
High Flow-Rate Sample Loading in Large Volume Whole Water Organic Trace Analysis Using Positive Pressure and Finely Ground Sand as a SPE-Column In-Line Filter


Abstract
:1. Introduction
2. Results
2.1. Absolute and Relative Recoveries for the Three Experiments
2.1.1. Recovery from Pure Water at Three Flow Rates
2.1.2. Method Validation. Recovery Evaluation at High and Low Concentrations in Matrix-Rich Pond Water at the Highest Flow Rate of 40 mL/min
2.1.3. Applying the Developed Method to Recipient Samples from Three Different Natural Locations
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Instrumentation
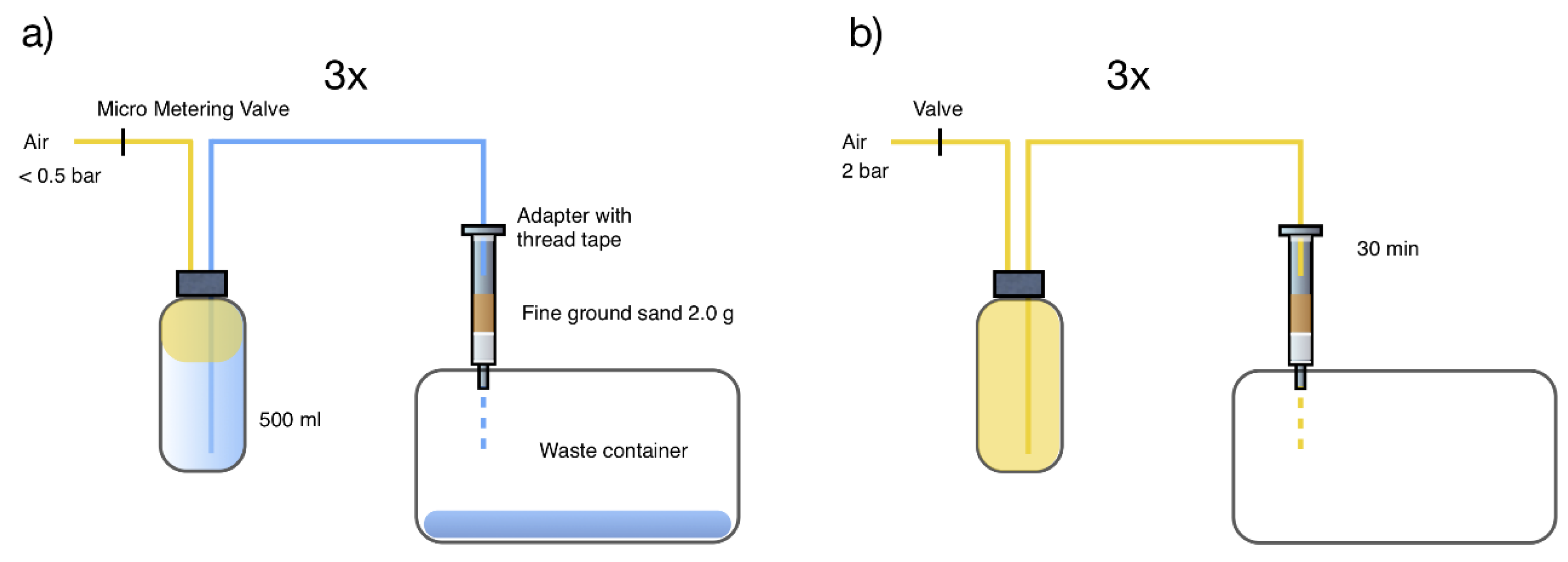
4.2.1. Sample Loader
4.2.2. Sample Loader Construction
4.3. Methods
4.3.1. Recovery from Pure Water at Three Flow Rates
4.3.2. Method Validation. Recovery from Matrix Containing Pond Water at High Flow Rate
4.3.3. Applying the Method to Recipient Samples from Three Different Natural Locations
4.3.4. Sample Loader Operation
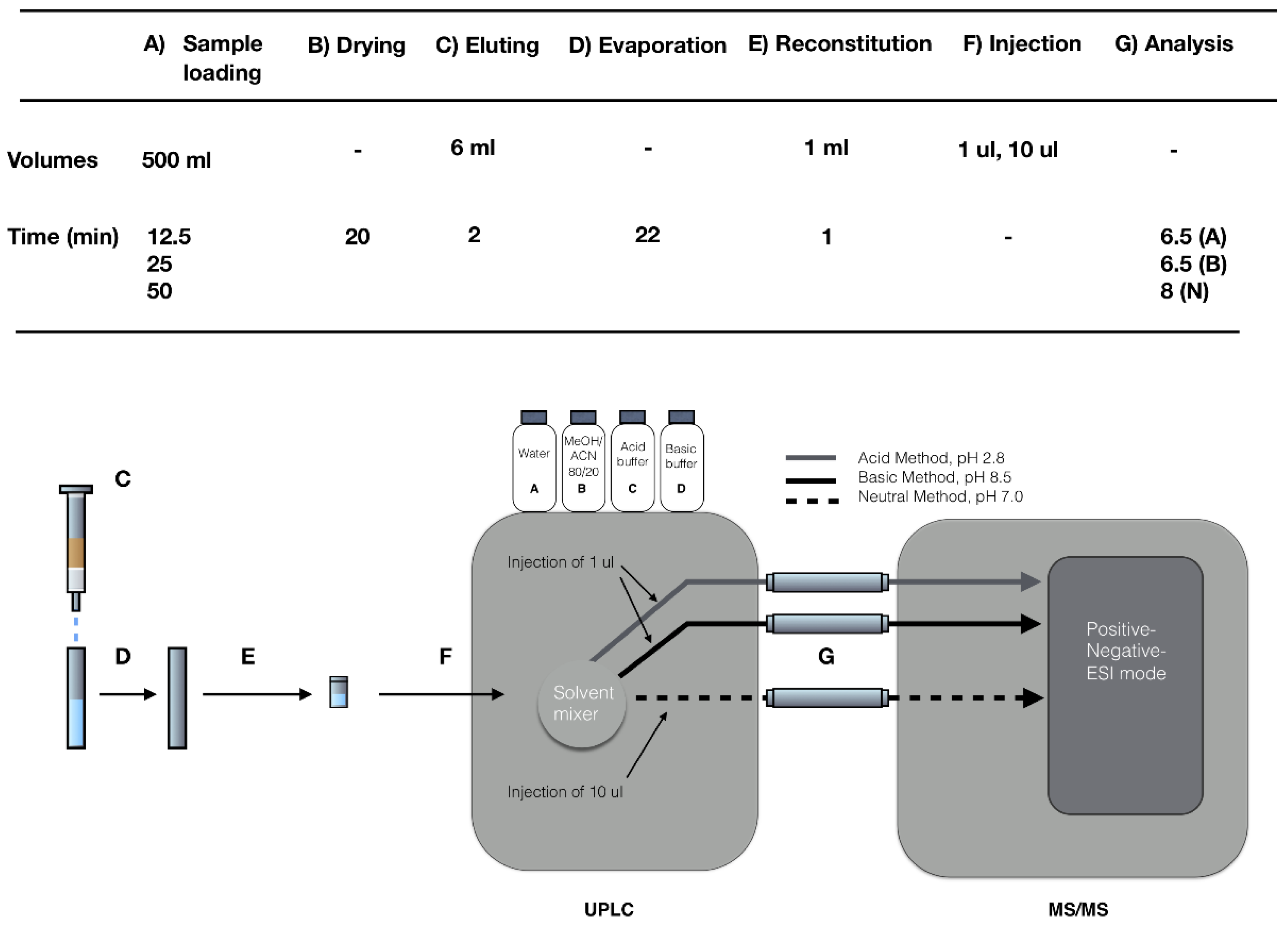
4.3.5. Drying Procedure, Eluting, Reconstituting, and Final Analysis
4.4. Calculations
4.4.1. Absolute Recovery
4.4.2. Relative Recovery
- An = the area of the daughter m/z for the native compound
- Al = the area of the daughter m/z for the labeled compound
- Cl = the amount of the labeled compound in the calibration standard (pg)
- Cn = the amount of the native compound in the calibration standard (pg)
4.4.3. ANOVA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Petrovic, M.; Farré, M.; de Alda, M.L.; Pérez, S.; Postigo, C.; Köck, M.; Radjenovic, J.; Gros, M.; Barcelo, D. Recent trends in the liquid chromatography-mass spectrometry analysis of organic contaminants in environmental samples. J. Chromatogr. A 2010, 1217, 4004–4017. [Google Scholar] [CrossRef] [PubMed]
- QuickStart Guide to SPE; Biotage: Uppsala, Sweden, 2013.
- Guide to Solid Phase Extraction. Bulletin 910; Supelco: Bellefonte, PA, USA, 1998.
- Beginners Guide to SPE; Waters: Milford, MA, USA, 2015.
- Lopez-Serna, R.L.; Petrovic, M.; Barcelo, D. Development of a fast instrumental method for the analysis of pharmaceuticals in environmental and wastewaters based on ultra high performance liquid chromatography (UHPLC)-tandem mass spectrometry (MS/MS). Chemosphere 2011, 85, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- COMMISSION IMPLEMENTING DECISION (EU) 2015/495 of 20 March. Establishing a Watch List of Substances for Union-Wide Monitoring in the Field of Water Policy Pursuant to Directive 2008/105/EC of the European Parliament and of the Council. L 78/40; Official Journal of the European Union, Publications Office of the European Union: Luxembourg, 2015.
- COMMISSION IMPLEMENTING DECISION (EU) 2018/840 of 5 June 2018. Establishing a Watch List of Substances for Union-Wide Monitoring in the Field of Water Policy Pursuant to DIRECTIVE 2008/105/EC of the European Parliament and of the Council. L 141/61; Official Journal of the European Union, Publications Office of the European Union: Luxembourg, 2018.
- Petrovic, M. Methodological challenges of multi-residue analysis of pharmaceuticals in environmental samples. Trends Environ. Anal. Chem. 2014, 1, e25–e33. [Google Scholar] [CrossRef]
- Svahn, O.; Björklund, E. Simple, fast and inexpensive large “whole water” volume sample SPE-loading using compressed air and finely ground sand. Anal. Methods VL IS 2019, 1431, 64–73. [Google Scholar] [CrossRef]
- Suri, R.P.S.; Singh, T.S.; Chimchirian, R.F. Effect of process conditions on the analysis of free and conjugated estrogen hormones by solid-phase extraction–gas chromatography/mass spectrometry (SPE–GC/MS). Environ. Monit. Assess. 2011, 184, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Wickramasekara, S.; Hernández-Ruiz, S.; Abrell, L.; Arnold, R.; Chorover, J. Natural dissolved organic matter affects electrospray ionization during analysis of emerging contaminants by mass spectrometry. Anal. Chim. Acta 2012, 717, 77–84. [Google Scholar] [CrossRef]
- Petrie, B.; Youdan, J.; Barden, R.; Kasprzyk-Hordern, B. Multi-residue analysis of 90 emerging contaminants in liquid and solid environmental matrices by ultra-high-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2016, 1431, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Lor, E.; Martínez, M.; Sancho, J.V.; Peñuela, G.; Hernández, F. Multi-class determination of personal care products and pharmaceuticals in environmental and wastewater samples by ultra-high performance liquid-chromatography-tandem mass spectrometry. Talanta 2012, 99, 1011–1023. [Google Scholar] [CrossRef] [Green Version]
- Hladik, M.L.; Kolpin, D.W.; Kuivila, K.M. Widespread occurrence of neonicotinoid insecticides in streams in a high corn and soybean producing region. USA. Environ. Pollut. 2014, 193, 189–196. [Google Scholar] [CrossRef]
- Baker, D.R.; Kasprzyk-Hordern, B. Critical evaluation of methodology commonly used in sample collection. storage and preparation for the analysis of pharmaceuticals and illicit drugs in surface water and wastewater by solid phase extraction and liquid chromatography-mass spectrometry. J. Chromatogr. A 2011, 1218, 8036–8059. [Google Scholar] [CrossRef] [PubMed]
- Öller SSinger, H.P.; Fässler, P.; Müller, S.R. Simultaneous quantification of neutral and acidic pharmaceuticals and pesticides at the low-ng/l level in surface and waste water. J. Chromatogr. A 2001, 911, 225–234. [Google Scholar] [CrossRef]
- Nowara, A.; Burhenne, J.; Spiteller, M. Binding of Fluoroquinolone Carboxylic Acid Derivatives to ClayMinerals. J. Agric. Food Chem. 1997, 1459–1463. [Google Scholar] [CrossRef]
- Pérez-Fernández, V.; Rocca, L.M.; Tomai, P.; Fanali, S.; Gentili, A. Recent advancements and future trends in environmental analysis: Sample preparation, liquid chromatography and mass spectrometry. Anal. Chim. Acta 2017, 983, 9–41. [Google Scholar] [CrossRef] [PubMed]
- Svahn, O.; Björklund, E. Increased electrospray ionization intensities and expanded chromatographic possibilities for emerging contaminants using mobile phases of different pH. J. Chromatogr. B 2016, 1033–1034, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Method 1694: Pharmaceuticals and Personal Care Products in Water, Soil, Sediment, and Biosolids by HPLC/MS/MS; EPA: Washington, DC, USA, 2007.
- Svahn, O. Tillämpad Miljöanalytisk Kemi för Monitorering Och Åtgärder av Antibiotika- Och Läkemedelsrester i Vattenriket; Lund University: Lund, Sweden, 2016. [Google Scholar]
- DURAN, Group. Available online: http://www.duran-group.com/en/products-solutions/laboratory-glassware/products/laboratory-glass-bottles/ (accessed on 10 March 2018).
- Płotka-Wasylka, J. A new tool for the evaluation of the analytical procedure: Green Analytical Procedure Index. Talanta 2018, 181, 204–209. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Compound | Absolute Recovery | Relative Recovery | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A (%) | RSD (%) | B (%) | RSD (%) | C (%) | RSD (%) | ANOVA, F | A (%) | RSD (%) | B (%) | RSD (%) | C (%) | RSD (%) | IS standard | Method | |
| Acetamiprid | 97.4 | 2.4 | 100.1 | 1.3 | 98.4 | 0.8 | 1.9 | 107.9 | 9.4 | 101.2 | 2.5 | 94.6 | 3.3 | Carbamazepine_D10 | Basic |
| Amoxicillin | 3.3 | 0.3 | 3.2 | 0.1 | 3.1 | 0.4 | 0.4 | 15.7 | 2.7 | 10.7 | 0.7 | 8.3 | 1.8 | Ciprofloxacin_D8 | Acid |
| Azithromycin | 3.5 | 1.7 | 4.0 | 0.4 | 9.6 | 0.8 | 28.1 | 19.9 | 11.8 | 15.0 | 2.4 | 27.8 | 3.5 | Ciprofloxacin_D8 | Acid |
| Carbamazepine | 93.2 | 3.6 | 93.4 | 2.5 | 92.0 | 0.7 | 0.3 | 103.0 | 5.5 | 94.5 | 4.7 | 88.5 | 1.8 | Carbamazepine_D10 | Basic |
| Carbamazepine_D10 | 90.7 | 8.1 | 98.9 | 3.5 | 104.0 | 2.8 | 4.7 | Basic/Acid* | |||||||
| Ciprofloxacin | 22.7 | 4.1 | 26.9 | 3.3 | 32.2 | 6.8 | 2.7 | 105.2 | 8.0 | 89.9 | 2.7 | 84.1 | 11.6 | Ciprofloxacin_D8 | Acid |
| Ciprofloxacin_D8 | 21.4 | 2.2 | 29.8 | 3.2 | 37.9 | 3.1 | 24.6 | Acid | |||||||
| Citalopram | 53.5 | 3.8 | 57.2 | 6.6 | 55.4 | 5.4 | 0.3 | 51.9 | 1.4 | 51.5 | 6.1 | 48.2 | 5.9 | Carbamazepine_D10 | Acid |
| Clarithromycin | 55.5 | 4.3 | 61.1 | 1.7 | 63.2 | 5.2 | 3.0 | 101.4 | 8.0 | 94.0 | 3.3 | 89.1 | 2.6 | Clarithromycin_D3 | Basic |
| Clarithromycin_D3 | 54.7 | 0.7 | 65.0 | 2.9 | 70.9 | 4.8 | 19.2 | Basic | |||||||
| Clothianidin | 101.9 | 1.6 | 101.5 | 4.1 | 102.3 | 1.8 | 0.1 | 99.1 | 6.9 | 91.4 | 5.5 | 89.0 | 2.3 | Carbamazepine_D10 | Acid |
| Diclofenac | 80.8 | 2.9 | 84.7 | 7.1 | 85.9 | 5.9 | 0.7 | 104.0 | 6.1 | 97.2 | 2.4 | 91.1 | 2.5 | Diclofenac_C6 | Basic/Acid* |
| Diclofenac_C6 | 77.9 | 6.9 | 87.1 | 5.3 | 94.4 | 8.3 | 4.3 | Basic | |||||||
| Doxycycline | 77.4 | 1.8 | 83.6 | 3.6 | 76.6 | 9.2 | 1.3 | 75.3 | 4.2 | 75.3 | 2.0 | 66.8 | 9.7 | Ciprofloxacin_D8 | Acid |
| Erythromycin | 21.2 | 0.1 | 27.8 | 1.4 | 37.7 | 3.4 | 45.5 | 38.7 | 0.5 | 42.7 | 0.7 | 53.2 | 4.0 | Clarithromycin_D10 | Basic |
| 17-Beta-estradiol (E2) | 57.6 | 2.5 | 62.6 | 7.2 | 63.1 | 3.4 | 1.2 | 101.5 | 6.7 | 95.7 | 4.9 | 90.1 | 1.7 | 17-Beta-estradiol (E2)_D5 | Neutral |
| 17-Beta-estradiol (E2)_D5 | 56.9 | 6.0 | 65.3 | 5.2 | 70.0 | 5.0 | 4.5 | Neutral | |||||||
| Estrone (E1) | 57.0 | 2.9 | 62.3 | 8.0 | 61.6 | 4.3 | 0.8 | 102.3 | 7.1 | 96.1 | 3.6 | 89.3 | 1.7 | Estrone (E1)_D4 | Neutral |
| Estrone (E1)_D4 | 56.0 | 6.4 | 64.8 | 6.8 | 69.0 | 6.0 | 3.2 | Neutral | |||||||
| 17-Alpha-ethinylestradiol (EE2) | 52.2 | 2.9 | 59.1 | 8.7 | 57.5 | 5.4 | 1.1 | 93.6 | 6.0 | 91.1 | 5.2 | 83.5 | 3.1 | Estrone (E1)_D4 | Neutral |
| Fluconazole | 96.4 | 0.7 | 96.3 | 1.1 | 97.7 | 0.8 | 2.2 | 106.9 | 9.3 | 97.4 | 3.4 | 93.9 | 2.6 | Carbamazepine_D10 | Basic |
| Imidacloprid | 97.3 | 0.2 | 98.0 | 1.2 | 97.9 | 1.7 | 0.3 | 107.8 | 10.4 | 99.2 | 2.8 | 94.2 | 2.0 | Carbamazepine_D10 | Basic |
| Metaflumizone | 64.1 | 12.3 | 69.9 | 5.2 | 89.2 | 16.5 | 3.5 | 84.1 | 9.3 | 80.0 | 2.1 | 98.0 | 13.8 | Diclofenac_C6 | Acid |
| Methiocarb | 68.2 | 4.0 | 69.8 | 5.6 | 69.4 | 3.1 | 0.1 | 102.0 | 6.0 | 94.0 | 5.5 | 87.4 | 3.6 | Methiocarb_D3 | Basic |
| Methiocarb_D3 | 67.1 | 6.5 | 74.3 | 4.2 | 79.5 | 5.3 | 3.9 | Basic | |||||||
| Metoprolol | 88.8 | 2.3 | 90.9 | 1.5 | 89.6 | 3.4 | 0.5 | 98.8 | 4.0 | 93.4 | 4.2 | 89.8 | 6.0 | Metoprolol_D7 | Basic |
| Metoprolol_D7 | 90.0 | 3.5 | 97.5 | 4.8 | 99.9 | 6.1 | 3.3 | Basic | |||||||
| 2-Ethylhexyl 4-methoxycinnamate | 16.5 | 4.9 | 16.2 | 0.6 | 29.0 | 7.5 | 6.0 | 15.9 | 4.1 | 14.6 | 0.4 | 25.3 | 7.1 | Carbamazepine_D10 | Acid |
| Oxadiazone | 48.8 | 3.9 | 53.3 | 4.1 | 52.9 | 8.8 | 0.5 | 47.3 | 1.3 | 48.0 | 3.9 | 46.1 | 8.7 | Carbamazepine_D10 | Acid |
| Oxazepame | 85.7 | 2.5 | 87.0 | 4.1 | 86.7 | 3.4 | 0.1 | 110.4 | 7.0 | 100.0 | 3.1 | 92.1 | 4.3 | Carbamazepine_D10 | Basic |
| Sulfamethoxazole | 96.2 | 1.8 | 95.5 | 0.4 | 93.5 | 3.2 | 1.2 | 102.1 | 6.8 | 94.0 | 3.4 | 88.0 | 3.3 | Sulfamethoxazole_C6 | Acid |
| Sulfamethoxazole_C6 | 94.4 | 4.8 | 101.6 | 3.8 | 106.3 | 0.8 | 8.5 | Acid | |||||||
| Thiamethoxam | 100.2 | 1.3 | 101.4 | 0.7 | 100.8 | 1.1 | 1.0 | 111.0 | 9.2 | 102.6 | 3.1 | 97.0 | 3.6 | Carbamazepine_D10 | Basic |
| Triallate | 9.9 | 0.7 | 9.8 | 0.9 | 10.4 | 2.4 | 0.1 | 55.7 | 11.4 | 36.8 | 4.9 | 29.5 | 2.4 | Carbamazepine_D10 | Acid |
| Trimethoprim | 88.3 | 0.9 | 87.2 | 0.2 | 86.1 | 1.4 | 3.9 | 97.9 | 9.6 | 88.2 | 3.0 | 82.9 | 3.5 | Carbamazepine_D10 | Basic |
| Mean: | 64.2 | 3.3 | 68.2 | 3.5 | 70.7 | 4.3 | F-crit 5.14 | 83.1 | 6.6 | 76.7 | 3.3 | 74.1 | 4.5 | ||
| Absolute Recovery | Aq-Matrix | Relative Recovery | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | High (%) | RSD (%) | Difference | Low (%) | RSD (%) | High (%) | RSD (%) | Low (ng) | High (ng) |
| Doxycycline | 198.3 | 36.7 | −121.7 | 30.4 | 2.3 | 436.7 | 97.8 | 5.0 | 50.0 |
| Azithromycin | 107.0 | 2.7 | −97.4 | 87.1 | 0.7 | 382.4 | 16.6 | 5.0 | 50.0 |
| Clothianidin | 86.7 | 2.8 | 2.3 | 98.9 | 2.2 | 125.8 | 3.7 | 1.0 | 10.0 |
| Sulfamethoxazole | 82.3 | 2.4 | 11.2 | 116.7 | 0.4 | 119.3 | 4.0 | 1.0 | 10.0 |
| Clarithromycin | 65.9 | 3.6 | −2.7 | 78.3 | 0.5 | 105.8 | 3.8 | 1.0 | 10.0 |
| Carbamazepine | 64.5 | 0.7 | 27.5 | 122.4 | 0.4 | 127.9 | 6.2 | 1.0 | 10.0 |
| Metoprolol | 63.4 | 1.6 | 26.2 | 94.4 | 0.5 | 100.7 | 2.9 | 1.0 | 10.0 |
| Erythromycin | 62.6 | 3.6 | −24.9 | 112.7 | 0.2 | 100.6 | 4.9 | 1.0 | 10.0 |
| Trimethoprim | 60.8 | 1.4 | 25.4 | 125.9 | 0.0 | 120.4 | 5.2 | 1.0 | 10.0 |
| Acetamiprid | 60.7 | 1.0 | 37.6 | 119.8 | 0.3 | 120.4 | 4.9 | 1.0 | 10.0 |
| Imidacloprid | 60.0 | 2.5 | 38.0 | 116.1 | 0.7 | 118.9 | 1.8 | 1.0 | 10.0 |
| Thiamethoxam | 58.0 | 1.3 | 42.8 | 105.4 | 0.2 | 115.0 | 3.9 | 1.0 | 10.0 |
| Oxazepame | 56.9 | 1.6 | 29.8 | 132.8 | 0.2 | 112.8 | 3.8 | 1.0 | 10.0 |
| Triallate | 50.9 | 3.9 | −40.5 | 92.5 | 0.6 | 182.1 | 17.2 | 1.0 | 10.0 |
| Methiocarb | 50.5 | 2.5 | 18.9 | 91.0 | 0.2 | 102.0 | 6.6 | 1.0 | 10.0 |
| Oxadiazone | 50.3 | 2.4 | 2.6 | 91.9 | 0.6 | 110.3 | 10.5 | 2.0 | 20.0 |
| Diclofenac | 42.3 | 3.0 | 43.6 | 115.4 | 1.3 | 105.1 | 2.3 | 1.0 | 10.0 |
| 2-Ethylhexyl 4-methoxycinnamate | 38.9 | 9.5 | −9.9 | 119.3 | 1.7 | 224.5 | 59.3 | 5.0 | 50.0 |
| Metaflumizone | 37.8 | 4.4 | 51.4 | 66.2 | 0.2 | 61.9 | 8.2 | 5.0 | 50.0 |
| Citalopram | 36.7 | 1.8 | 18.6 | 68.3 | 0.1 | 80.7 | 8.0 | 1.0 | 10.0 |
| 17-Beta-estradiol (E2) | 34.8 | 1.1 | 28.3 | 102.3 | 0.8 | 105.6 | 7.1 | 50.0 | 500.0 |
| Fluconazole | 34.3 | 1.2 | 63.3 | 65.0 | 0.1 | 68.0 | 1.6 | 1.0 | 10.0 |
| 17-Alpha-ethinylestradiol (EE2) | 31.6 | 1.0 | 25.9 | 91.9 | 0.9 | 100.4 | 5.5 | 100.0 | 1000.0 |
| Ciprofloxacin | 30.1 | 3.3 | 0.12 | 26.6 | 0.6 | 108.4 | 13.5 | 5.0 | 50.0 |
| Estrone (E1) | 28.8 | 1.2 | 32.8 | 88.8 | 0.3 | 91.7 | 2.7 | 10.0 | 100.0 |
| Amoxicillin | 1.3 | 0.3 | 1.9 | 137.6 | 3.1 | 44.6 | 9.8 | 10.0 | 100.0 |
| Sulfamethoxazole_C6 | 68.5 | 4.0 | 37.8 | ||||||
| Metoprolol_D7 | 62.8 | 2.9 | 37.1 | ||||||
| Clarithromycin_D3 | 62.1 | 4.7 | 8.8 | ||||||
| Carbamazepine_D10 | 50.4 | 2.8 | 53.6 | ||||||
| Methiocarb_D3 | 49.5 | 3.2 | 29.9 | ||||||
| Diclofenac_C6 | 40.2 | 2.1 | 54.3 | ||||||
| 17-Beta-estradiol (E2)_D5 | 33.0 | 2.8 | 37.0 | ||||||
| Estrone (E1)_D4 | 31.4 | 2.2 | 37.6 | ||||||
| Ciprofloxacin_D8 | 27.4 | 0.5 | 0.01 | ||||||
| Mean: | 54.7 | 3.6 | |||||||
| Absolute Recovery | Relative Recovery | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Degeberga (%) | RSD (%) | St Olof (%) | RSD (%) | Pynten (%) | RSD (%) | Degeberga (%) | RSD (%) | St Olof (%) | RSD (%) | Pynten (%) | RSD (%) |
| Acetamiprid | nd | - | nd | - | 0.2 | 13.9 | ||||||
| Thiamethoxam | nd | - | nd | - | 0.9 | 23.9 | ||||||
| Estrone (E1) | 0.1 | 3.6 | 0.7 | 2.2 | 2.2 | 5.5 | ||||||
| Imidacloprid | 0.2 | 12.8 | nd | - | 3.0 | 2.3 | ||||||
| Clarithromycin | nd | - | 0.04 | 2.9 | 12.6 | 4.9 | ||||||
| Azithromycin | nd | - | nd | - | 15.8 | 1.4 | ||||||
| Fluconazole | 1.1 | 13.0 | 0.5 | 9.5 | 29.7 | 3.8 | ||||||
| Citalopram | 2.7 | 17.0 | 3.4 | 3.4 | 58.0 | 7.5 | ||||||
| Erythromycin | 1.2 | 11.9 | 0.7 | 9.7 | 62.7 | 2.5 | ||||||
| Trimethoprim | 0.2 | 3.6 | 3.6 | 1.7 | 75.2 | 4.9 | ||||||
| Sulfamethoxazole | 2.5 | 12.3 | 4.8 | 3.5 | 124.0 | 4.4 | ||||||
| Carbamazepine | 79.1 | 12.7 | 0.6 | 10.3 | 224.3 | 3.2 | ||||||
| Diclofenac | 30.3 | 13.1 | 20.7 | 1.8 | 374.5 | 3.5 | ||||||
| Metoprolol | 2.5 | 10.7 | 33.6 | 3.0 | 422.5 | 5.1 | ||||||
| Oxazepame | 20.8 | 14.8 | 14.1 | 6.8 | 473.0 | 4.1 | ||||||
| Amoxicillin | nd | - | nd | - | nd | - | ||||||
| Ciprofloxacin | nd | - | nd | - | nd | - | ||||||
| Clothianidin | nd | - | nd | - | nd | - | ||||||
| Doxycycline | nd | - | nd | - | nd | - | ||||||
| 17-Beta-estradiol (E2) | nd | - | nd | - | nd | - | ||||||
| 17-Alpha-ethinylestradiol (EE2) | nd | - | nd | - | nd | - | ||||||
| Metaflumizone | nd | - | nd | - | nd | - | ||||||
| Methiocarb | nd | - | nd | - | nd | - | ||||||
| 2-Ethylhexyl 4-methoxycinnamate | nd | - | nd | - | nd | - | ||||||
| Oxadiazone | nd | - | nd | - | nd | - | ||||||
| Triallate | nd | - | nd | - | nd | - | ||||||
| Carbamazepine-D10 | 63.6 | 8.1 | 51.9 | 0.8 | 33.8 | 1.1 | ||||||
| Ciprofloxacin_D8 | 7.8 | 0.6 | 4.3 | 2.1 | 3.1 | 1.1 | ||||||
| Clarithromycin_D3 | 51.5 | 5.6 | 47.8 | 1.8 | 60.7 | 2.3 | ||||||
| Diclofenac_C6 | 51.3 | 8.0 | 40.2 | 3.3 | 30.1 | 1.1 | ||||||
| 17-Beta-estradiol (E2)_D5 | 42.2 | 4.6 | 39.9 | 2.2 | 38.6 | 2.0 | ||||||
| Estrone (E1)_D4 | 40.5 | 4.7 | 39.3 | 3.1 | 32.1 | 1.1 | ||||||
| Methiocarb_D3 | 62.5 | 8.0 | 57.2 | 4.8 | 48.0 | 1.1 | ||||||
| Metoprolol_D7 | 64.9 | 7.2 | 55.7 | 0.8 | 41.5 | 1.8 | ||||||
| Sulfamethoxazole_C6 | 99.7 | 12.8 | 70.9 | 3.6 | 45.7 | 1.8 | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Svahn, O.; Björklund, E. High Flow-Rate Sample Loading in Large Volume Whole Water Organic Trace Analysis Using Positive Pressure and Finely Ground Sand as a SPE-Column In-Line Filter. Molecules 2019, 24, 1426. https://doi.org/10.3390/molecules24071426
Svahn O, Björklund E. High Flow-Rate Sample Loading in Large Volume Whole Water Organic Trace Analysis Using Positive Pressure and Finely Ground Sand as a SPE-Column In-Line Filter. Molecules. 2019; 24(7):1426. https://doi.org/10.3390/molecules24071426
Chicago/Turabian StyleSvahn, Ola, and Erland Björklund. 2019. "High Flow-Rate Sample Loading in Large Volume Whole Water Organic Trace Analysis Using Positive Pressure and Finely Ground Sand as a SPE-Column In-Line Filter" Molecules 24, no. 7: 1426. https://doi.org/10.3390/molecules24071426
APA StyleSvahn, O., & Björklund, E. (2019). High Flow-Rate Sample Loading in Large Volume Whole Water Organic Trace Analysis Using Positive Pressure and Finely Ground Sand as a SPE-Column In-Line Filter. Molecules, 24(7), 1426. https://doi.org/10.3390/molecules24071426