, A.G.; Hodson, M.J. Some factors influencing silica transport and deposition in grasses. Am. J. Bot. 1986, 73, 642–643. [Google Scholar]
- Fu, F.F.; Akagi, T.; Yabuki, S. Origin of silica particles found in the cortex of Matteuccia roots. Soil Sci. Soc. Am. J. 2002, 66, 1265–1271. [Google Scholar] [CrossRef]
- Kinrade, S.D.; Del Nin, J.W.; Schach, A.S.; Sloan, T.A.; Wilson, K.L.; Knight, C. Stable five–and six–coordinated silicate anions in aqueous solution. Science 1999, 285, 1542–1545. [Google Scholar] [CrossRef]
- Kinrade, S.D.; Hamilton, R.J.; Schach, A.S.; Knight, C. Aqueous hypervalent silicon complexes with aliphatic sugar acids. J. Chem. Soc. Dalton. 2001, 7, 961–963. [Google Scholar] [CrossRef]
- Lambert, J.B.; Lu, G.; Singer, S.R.; Kolb, V.M. Silicate complexes of sugars in aqueous solution. J. Am. Chem. Soc. 2004, 126, 9611–9625. [Google Scholar] [CrossRef]
- Kinrade, S.D.; Deguns, E.W.; Gillson, A.M.E.; Knight, C. Complexes of pentaoxo and hexaoxo silicon with furanoidic vicinal cisdiols in aqueous solution. J. Chem. Soc. Dalton Trans. 2003, 19, 3713–3716. [Google Scholar] [CrossRef]
- Yoshimura, K.; Miyazaki, Y.; Ota, F.; Matsuoka, S.; Sakashita, H. Complexation of boric acid with the N–methyl–D–glucamine group insolution and in crosslinked polymer. J. Chem. Soc. Faraday Trans. 1998, 94, 683–689. [Google Scholar] [CrossRef]
- Mitzutani, A.J.; Nagase, H.; Fujiwara, N.; Ogoshi, H. Silicic acid polymerization catalyzed by amines and polyamines. Chem. Soc. Jpn. 1998, 71, 2017–2022. [Google Scholar] [CrossRef]
- Pohnert, G. Biomineralization in diatoms mediated through peptide– and polyamine–assisted condensation of silica. Angew. Chem. Int. Edit. 2002, 41, 3167–3169. [Google Scholar] [CrossRef]
- Love, G.D.; Grosjean, E.; Stalvies, C.; Fike, D.A.; Grotzinger, J.P.; Bradley, A.S.; Kelly, A.E.; Bhatia, M.; Meredith, W.; Snape, C.E.; et al. Fossil steroids record the appearance of Demospongiae during the Cryogenian period. Nature 2009, 457, 718–722. [Google Scholar] [CrossRef]
- Frohlich, H.; Barthel, D. Silica uptake of the marine sponge Halichondria panicea in Kiel Bight. Mar. Bio. 1997, 128, 115–125. [Google Scholar] [CrossRef]
- Siever, R. The silica cycle in the precambrian. Geochim. Cosmochim. Acta 1992, 56, 3265–3272. [Google Scholar] [CrossRef]
- Maldonado, M.; Carmona, M.C.; Uriz, M.J.; Cruzado, A. Decline in Mesozoic reef building sponges explained by silicon limitation. Nature 1999, 401, 785–788. [Google Scholar] [CrossRef]
- Tatzel, M.; von Blanckenburg, F.; Oelze, M.; Bouchez, J.; Hippler, D. Late Neoproterozoic seawater oxygenation by siliceous sponges. Nat. Commun. 2017, 8, 621. [Google Scholar] [CrossRef]
- Cassarino, L.; Hendry, K.R.; Meredith, M.P.; Venables, H.J.; De La Rocha, C.L. Silicon isotope and silicic acid uptake in surface waters of Marguerite Bay, West Antarctic Peninsula. Deep Sea Res. Part II 2017, 139, 143–150. [Google Scholar] [CrossRef]
- Reynolds, B.C.; Frank, M.; Halliday, A.N. Silicon isotope fractionation during nutrient utilization in the North Pacific. Earth Planet. Sci. Lett. 2006, 244, 431–443. [Google Scholar] [CrossRef]
- Beucher, C.P.; Brzezinski, M.A.; Jones, J.L. Sources and biological fractionation of Silicon isotopes in the Eastern Equatorial Pacific. Geochim. Cosmochim. Acta 2008, 72, 3063–3073. [Google Scholar] [CrossRef]
- Hendry, K.R.; Brzezinski, M.A. Using silicon isotopes to understand the role of the Southern Ocean in modern and ancient biogeochemistry and climate. Quat. Sci. Rev. 2014, 89, 13–26. [Google Scholar] [CrossRef]
- Ehlert, C.; Doering, K.; Wallmann, K.; Scholz, F.; Sommer, S.; Grasse, P.; Geilert, S.; Frank, M. Stable silicon isotope signatures of marine pore waters–Biogenic opal dissolution versus authigenic clay mineral formation. Geochim. Cosmochim. Acta 2016, 191, 102–117. [Google Scholar] [CrossRef]
- Fontorbe, G.; Frings, P.J.; De La Rocha, C.L.; Hendry, K.R.; Conley, D.J. A silicon depleted North Atlantic since the Palaeogene: Evidence from sponge and radiolarian silicon isotopes. Earth Planet. Sci. Lett. 2016, 453, 67–77. [Google Scholar] [CrossRef]
- De La Rocha, C.L.; Bickle, M.J. Sensitivity of silicon isotopes to whole–ocean changes in the silica cycle. Mar. Geol. 2005, 217, 267–282. [Google Scholar] [CrossRef]
- Horn, M.G.; Beucher, C.; Robinson, R.S.; Brzezinski, M.A. Southern Ocean nitrogen and silicon dynamics during the last deglaciation. Earth Planet. Sci. Lett. 2011, 310, 334–339. [Google Scholar] [CrossRef]
- Marin–Carbonnea, J.; Robert, F.; Chaussidon, M. The silicon and oxygen isotope compositions of Precambrian cherts: A record of oceanic paleo–temperatures? Precambrian Res. 2014, 247, 223–234. [Google Scholar] [CrossRef]
- Ding, T.P. Analytical methods for silicon isotope determinations. In Handbook of Stable Isotope Analytical Techniques; Elsevier: Amsterdam, The Netherlands, 2004; Volume 1, pp. 523–537. [Google Scholar]
- De La Rocha, C.L.; Bescont, P.; Croguennoc, A.; Ponzevera, E. The silicon isotopic composition of surface waters in the Atlantic and Indian sectors of the Southern Ocean. Geochim. Cosmochim. Acta 2011, 75, 5283–5295. [Google Scholar] [CrossRef]
- Bayon, G.; Delvigne, C.; Ponzevera, E.; Borges, A.V.; Darchambeau, F.; Deckker, P.D.; Lambert, T.; Monin, L.; Toucanne, S.; André, L. The silicon isotopic composition of fine–grained river sediments and its relation to climate and lithology. Geochim. Cosmochim. Acta 2018, 229, 147–161. [Google Scholar] [CrossRef]
- Sun, X.L.; Morth, C.M.; Porcelli, D.; Kutscher, L.; Hirst, C.; Murphy, M.; Maximov, T.; Petrov, R.E.; Humborg, C.; Schmitt, M.; et al. Stable silicon isotopic compositions of the Lena River and its tributaries: Implications for silicon delivery to the Arctic Ocean. Geochim. Cosmochim. Acta 2018, 241, 120–133. [Google Scholar] [CrossRef]
- Fripiat, F.; Cardinal, D.; Tison, J.L.; Worby, A.; Andre, L. Diatom–induced silicon isotopic fractionation in Antarctic sea ice. J. Geophys Res. 2007, 112. [Google Scholar] [CrossRef]
- Armytage, R.M.G.; Georg, R.B.; Williams, H.M.; Halliday, A.N. Silicon isotopes in lunar rocks: Implications for the Moon’s formation and the early history of the Earth. Geochim. Cosmochim. Acta 2012, 77, 504–514. [Google Scholar] [CrossRef]
- Poirier, J.P. Light elements in the Earth’s outer core: a critical review. Phys. Earth Planet. 1994, 85, 319–337. [Google Scholar] [CrossRef]
- Allegre, C.J.; Lewin, E. Isotopic systems and stirring times of the earth’s mantle. Earth Planet. Sci. Lett. 1995, 136, 629–646. [Google Scholar] [CrossRef]
- Drake, M.J.; Righter, K. Determining the composition of the Earth. Nature 2002, 416, 39–44. [Google Scholar] [CrossRef]
- Kilburn, M.R.; Wood, B.J. Metal–silicate partitioning and the incompatibility of S and Si during core formation. Earth Planet. Sci. Lett. 1997, 152, 139–148. [Google Scholar] [CrossRef]
- Gessmann, C.K.; Wood, B.J.; Rubie, D.C.; Kilburn, M.R. Solubility of silicon in liquid metal at high pressure: Implications for the composition of the Earth’s core. Earth Planet. Sci. Lett. 2001, 184, 367–376. [Google Scholar] [CrossRef]
- Wade, J.; Wood, B.J. Core formation and the oxidation state of the Earth. Earth Planet. Sci. Lett. 2005, 236, 78–95. [Google Scholar] [CrossRef]
- Kempl, J.; Vroon, P.Z.; Zinngrebe, E.; van Westrenen, W. Si isotope fractionation between Si–poor metal and silicate melt at pressure–temperature conditions relevant to metal segregation in small planetary bodies. Earth Planet. Sci. Lett. 2013, 368, 61–68. [Google Scholar] [CrossRef]
- Palme, H.; O’Neill, H.S.C. Cosmochemical estimates of mantle composition. In Treatise on Geochemistry; Heinrich, D.H., Karl, K.T., Eds.; Pergamon: Oxford, UK, 2003. [Google Scholar]
- Schauble, E.A.; Tonui, E.; Georg, R.B.; Young, E.D.; Halliday, A.N. Estimating magnesium and silicon isotope fractionation with first–principles lattice dynamics. Geochim. Cosmochim. Acta 2007, 71, A884. [Google Scholar]
- Shahar, A.; Ziegler, K.; Young, E.D.; Ricolleau, A.; Schauble, E.A.; Fei, Y.W. Experimentally determined Si isotope fractionation between silicate and Fe metal and implications for Earth’s core formation. Earth Planet. Sci. Lett. 2009, 288, 228–234. [Google Scholar] [CrossRef]
- Corgne, A.; Keshav, S.; Wood, B.J.; McDonough, W.F.; Fei, Y.W. Metal–silicate partitioning and constraints on core composition and oxygen fugacity during Earth accretion. Geochim. Cosmochim. Acta 2008, 72, 574–589. [Google Scholar] [CrossRef]
- Young, E.D.; Manning, C.E.; Schauble, E.A.; Shahar, A.; Macris, C.A.; Lazar, C.; Jordan, M. High–temperature equilibrium isotope fractionation of non–traditional stable isotopes: Experiments, theory, and applications. Chem. Geol. 2015, 395, 176–195. [Google Scholar] [CrossRef]
- Holland, H.D. The Chemical Evolution of the Atmosphere and Oceans; Princeton Univ. Press: Princeton, NJ, USA, 1984; pp. 86–189. [Google Scholar]
- André, L.; Cardinal, D.; Alleman, L.Y.; Moorbath, S. Silicon isotopes in ~3.8 Ga West Greenland rocks as clues to the Eoarchaean supracrustal Si cycle. Earth Planet. Sci. Lett. 2006, 245, 162–173. [Google Scholar] [CrossRef]
- Li, Y.H.; Hou, K.J.; Wan, D.F.; Zhang, Z.J.; Yue, G.L. Precambrian banded iron formations in the North China Craton: Silicon and oxygen isotopes and genetic implications. Ore Geol. Rev. 2014, 57, 299–307. [Google Scholar] [CrossRef]
- van den Boorn, S.H.J.M.; Sander, H.J.M.; van, B.; Manfred, J.N.; Wouter, V.; Pieter, Z. Dual role of seawater and hydrothermal fluids in Early Archean chert formation: Evidence from silicon isotopes. Geology 2007, 35, 939. [Google Scholar] [CrossRef]
- Marin–Carbonne, J.; Chaussidon, M.; Robert, F. Micrometer–scale chemical and isotopic criteria (O and Si) on the origin and history of Precambrian cherts: Implications for paleo–temperature reconstructions. Geochim. Cosmochim. Acta 2012, 92, 129–147. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Knoll, H.A.; Jacobsen, B.S.; Fischer, W.W. Si isotope variability in Proterozoic cherts. Geochim. Cosmochim. Acta 2012, 91, 187–201. [Google Scholar] [CrossRef]
- Marin–Carbonne, J.; Chaussidon, M.; Boiron, M.C.; Robert, F. A combined in situ oxygen, silicon isotopic and fluid inclusion study of a chert sample from Onverwacht Group (3.35 Ga, South Africa): New constraints on fluid circulation. Chem. Geol. 2011, 286, 59–71. [Google Scholar]
- Steinhoefel, G.; Horn, I.; von Blanckenburg, F. Micro–scale tracing of Fe and Si isotope signatures in banded iron formation using femtosecond laser ablation. Geochim. Cosmochim. Acta 2009, 73, 5343–5360. [Google Scholar] [CrossRef]
- Jiang, S.Y.; Palmer, M.R.; Li, Y.H.; Xue, C.J. Chemical compositions of tourmaline in the Yindongzi–Tongmugou Pb–Zn deposits, Qinling, China: implications for hydrothermal ore–forming processes. Miner. Depos. 1995, 30, 225–234. [Google Scholar] [CrossRef]
- Fu, M.; Changkakoti, A.; Krouse, H.R.; Gray, J.; Kwak, T.A.P. An oxygen, hydrogen, sulfur, and carbon isotope study of carbonate–replacement (skarn) tin deposits of the Dachang tin field, China. Econ. Geol. 1991, 86, 1683–1703. [Google Scholar] [CrossRef]
- Han, F.; Hutchinson, R.W. Synthetic studies on the origin of the Dachang tin–polymetallic deposits and their metallogenetic model. Chinese Acad. Geol. Sci. Bull. 1991, 22, 61–80. (in Chinese). [Google Scholar]
- Slack, J.F.; Palmer, M.R.; Stevens, B.P.J.; Barnes, R.G. Origin and significance of tourmaline rich rocks in Broken Hill district, Australia. Econ. Geol. 1993, 88, 505–541. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biotic and Abiotic Material | Δ30Si (‰) | αsolid-solution | References |
|---|---|---|---|
| Biological Uptake | |||
| Sugar | −0.26 to −2.09 | 0.9980 | [98] |
| Rice | −0.5 to −1.6 | 0.9995–0.9984 | [21] |
| Bamboo | −0.8 to −1.4 | 0.9992–0.9986 | [22] |
| Banana | −0.6 to −1.0 | 0.9994–0.9990 | [23] |
| Diatoms | −0.7 to −1.5 | 0.9993–0.9985 | [16,19,20] |
| −1.3 to −0.9 | 0.9987–0.9991 | [16] | |
| Sponge | −0.8 to −2.1 | [19,20] | |
| −2.5 to −5.3 | 0.9960–0.9993 | [24] | |
| −1.32 to −6.52 | 0.9934–0.9986 | [25] | |
| Adsorption Process | |||
| Fe–oxide | −0.73 to −1.09 | 0.9992–0.9995 | [112] |
| Gibbsite (γ–Al(OH)3) | 0.9970–0.9982 | [11] | |
| Clay–bulk soil | −0.29 to −1.74 | 0.9993–0.9997 | [12,13] |
| Precipitation | |||
| Abiotic silica | −1.3 to −3.8 | 0.9990–0.9996 | [102] |
| Biogenic silica | −1.93 to −1.33 | [139] | |
| Euhedral megaquartz | −1.8 to −2.1 | [105] | |
| Chemical Weathering | |||
| Smectite | −0.16 to −0.52 | [51] | |
| Kaolinite | −2.2 | [26,118] | |
| −1.9 to +0.1 | [10] | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Wei, H.-Z.; Jiang, S.-Y.; Liu, X.; Lei, F.; Lin, Y.-B.; Zhao, Y. Silicon Isotope Geochemistry: Fractionation Linked to Silicon Complexations and Its Geological Applications. Molecules 2019, 24, 1415. https://doi.org/10.3390/molecules24071415
Wang W, Wei H-Z, Jiang S-Y, Liu X, Lei F, Lin Y-B, Zhao Y. Silicon Isotope Geochemistry: Fractionation Linked to Silicon Complexations and Its Geological Applications. Molecules. 2019; 24(7):1415. https://doi.org/10.3390/molecules24071415
Chicago/Turabian StyleWang, Wei, Hai-Zhen Wei, Shao-Yong Jiang, Xi Liu, Fang Lei, Yi-Bo Lin, and Yao Zhao. 2019. "Silicon Isotope Geochemistry: Fractionation Linked to Silicon Complexations and Its Geological Applications" Molecules 24, no. 7: 1415. https://doi.org/10.3390/molecules24071415
APA StyleWang, W., Wei, H. -Z., Jiang, S. -Y., Liu, X., Lei, F., Lin, Y. -B., & Zhao, Y. (2019). Silicon Isotope Geochemistry: Fractionation Linked to Silicon Complexations and Its Geological Applications. Molecules, 24(7), 1415. https://doi.org/10.3390/molecules24071415