
Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases


Abstract
:1. Introduction
1.1. Role of FUS in RNA Metabolism
1.2. Role of FUS in DNA Damage Repair
1.3. The Link between FUS and Neurodegenerative Disease
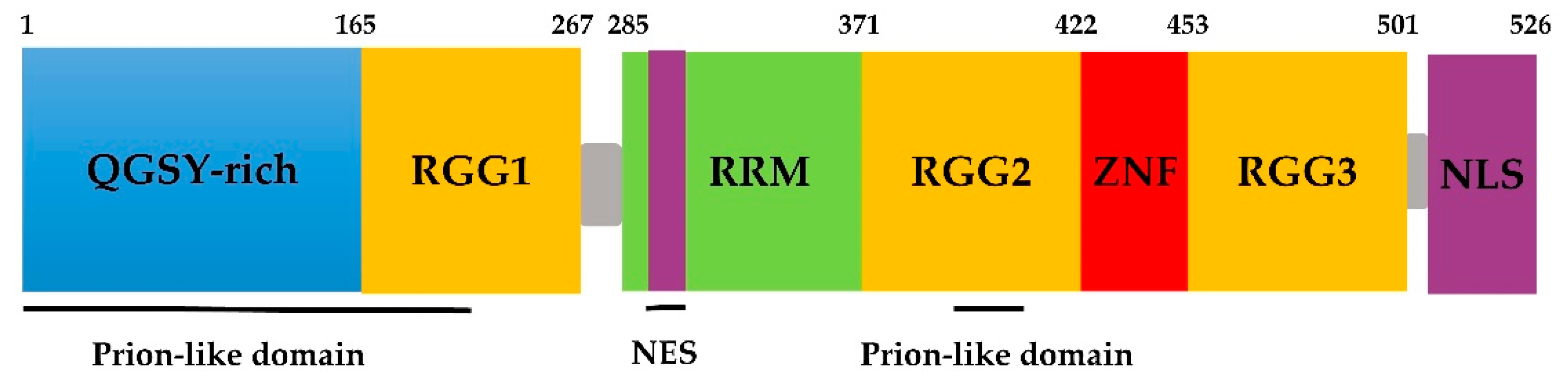
2. Domains of the FUS Protein
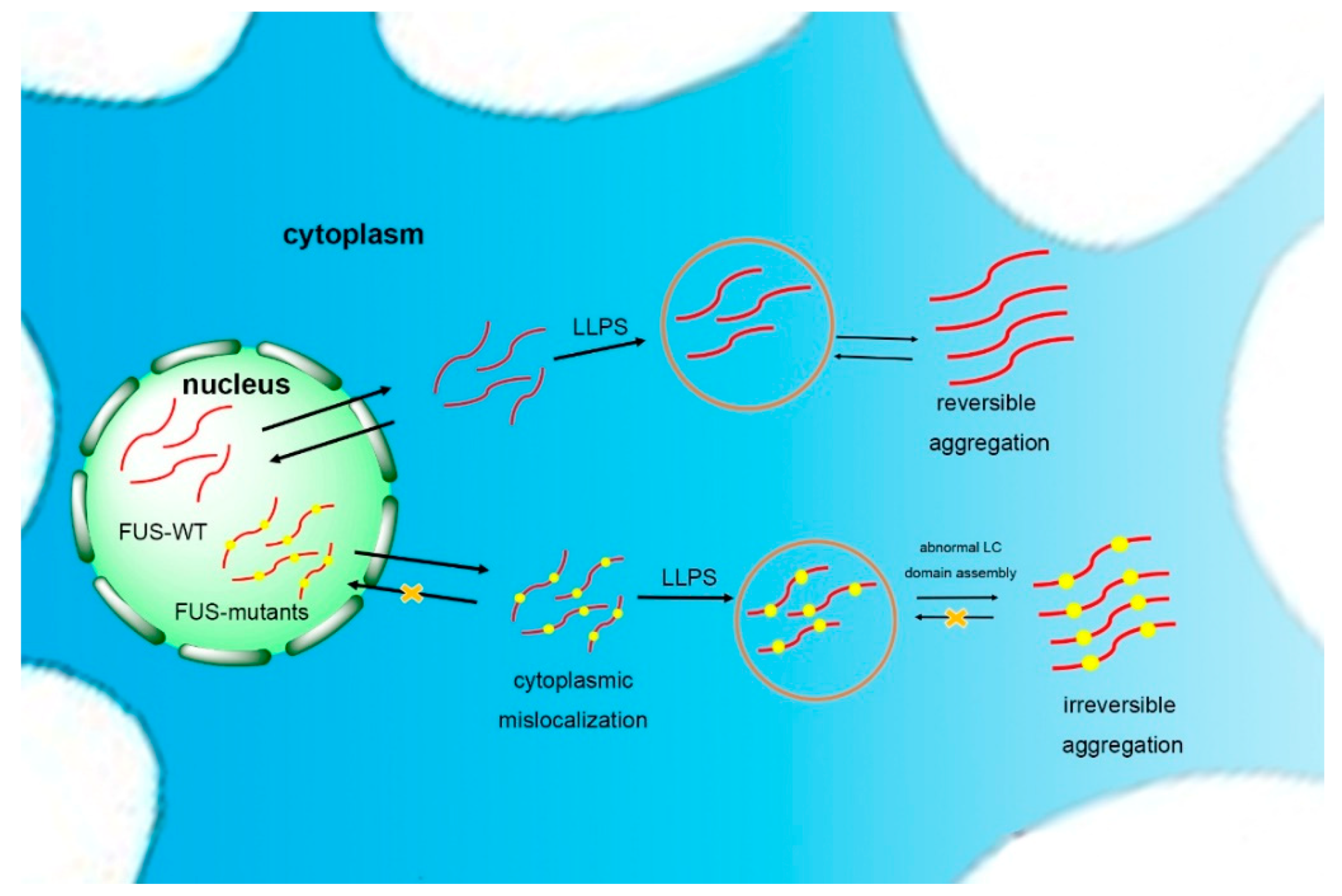
3. FUS Self-Assembly
3.1. Prion-Like Domains and Self-Assembly of FUS
3.2. Amyloid Core in LC Domains of FUS Regulates Its Self-Assembly
4. Regulation Factors of FUS Self-Assembly
5. Expectations
Author Contributions
Funding
Conflicts of Interest
References
- Crozat, A.; Aman, P.; Mandahl, N.; Ron, D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993, 363, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Lagiertourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416. [Google Scholar] [CrossRef]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Invest. 2014, 124, 981–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, J.C.; Cech, T.R.; Parker, R.R. Biochemical Properties and Biological Functions of FET Proteins. Annu. Rev. Biochem. 2015, 84, 355. [Google Scholar] [CrossRef]
- Tan, A.Y.; Manley, J.L. The TET Family of Proteins: Functions and Roles in Disease. J. Mol. Cell Biol. 2009, 1, 82. [Google Scholar] [CrossRef]
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 1997, 110, 1741–1750. [Google Scholar] [PubMed]
- Calvio, C.; Neubauer, G.; Mann, M.; Lamond, A.I. Identification of hnRNP P2 as TLS/FUS using electrospray mass spectrometry. Rna—Publ. Rna Soc. 1995, 1, 724–733. [Google Scholar]
- Tan, A.Y.; Manley, J.L. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc. Natl Acad. Sci. USA 2012, 109, 6030–6035. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.Y.; Manley, J.L. TLS inhibits RNA polymerase III transcription. Mol. Cell. Biol. 2010, 30, 186–196. [Google Scholar] [CrossRef]
- Hallier, M.; Lerga, A.; Barnache, S.; Tavitian, A.; Moreau-Gachelin, F. The transcription factor Spi-1/PU.1 interacts with the potential splicing factor TLS. J. Biol. Chem. 1998, 273, 4838–4842. [Google Scholar] [CrossRef]
- Uranishi, H.; Tetsuka, T.; Yamashita, M.; Asamitsu, K.; Shimizu, M.; Itoh, M.; Okamoto, T. Involvement of the pro-oncoprotein TLS (translocated in liposarcoma) in nuclear factor-kappa B p65-mediated transcription as a coactivator. J. Biol. Chem. 2001, 276, 13395–13401. [Google Scholar] [CrossRef]
- Li, X.; Decker, M.; Westendorf, J.J. TEThered to Runx: Novel binding partners for runx factors. Blood Cells Mol. Dis. 2010, 45, 82–85. [Google Scholar] [CrossRef]
- Bertolotti, A.; Lutz, Y.; Heard, D.J.; Chambon, P.; Tora, L. hTAF(II)68, a novel RNA/ssDNA-binding protein with homology to the pro-oncoproteins TLS/FUS and EWS is associated with both TFIID and RNA polymerase II. Embo J. 1996, 15, 5022–5031. [Google Scholar] [CrossRef]
- Schwartz, J.; R Cech, T.; R Parker, R. Biochemical Properties and Biological Functions of FET Proteins. Ann. Rev. Biochem. 2015, 84, 355–379. [Google Scholar] [CrossRef]
- Fay, R.M.; Walker, C.S.; Powers, J.M. Discoloration of a compomer by stains. J. Gt Houst Dent. Soc. 1998, 69, 12–13. [Google Scholar]
- Immanuel, D.; Zinszner, H.; Ron, D. Association of SARFH (sarcoma-associated RNA-binding fly homolog) with regions of chromatin transcribed by RNA polymerase II. Mol. Cell. Biol. 1995, 15, 4562–4571. [Google Scholar] [CrossRef]
- Kuroda, M.; Sok, J.; Webb, L.; Baechtold, H.; Urano, F.; Yin, Y.; Chung, P.; Rooij, D.G.D.; Akhmedov, A.; Ashley, T. Male sterility and enhanced radiation sensitivity in TLS−/−mice. Embo J. 2014, 19, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Alliegro, M.C.; Alliegro, M.A. A nuclear protein regulated during the transition from active to quiescent phenotype in cultured endothelial cells. Dev. Biol. 1996, 174, 288–297. [Google Scholar] [CrossRef]
- Fujii, R.; Okabe, S.; Urushido, T.; Inoue, K.; Yoshimura, A.; Tachibana, T.; Nishikawa, T.; Hicks, G.G.; Takumi, T. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr. Biol. 2005, 15, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Yan, K.P. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef]
- Wen-Yuan, W.; Ling, P.; Su, S.C.; Quinn, E.J.; Megumi, S.; Jimenez, J.C.; Mackenzie, I.R.A.; Huang, E.J.; Li-Huei, T. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurol. 2013, 16, 1383. [Google Scholar]
- Mastrocola, A.S.; Sang, H.K.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding Protein Fused in Sarcoma (FUS) Functions Downstream of Poly(ADP-ribose) Polymerase (PARP) in Response to DNA Damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [PubMed]
- Qiudong, D.; Holler, C.J.; Georgia, T.; Hudson, K.F.; William, W.; Marla, G.; Daisuke, I.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gómez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef]
- Baechtold, H.; Kuroda, M.; Sok, J.; Ron, D.; Lopez, B.S.; Akhmedov, A.T. Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J. Biol. Chem. 1999, 274, 34337–34342. [Google Scholar] [CrossRef] [PubMed]
- Mary, G.; Rachel, T.; Franck, V.; Nicholas, A.M.; John, R. Identification and characterization of FUS/TLS as a new target of ATM. Biochem. J. 2008, 415, 297–307. [Google Scholar] [Green Version]
- Xiangting, W.; Shigeki, A.; Xiaoyuan, S.; Donna, R.; Kun, D.; Gabriel, P.; Paul, T.; Rosenfeld, M.G.; Glass, C.K.; Riki, K. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126. [Google Scholar]
- Charcot, J.M. Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux anterolateraux de la moelle epiniere. Arch. Pathol. Norm. Pathol. 1869, 2, 744–760. [Google Scholar]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef]
- Neumann, M.; Roeber, S.; Kretzschmar, H.A.; Rademakers, R.; Baker, M.; Mackenzie, I.R. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009, 118, 605–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, H.; Koyano, S.; Suzuki, Y.; Nukina, N.; Kuroiwa, Y. The RNA-binding protein FUS/TLS is a common aggregate-interacting protein in polyglutamine diseases. Neurosci. Res. 2010, 66, 131–133. [Google Scholar] [CrossRef]
- Woulfe, J.; Gray, D.A.; Mackenzie, I.R. FUS-immunoreactive intranuclear inclusions in neurodegenerative disease. Brain Pathol. 2010, 20, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Girard, S.L.; Catoire, H.; Bourassa, C.V.; Belzil, V.V.; Rivière, J.B.; Hince, P.; Levert, A.; Dionnelaporte, A.; Spiegelman, D. Exome sequencing identifies FUS mutations as a cause of essential tremor. Am. J. Hum. Genet. 2012, 91, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.; Rayaprolu, S.; Soto-Ortolaza, A.; Ogaki, K.; Uitti, R.J.; Wszolek, Z.K.; Ross, O.A. Investigating FUS variation in Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20, S147. [Google Scholar] [CrossRef]
- Svetoni, F.; Frisone, P.; Paronetto, M.P. Role of FET proteins in neurodegenerative disorders. Rna Biol. 2016, 13, 1089–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.; Bentmann, E.; Dormann, D.; Jawaid, A.; DeJesus-Hernandez, M.; Ansorge, O.; Roeber, S.; Kretzschmar, H.A.; Munoz, D.G.; Kusaka, H.; et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain J. Neurol. 2011, 134, 2595–2609. [Google Scholar] [CrossRef] [Green Version]
- Caroline, V.; Emma, L.S.; Agnes, L.N.; Claire, T.; Jacqueline, C.M.; Claudia, K.; Hazel, U.; Catherine, M.; Christopher, C.M.; Tibor, H.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [Green Version]
- Bosco, D.A.; Nathan, L.; Hae Kyung, K.; Hongru, Z.; Chris, B.; Kwiatkowski, T.J.; Peter, S.; Diane, M.K.Y.; Brown, R.H.; Hayward, L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef]
- Sama, R.R.; Ward, C.L.; Kaushansky, L.J.; Lemay, N.; Ishigaki, S.; Urano, F.; Bosco, D.A. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J. Cell. Phys. 2013, 228, 2222–2231. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell. Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.B.; Almeida, S.; Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J. 2017, 36, e201797342. [Google Scholar] [CrossRef]
- Ramaswami, M.; Taylor, J.P.; Parker, R. Altered Ribostasis: RNA-Protein Granules in Degenerative Disorders. Cell 2013, 154, 727–736. [Google Scholar] [CrossRef]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; Mackenzie, I.R.A. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 2009, 132, 2922–2931. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; Huang, E.J. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 2016, 1647, 65–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, A.A. Neuronal cytoplasmic inclusions in tau, TDP-43, and FUS molecular subtypes of frontotemporal lobar degeneration share similar spatial patterns. Folia Neuropathol. 2017, 55, 185–192. [Google Scholar]
- Morohoshi, F.; Ootsuka, Y.; Arai, K.; Ichikawa, H.; Mitani, S.; Munakata, N.; Ohki, M. Genomic structure of the human RBP56/hTAFII68 and FUS/TLS genes. Gene 1998, 221, 191–198. [Google Scholar] [CrossRef]
- Lanson, N.A., Jr.; Pandey, U.B. FUS-related proteinopathies: lessons from animal models. Brain Res. 2012, 1462, 44–60. [Google Scholar]
- Guerrero, E.N.; Wang, H.; Mitra, J.; Hegde, P.M.; Stowell, S.E.; Liachko, N.F.; Kraemer, B.C.; Garruto, R.M.; Rao, K.S.; Hegde, M.L. TDP-43/FUS in motor neuron disease: Complexity and challenges. Prog. Neurobiol. 2016, 145, 78–97. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular Determinants and Genetic Modifiers of Aggregation and Toxicity for the ALS Disease Protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef]
- Iko, Y.; Kodama, T.S.; Kasai, N.; Oyama, T.; Morita, E.H.; Muto, T.; Okumura, M.; Fujii, R.; Takumi, T.; Tate, S. Domain Architectures and Characterization of an RNA-binding Protein, TLS. J. Biol. Chem. 2004, 279, 44834. [Google Scholar] [CrossRef] [PubMed]
- Lerga, A.; Hallier, M.; Delva, L.; Orvain, C.; Gallais, I.; Marie, J.; Moreaugachelin, F. Identification of an RNA binding specificity for the potential splicing factor TLS. J. Biol. Chem. 2001, 276, 6807–6816. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y.; Ling, S.C.; Qiu, J.S.; Albuquerque, C.P.; Zhou, Y.; Tokunaga, S.; Li, H.R.; Qiu, H.Y.; Bui, A.; Yeo, G.W.; et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat. Commun. 2015, 6, 6171. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.J.; Cansizoglu, A.E.; Süel, K.E.; Louis, T.H.; Zhang, Z.; Chook, Y.M. Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell 2006, 126, 543–558. [Google Scholar] [CrossRef]
- Zhang, Z.C.; Chook, Y.M. Structural and energetic basis of ALS-causing mutations in the atypical proline-tyrosine nuclear localization signal of the Fused in Sarcoma protein (FUS). Proc. Natl. Acad. Sci. USA 2012, 109, 12017–12021. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J.; Lindquist, S. Prions as adaptive conduits of memory and inheritance. Nat. Rev. Genet. 2005, 6, 435–450. [Google Scholar] [CrossRef]
- Shorter, J. Emergence and natural selection of drug-resistant prions. Mol. Biosyst. 2010, 6, 1115–1130. [Google Scholar] [CrossRef] [Green Version]
- Wiltzius, J.J.; Landau, M.; Nelson, R.; Sawaya, M.R.; Apostol, M.I.; Goldschmidt, L.; Soriaga, A.B.; Cascio, D.; Rajashankar, K.; Eisenberg, D. Molecular mechanisms for protein-encoded inheritance. Nat. Struct. Mol. Biol. 2009, 16, 973–978. [Google Scholar] [CrossRef] [Green Version]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef] [Green Version]
- Udan, M.; Baloh, R.H. Implications of the prion-related Q/N domains in TDP-43 and FUS. Prion 2011, 5, 1–5. [Google Scholar] [CrossRef] [Green Version]
- March, Z.M.; King, O.D.; Shorter, J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016, 1647, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniecka, Z.; Polymenidou, M. From nucleation to widespread propagation: A prion-like concept for ALS. Virus Res. 2015, 207, 94–105. [Google Scholar] [CrossRef] [Green Version]
- Banfi, S.; Servadio, A.; Chung, M.Y.; Kwiatkowski, T.J., Jr.; Mccall, A.E.; Duvick, L.A.; Shen, Y.; Roth, E.J.; Orr, H.T.; Zoghbi, H.Y. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat. Genet. 1994, 7, 513–520. [Google Scholar] [CrossRef]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; Defranco, D.B.; Orr, H.T.; Zoghbi, H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Shorter, J. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion 2011, 5, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Cushman, M.; Johnson, B.S.; King, O.D.; Gitler, A.D.; Shorter, J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J. Cell. Sci. 2010, 123, 1191–1201. [Google Scholar] [CrossRef] [Green Version]
- King, O.D.; Gitler, A.D.; Shorter, J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012, 1462, 61–80. [Google Scholar] [CrossRef] [Green Version]
- Han, T.; Kato, M.; Xie, S.; Wu, L.; Mirzaei, H.; Pei, J.; Chen, M.; Xie, Y.; Allen, J.; Xiao, G. Cell-free Formation of RNA Granules: Bound RNAs Identify Features and Components of Cellular Assemblies. Cell 2012, 149, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryndushkin, D.; Wickner, R.B.; Shewmaker, F. FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast model of amyotrophic lateral sclerosis. Protein Cell 2011, 2, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, S.; Tardiff, D.F.; Han, H.; Divya, K.; Quan, Z.; Maquat, L.E.; Bosco, D.A.; Hayward, L.J.; Brown, R.H., Jr.; Lindquist, S. A Yeast Model of FUS/TLS-Dependent Cytotoxicity. PLoS Biol. 2011, 9, e1001052. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Wang, X.; Podell, E.R.; Cech, T.R. RNA seeds higher-order assembly of FUS protein. Cell Rep. 2013, 5, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Kato, M.; Xiang, S.; Wu, L.; Theodoropoulos, P.; Mirzaei, H.; Han, T.; Xie, S.; Corden, J.; Mcknight, S. Phosphorylation-Regulated Binding of RNA Polymerase II to Fibrous Polymers of Low-Complexity Domains. Cell 2013, 155, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Han, T.N.W.; Xie, S.H.; Shi, K.; Du, X.L.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.M.; et al. Cell-free Formation of RNA Granules: Low Complexity Sequence Domains Form Dynamic Fibers within Hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 2018, 173, 706–719. [Google Scholar] [CrossRef]
- Broide, M.L.; Berland, C.R.; Pande, J.; Ogun, O.O.; Benedek, G.B. Binary-liquid phase separation of lens protein solutions. Proc. Natl. Acad. Sci. USA 1991, 88, 5660–5664. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.A.; Janke, A.M.; Rhine, C.L.; Fawzi, N.L. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol. Cell. 2015, 60, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Kriwacki, R.W. Phase separation in biology; functional organization of a higher order. Cell Commun. Sign. 2016, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutertre, M.; Lambert, S.; Carreira, A.; Amor-Guéret, M.; Vagner, S. DNA damage: RNA-binding proteins protect from near and far. Trends Biochem. Sci. 2014, 39, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Hyman, A.A.; Brangwynne, C.P. Beyond Stereospecificity: Liquids and Mesoscale Organization of Cytoplasm. Dev. Cell 2011, 21, 14–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Banjade, S.; Cheng, H.C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F. Phase Transitions in the Assembly of Multi-Valent Signaling Proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dormann, D.; Haass, C. TDP-43 and FUS: a nuclear affair. Trends Neurosci. 2011, 34, 339–348. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: from genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.K.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.S.; Michel, C.H. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Hyman, A.A. Are aberrant phase transitions a driver of cellular aging? Bioessays 2016, 38, 959–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, P.R.; Milin, A.N.; Moosa, M.M.; Onuchic, P.L.; Deniz, A.A. Reentrant Phase Transition Drives Dynamic Substructure Formation in Ribonucleoprotein Droplets. Angew. Chem. Int. Ed. 2017, 56, 11354–11359. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Hu, K.N.; Qiang, W.; Tycko, R. A general Monte Carlo/simulated annealing algorithm for resonance assignment in NMR of uniformly labeled biopolymers. J. Biomol. NMR 2011, 50, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; Mcknight, S.L.; Tycko, R. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell 2017, 171, 615–627. [Google Scholar] [CrossRef]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ø.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The Amyloid State of Proteins in Human Diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef]
- Rambaran, R.N.; Serpell, L.C. Amyloid fibrils Abnormal protein assembly. Prion 2008, 2, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.W.; McFarlane, H.T. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef]
- Thirumalai, D.; Reddy, G.; Straub, J.E. Role of water in Protein Aggregation and Amyloid Polymorphism. Acc. Chem. Res. 2011, 45, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Gui, X.; Zhou, H.; Gu, J.; Li, Y.; Liu, X.; Zhao, M.; Li, D.; Li, X.; Liu, C. Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat. Struct. Mol. Biol. 2018, 25, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.; Eisenberg, D. Recent atomic models of amyloid fibril structure. Curr. Opin. Struct. Biol. 2006, 16, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227–235. [Google Scholar] [CrossRef]
- Amen, T.; Kaganovich, D. Dynamic droplets: the role of cytoplasmic inclusions in stress, function, and disease. Cell. Mol. Life Sci. 2015, 72, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, S.N.; Monahan, Z.T.; Yee, D.S.; Shewmaker, F.P. The Role of Post-Translational Modifications on Prion-Like Aggregation and Liquid-Phase Separation of FUS. Int. J. Mol. Sci. 2018, 19, 886–905. [Google Scholar] [CrossRef]
- Monahan, Z.; Ryan, V.H.; Janke, A.M.; Burke, K.A.; Rhoads, S.N.; Zerze, G.H.; O’Meally, R.; Dignon, G.L.; Conicella, A.E.; Zheng, W.; et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 2017, 36, 2951–2967. [Google Scholar] [CrossRef]
- Takanashi, K.; Yamaguchi, A. Aggregation of ALS-linked FUS mutant sequesters RNA binding proteins and impairs RNA granules formation. Biochem. Biophys. Res. Commun. 2014, 452, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Qamar, S.; Wang, G.; Randle, S.J.; Ruggeri, F.S.; Varela, J.A.; Lin, J.Q.; Phillips, E.C.; Miyashita, A.; Williams, D.; Ströhl, F. FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-π Interactions. Cell 2018, 173, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Chiara, S.; John, M.; Carmelo, M.; Lanson, N.A.; Astha, M.; Tanya, A.; Ian, C.; Fackelmayer, F.O.; Maria, P.; Udai Bhan, P. Protein arginine methyltransferase 1 and 8 interact with FUS to modify its sub-cellular distribution and toxicity in vitro and in vivo. PLoS ONE 2013, 8, e61576. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Clancy, K.W.; Thompson, P.R. Chemical biology of protein arginine modifications in epigenetic regulation. Chem. Rev. 2015, 115, 5413–5461. [Google Scholar] [CrossRef]
- Dormann, D.; Madl, T.; Valori, C.F.; Bentmann, E.; Tahirovic, S.; Abou-Ajram, C.; Kremmer, E.; Ansorge, O.; Mackenzie, I.R.; Neumann, M.; Haass, C. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012, 31, 4258–4275. [Google Scholar] [CrossRef] [Green Version]
- Yoshizawa, T.; Ali, R.; Jiou, J.; Fung, H.; Burke, K.A.; Kim, S.J.; Lin, Y.; Peeples, W.B.; Saltzberg, D.; Soniat, M. Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell 2018, 173, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Hong, J.K.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 2018, 173, 677–692. [Google Scholar] [CrossRef]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Watanabe, S.; Kaneko, K.; Yamanaka, K.; Nukina, N.; Furukawa, Y. Intranuclear Aggregation of Mutant FUS/TLS as a Molecular Pathomechanism of Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2014, 289, 1192–1202. [Google Scholar] [CrossRef]
- Belzil, V.V.; St-Onge, J.; Daoud, H.; Desjarlais, A.; Bouchard, J.P.; Dupré, N.; Camu, W.; Dion, P.A.; Rouleau, G.A. Identification of a FUS splicing mutation in a large family with amyotrophic lateral sclerosis. J. Hum. Genet. 2011, 56, 247. [Google Scholar] [CrossRef]
- Belzil, V.V.; Valdmanis, P.N.; Dion, P.A.; Daoud, H.; Kabashi, E.; Noreau, A.; Gauthier, J.; Hince, P.; Desjarlais, A.; Desjarlais, J.-P.; et al. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology 2009, 73, 1176–1179. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Deng, H.X.; Siddique, N.; Fecto, F.; Chen, W.; Yang, Y.; Liu, E.; Donkervoort, S.; Zheng, J.G.; Shi, Y. Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 2010, 75, 807–814. [Google Scholar] [CrossRef]
- Marka, V.B.; Van Es, M.A.; Hennekam, E.A.M.; Dennis, D.; Wouter, V.R.; Jelena, M.; Bourque, P.R.; Schelhaas, H.J.; Van Der Kooi, A.J.; Marianne, D.V. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 3776–3784. [Google Scholar]
- Brown, J.A.; Min, J.; Staropoli, J.F.; Collin, E.; Bi, S.; Feng, X.; Barone, R.; Cao, Y.; O’Malley, L.; Xin, W. SOD1, ANG, TARDBP and FUS mutations in amyotrophic lateral sclerosis: a United States clinical testing lab experience. Amyotroph. Lateral Scler. 2012, 13, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Ticozzi, N.; Silani, V.; Leclerc, A.L.; Keagle, P.; Gellera, C.; Ratti, A.; Taroni, F.; Kwiatkowski, T.J.; Mckenna-Yasek, D.M.; Sapp, P.C. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 2009, 73, 1180–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucia, C.; Roberto, D.B.; Barbara, C.; Antonia, R.; Cristina, C.; Silvana, P.; Gianni, S.; Yari, C.; Serena, G.; Viviana, P. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J. Med. Genetics 2010, 47, 190–194. [Google Scholar]
- Kwon, M.J.; Baek, W.; Ki, C.S.; Kim, H.Y.; Koh, S.H.; Kim, J.W.; Kim, S.H. Screening of the SOD1, FUS, TARDBP, ANG, and OPTN mutations in Korean patients with familial and sporadic ALS. Neurobio. Aging 2012, 33, 1017.e17–1017.e23. [Google Scholar] [CrossRef] [PubMed]
- Tarlarini, C.; Lunetta, C.; Mosca, L.; Avemaria, F.; Riva, N.; Mantero, V.; Maestri, E.; Quattrini, A.; Corbo, M.; Melazzini, M.G. Novel FUS mutations identified through molecular screening in a large cohort of familial and sporadic amyotrophic lateral sclerosis. Eur. J. Neurol. 2015, 22, 1474–1481. [Google Scholar] [CrossRef]
- Murni, T.; Wen, R.; Lin, L.Y.; Wang, H.; Ling, S.C.; Yi, Z.; Tan, E.K. FUS-linked essential tremor associated with motor dysfunction inDrosophila. Hum. Genet. 2016, 135, 1223–1232. [Google Scholar]
- Rajput, A.; Rajput, A.H.; Rajput, M.L.; Encarnacion, M.; Bernales, C.Q.; Ross, J.P.; Farrer, M.J.; Vilariñogüell, C. Identification of FUS p.R377W in essential tremor. Eur. J. Neurol. 2014, 21, 361–363. [Google Scholar] [CrossRef]
- Gao, K.; Zheng, W.; Deng, X.; Xiong, W.; Song, Z.; Yang, Y.; Deng, H. Genetic analysis of the fused in sarcoma gene in Chinese Han patients with Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20, 119–121. [Google Scholar] [CrossRef]
- Mariely, D.J.H.; Jannet, K.; Nicole, F.; Richard, C.; Matt, B.; Pamela, D.; Amelia, J.; Nicola, R.; Aleksandra, W.; Kathleen, K. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum. Mutat. 2010, 31, E1377–E1389. [Google Scholar]
- Labbé, C.; Sotoortolaza, A.I.; Rayaprolu, S.; Harriott, A.M.; Strongosky, A.J.; Uitti, R.J.; Gerpen, J.A.V.; Wszolek, Z.K.; Ross, O.A. Investigating the role of FUS exonic variants in Essential Tremor. Parkinsonism Relat. Disord. 2013, 19, 755–757. [Google Scholar] [CrossRef] [Green Version]
- Groen, E.J.; Van, E.M.V.P. FUS mutations in familial amyotrophic lateral sclerosis in the Netherlands. Arch. Neurol. 2010, 67, 224–230. [Google Scholar] [CrossRef]
- Nagayama, S.; Minato-Hashiba, N.; Nakata, M.; Kaito, M.; Nakanishi, M.; Tanaka, K.; Arai, M.; Akiyama, H.; Matsui, M. Novel FUS mutation in patients with sporadic amyotrophic lateral sclerosis and corticobasal degeneration. J. Clin. Neurol. 2012, 19, 1738–1739. [Google Scholar] [CrossRef]
- Hara, M.; Minami, M.; Suzuki, N.; Kato, M.; Aoki, M. Lower motor neuron disease caused by a novel FUS/TLS gene frameshift mutation. J. Neurol. 2012, 259, 2237–2239. [Google Scholar] [CrossRef]
- Liu, Z.J.; Lin, H.X.; Liu, G.L.; Tao, Q.Q.; Ni, W.; Xiao, B.G.; Wu, Z.Y. The investigation of genetic and clinical features in Chinese patients with juvenile amyotrophic lateral sclerosis. Clin. Genet. 2017, 92, 267–273. [Google Scholar] [CrossRef]
- Satoshi, Y.; Akira, M.; Hideya, S.; Tomohiro, S.; Daijirou, I.; Akihiko, U.; Taro, Y.; Yasushi, M.; Makoto, U.; Teruyuki, H. Sporadic juvenile amyotrophic lateral sclerosis caused by mutant FUS/TLS: possible association of mental retardation with this mutation. J. Neurol. 2012, 259, 1039–1044. [Google Scholar]
- Zou, Z.Y.; Cui, L.Y.; Sun, Q.; Li, X.G.; Liu, M.S.; Xu, Y.; Zhou, Y.; Yang, X.Z. De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurol. Aging 2013, 34, 1312.e1–1312.e8. [Google Scholar] [CrossRef]
- Waibel, S.; Neumann, M.; Rabe, M.; Meyer, T.; Ludolph, A.C. Novel missense and truncating mutations in FUS/TLS in familial ALS. Neurology 2010, 75, 815–817. [Google Scholar] [CrossRef]
- Belzil, V.V.; Jean-Sébastien, L.; Hussein, D.; Dion, P.A.; Bernard, B.; Rouleau, G.A. Novel FUS deletion in a patient with juvenile amyotrophic lateral sclerosis. Arch. Neurol. 2012, 69, 653–656. [Google Scholar]
- Sproviero, W.; Bella, V.L.; Mazzei, R.; Valentino, P.; Rodolico, C.; Simone, I.L.; Logroscino, G.; Ungaro, C.; Magariello, A.; Patitucci, A. FUS mutations in sporadic amyotrophic lateral sclerosis: Clinical and genetic analysis. Neurobiol. Aging 2012, 33, 837.e1–837.e5. [Google Scholar] [CrossRef] [Green Version]
- Christopher, H.; Janine, K.; Highley, J.R.; Hartley, J.A.; Rachael, H.; Hollinger, H.C.; Williams, T.L.; Ince, P.G.; Mcdermott, C.J.; Shaw, P.J. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 455–461. [Google Scholar]
- Suzuki, M.; Mikami, H.; Watanabe, T.; Yamano, T.; Yamazaki, T.; Nomura, M.; Yasui, K.; Ishikawa, H.; Ono, S. Increased expression of TDP-43 in the skin of amyotrophic lateral sclerosis. Acta Neurol. Scand. 2010, 122, 367–372. [Google Scholar] [CrossRef]
- Syriani, E.; Morales, M.; Gamez, J. FUS/TLS gene mutations are the second most frequent cause of familial ALS in the Spanish population. Amyotroph. Lateral Scler. 2011, 12, 118–123. [Google Scholar] [CrossRef]
- Chiò, A.; Mora, G.; Calvo, A.; Mazzini, L.; Bottacchi, E.; Mutani, R. Epidemiology of ALS in Italy: A 10-year prospective population-based study. Neurology 2009, 72, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chio, A.; Restagno, G.M. Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol. Aging 2009, 30, 1272–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stéphanie, M.; Francois, S.; Cécile, C.; Paul, G.; Bernard, B.; Agnès, C.; Léna, G.N.L.; Odile, R.; Gaelle, B.; Pierre-Francois, P.; et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: Genotype-phenotype correlations. J. Med. Genet. 2010, 47, 554–560. [Google Scholar]
- Robertson, J.; Bilbao, J.; Zinman, L.; Hazrati, L.-N.; Tokuhiro, S.; Sato, C.; Moreno, D.; Strome, R.; Mackenzie, I.R.; Rogaeva, E. A novel double mutation in FUS gene causing sporadic ALS. Neurobiol. Aging 2011, 32, 553.e27–553.e30. [Google Scholar] [CrossRef] [PubMed]
- Ching-Paio, T.; Bing-Wen, S.; Kon-Ping, L.; Pang-Hsien, T.; Jer-Li, L.; Yi-Chung, L. FUS, TARDBP, and SOD1 mutations in a Taiwanese cohort with familial ALS. Neurobiol. Aging 2011, 32, 553.e13–553.e21. [Google Scholar]
- BaUmer, D.; Hilton, D.; Paine, S.M.L.; Turner, M.R.; Lowe, J.; Talbot, K.; Ansorge, O. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology 2010, 75, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Blair, I.P.; Williams, K.L.; Warraich, S.T.; Durnall, J.C.; Thoeng, A.D.; Jim, M.; Blumbergs, P.C.; Steve, V.; Kiernan, M.C.; Nicholson, G.A. FUS mutations in amyotrophic lateral sclerosis: Clinical, pathological, neurophysiological and genetic analysis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 639–645. [Google Scholar] [CrossRef]
- Yamamoto-Watanabe, Y.; Watanabe, M.; Okamoto, K.; Fujita, Y.; Jackson, M.; Ikeda, M.; Nakazato, Y.; Ikeda, Y.; Matsubara, E.; Kawarabayashi, T. A Japanese ALS6 family with mutation R521C in the FUS/TLS gene: A clinical, pathological and genetic report. J. Neurol. Sci. 2010, 296, 59–63. [Google Scholar] [CrossRef]
- Broustal, O.; Camuzat, A.; Guillot-Noël, L.; Guy, N.; Millecamps, S.; Deffond, D.; Lacomblez, L.; Golfier, V.; Hannequin, D.; Salachas, F. FUS mutations in frontotemporal lobar degeneration with amyotrophic lateral sclerosis. J. Alzheimers Dis. 2010, 22, 765–769. [Google Scholar] [PubMed]
- Damme, P.V.; Goris, A.V.; Hersmus, N.; Dubois, B.; Bosch, L.V.; Matthijs, G.; Robberecht, W. The occurrence of mutations in FUS in a Belgian cohort of patients with familial ALS. Eur. J. Neurol. 2010, 17, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Eysel, K. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology 2012, 79, 66–72. [Google Scholar]
- Hou, L.; Jiao, B.; Xiao, T.; Zhou, L.; Zhou, Z.; Du, J.; Yan, X.; Wang, J.; Tang, B.; Shen, L. Screening of SOD1, FUS and TARDBP genes in patients with amyotrophic lateral sclerosis in central-southern China. Sci. Rep. 2016, 6, 32478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimoto, Y.; Nakagawa, S.; Hirose, T.; Okano, H.J.; Takao, M.; Shibata, S.; Suyama, S.; Kuwako, K.; Imai, T.; Murayama, S. The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 2013, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Shelkovnikova, T.A.; Peters, O.M.; Deykin, A.V.; Connorrobson, N.; Robinson, H.; Ustyugov, A.A.; Bachurin, S.O.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R. Fused in Sarcoma (FUS) Protein Lacking Nuclear Localization Signal (NLS) and Major RNA Binding Motifs Triggers Proteinopathy and Severe Motor Phenotype in Transgenic Mice. J. Biol. Chem. 2013, 288, 25266–25274. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| NO. | Exon | Domain/Region | Nucleotide Change | Amino Acid Mutation | ALS | References |
|---|---|---|---|---|---|---|
| 1 | 3 | PrLDs | c.52 C > T | p.P18S | ALS | [111] |
| 2 | 3 | PrLDs | c.170_172delCTT | p.S57del | ALS | [112] |
| 3 | 4 | PrLDs | c.287_291delCCTACinsAT | p.S96del | ALS | [113] |
| 4 | 4 | PrLDs | c.344 G > A | p.S115N | ALS | [114] |
| 5 | 5 | PrLDs | c.430_447delGG | p.G144Y149del | ALS | [111,115] |
| 6 | 5 | PrLDs | c.467 G > A | p.G156E | ALS | [116] |
| 7 | 6 | PrLDs | c.518_523del GAGGTG | p.G174_G175del | AlS/FTLD | [30,113] |
| 8 | 6 | RGG1 | c.616 G > A | p.G206S | AlS/FTLD | [113] |
| 9 | 6 | RGG1 | c.646 C > T | p.R216C | ALS | [117] |
| 10 | 6 | RGG1 | c.674 G > T | p.G225V | ALS | [117] |
| 11 | 6 | RGG1 | c.681_684delGGC | p.G230delG | ALS | [118] |
| 12 | 6 | RGG1 | c.688 G > T | p.G230C | ALS | [117] |
| 13 | 6 | RGG1 | c.700 C > T | p.R234C | ALS | [117] |
| 14 | 6 | RGG1 | c.701 G > T | p.R234L | ALS | [116] |
| 15 | 6 | RGG1 | c.730 C > T | p.R244C | ALS | [30] |
| 16 | 6 | RGG1 | c.734 G > T | p.G245V | ALS | [119] |
| 17 | 9 | RRM | c.868 C > T | p.Q290X | ET | [120] |
| 18 | 11 | RGG2 | c.1129 C > T | p.R377W | ET | [121] |
| 19 | 12 | PrLDs | c.1176 G > A | p.M392I | ET | [122] |
| 20 | 12 | PrLDs | c.1196 G > T | p.G399V | ALS | [118] |
| 21 | 12 | RGG2 | c.1204 1232 delinsGGAGGTGGAGG | p.S402P | ALS | [123] |
| 22 | 13 | ZNF | c.1292 C > T | p.P431L | ET | [124] |
| 23 | 13 | RGG3 | c.1385 C > T | p.S462F | ALS | [125] |
| 24 | 13 | RGG3 | c.1392 G > T | M464I | ALS | [126] |
| 25 | 14 | RGG3 | c.1394_1541del | p.G466VfsX14 | ALS | [123] |
| 26 | 14 | RGG3 | c.1420_1421insGT | p.G472VfsX57 | ALS | [127] |
| 27 | 14 | RGG3 | c.1449_1488del | p.Y485AfsX514 | ALS | [113] |
| 28 | 14 | RGG3 | c.1456_1457delGG | p.G486PfsX30 | ALS | [128] |
| 29 | 14 | RGG3 | c.1459 C > T | p.R487C | ALS | [114] |
| 30 | 14 | RGG3 | c.1464 C > T | p.G488G | ALS | [119] |
| 31 | 14 | RGG3 | c.1475delG | p.G492EfsX527 | ALS | [129] |
| 32 | 14 | RGG3 | c.1483delC | p.R495EfsX527 | ALS | [113] |
| 33 | 14 | RGG3 | c.1483 C > T | p.R495X | ALS | [39,130,131] |
| 34 | 14 | RGG3 | c.1484delG | p.R495QfsX527 | ALS | [132] |
| 35 | 14 | RGG3 | c.1485delA | p.G497AfsX527 | ALS | [113] |
| 36 | 14 | RGG3 | c.1489_1490dupGG | p.R498AfsX32 | ALS | [128] |
| 37 | 14 | - | c.1506dupA | p.R502fsX15 | ALS | [111] |
| 38 | 14 | - | c.1507_1508delAG | p.G503WfsX12 | ALS | [118] |
| 39 | 14 | - | c.1509_1510delAG | p.G504WfsX12 | ALS | [130] |
| 40 | 14 | - | c.1520 G > A | p.G507D | ALS | [117,133,134] |
| 41 | 14 | NLS | c.1526 G > A | p.G509D | ALS | [119] |
| 42 | 14 | NLS | c.1527_1528insTGCC | p.K510WfsX517 | ALS | [113] |
| 43 | 14 | NLS | c.1528 A > G | p.K510E | ALS | [135,136] |
| 44 | 14 | NLS | c.1529 A > G | p.K510R | ALS | [131] |
| 45 | 14 | NLS | c.1537 T > C | p.S513P | ALS | [135] |
| 46 | 14 | NLS | c.1540 A > G | p.R514G | ALS | [88] |
| 47 | 14 | NLS | c.1542 G > C | p.R514S | ALS | [30,137,138,139,140] |
| 48 | 15 | NLS | c.1543 G > T | p.G515C | ALS | [30] |
| 49 | 15 | NLS | c.1544delG | p.G515VfsX14 | ALS | [128] |
| 50 | 15 | NLS | c.1547 A > T | p.E516V | ALS | [140] |
| 51 | 15 | NLS | c.1549 C > G | p.H517D | ALS | [141] |
| 52 | 15 | NLS | c.1550 A > C | p.H517P | ALS | [135] |
| 53 | 15 | NLS | c.1551 C > G | p.H517Q | ALS | [30] |
| 54 | 15 | NLS | c.1552 A > G | p.R518G | ALS | [104] |
| 55 | 15 | NLS | c.1553 G > A | p.R518K | ALS | [30] |
| 56 | 15 | NLS | c.1554_1557delACAG | p.R518del | ALS | [142] |
| 57 | 15 | NLS | c.1555 C > T | p.Q519X | ALS | [112] |
| 58 | 15 | NLS | c.1561C > T | p.R521C | ALS | [112,143,144] |
| 59 | 15 | NLS | c.1561 C > G | p.R521G | ALS | [115,133] |
| 60 | 15 | NLS | c.1561 C > A | p.R521S | ALS | [139] |
| 61 | 15 | NLS | c.1562 G > A | p.R521H | ALS | [112,139,145,146] |
| 62 | 15 | NLS | c.1562 G > T | p.R521L | ALS | [113,139,147] |
| 63 | 15 | NLS | c.1564 C > T | p.R522G | ALS | [30] |
| 64 | 15 | NLS | c.1570 A > T | p.R524W | ALS | [134] |
| 65 | 15 | NLS | c.1571 G > C | p.R524T | ALS | [30] |
| 66 | 15 | NLS | c.1572 G > C | p.R524S | ALS | [30,113] |
| 67 | 15 | NLS | c.1574 C > T | p.525L | ALS | [142] |
| 68 | 15 | NLS | c.1574 C > G | p.525R | ALS | [142] |
| 69 | 15 | NLS | c.1575_1576insTAT | p.P525_Y526ins | ALS | [148] |
| 70 | 15 | NLS | c.1581delA | p.X527YextX | ALS | [118] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Ding, X.; Akram, N.; Xue, S.; Luo, S.-Z. Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases. Molecules 2019, 24, 1622. https://doi.org/10.3390/molecules24081622
Chen C, Ding X, Akram N, Xue S, Luo S-Z. Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases. Molecules. 2019; 24(8):1622. https://doi.org/10.3390/molecules24081622
Chicago/Turabian StyleChen, Chen, Xiufang Ding, Nimrah Akram, Song Xue, and Shi-Zhong Luo. 2019. "Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases" Molecules 24, no. 8: 1622. https://doi.org/10.3390/molecules24081622
APA StyleChen, C., Ding, X., Akram, N., Xue, S., & Luo, S. -Z. (2019). Fused in Sarcoma: Properties, Self-Assembly and Correlation with Neurodegenerative Diseases. Molecules, 24(8), 1622. https://doi.org/10.3390/molecules24081622