
Encoded Library Technologies as Integrated Lead Finding Platforms for Drug Discovery

Abstract
:1. Introduction
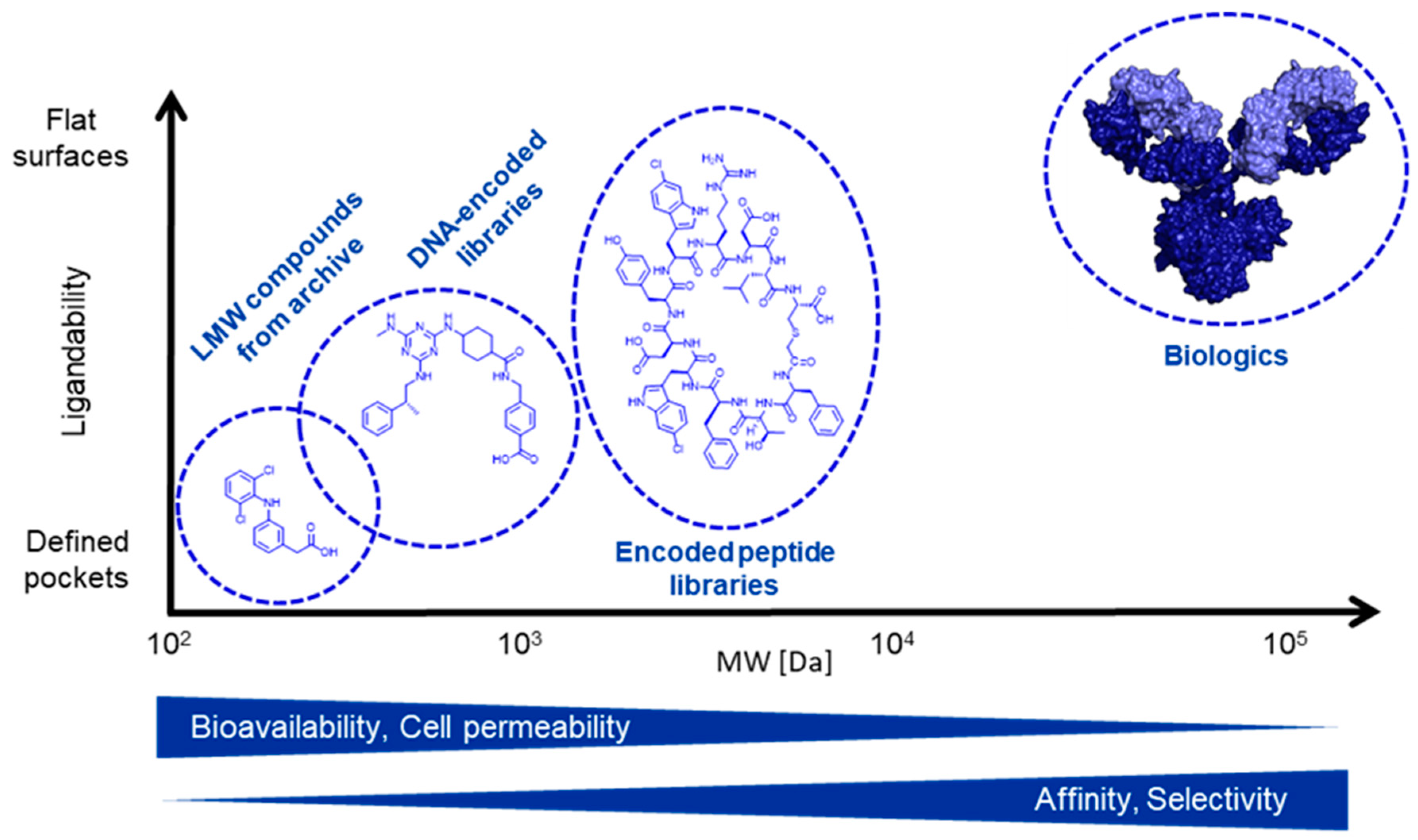
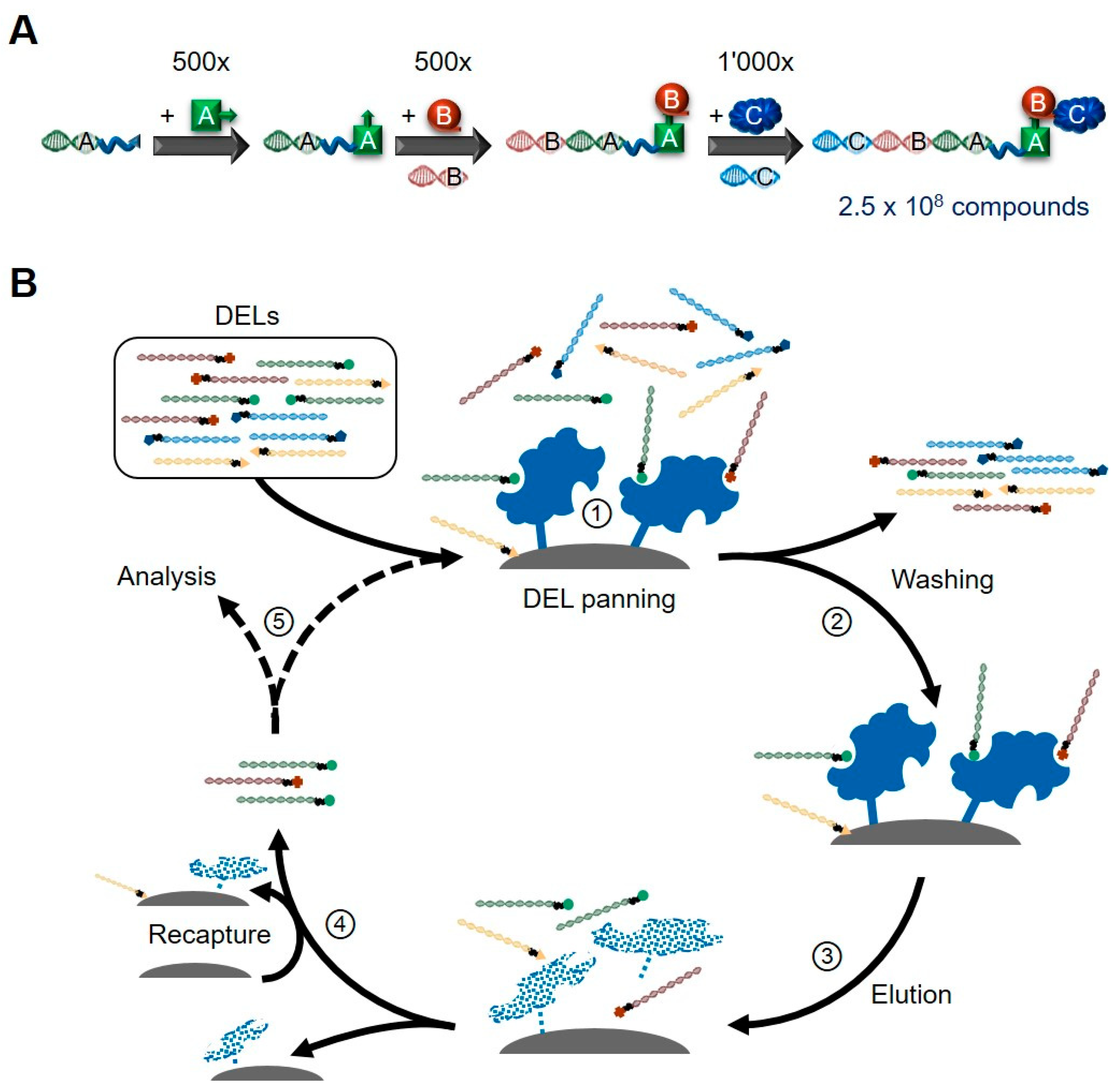
2. DNA-Encoded Libraries
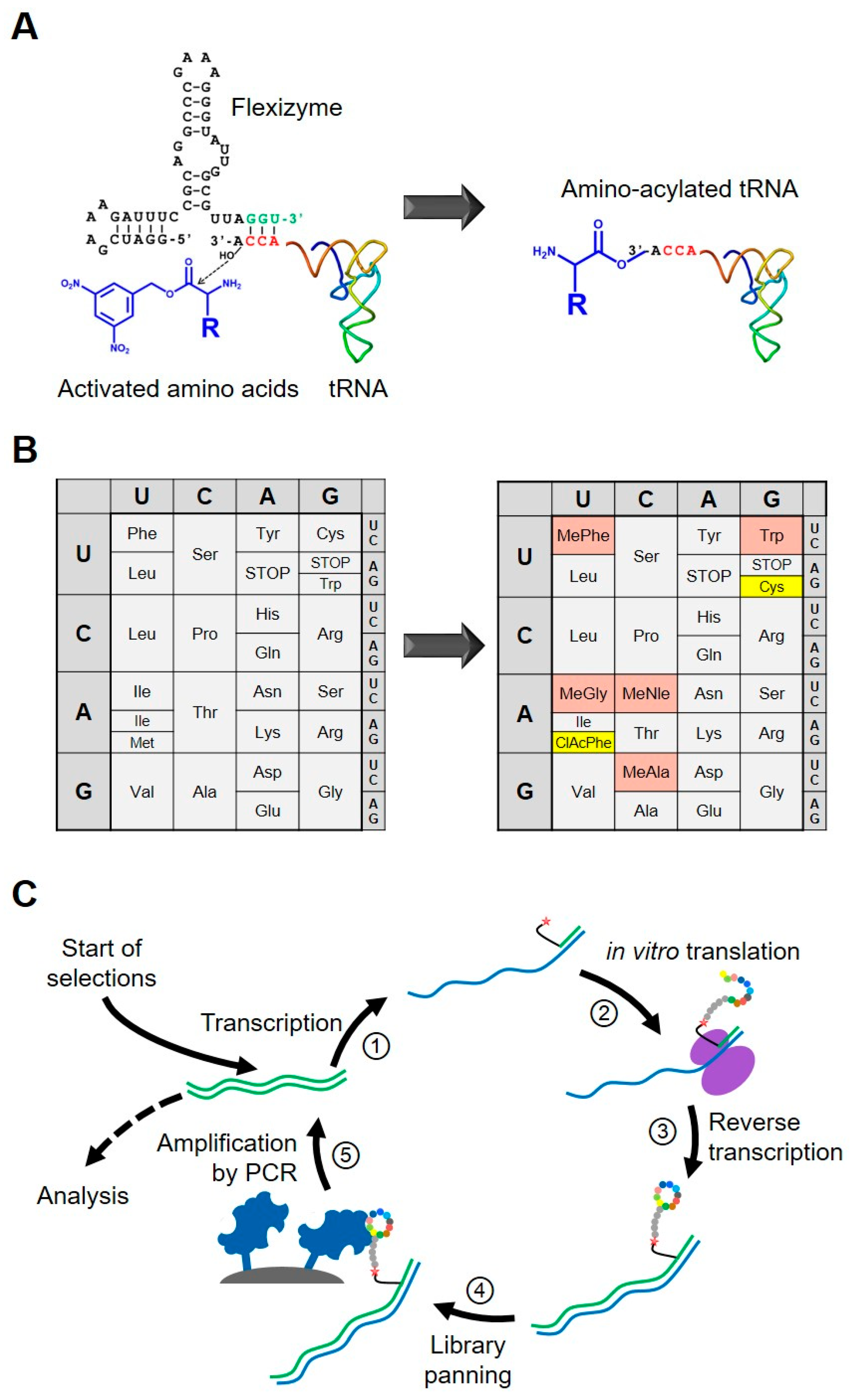
3. Peptide Discovery Platform
4. Advantages and Challenges of Encoded Library Technologies
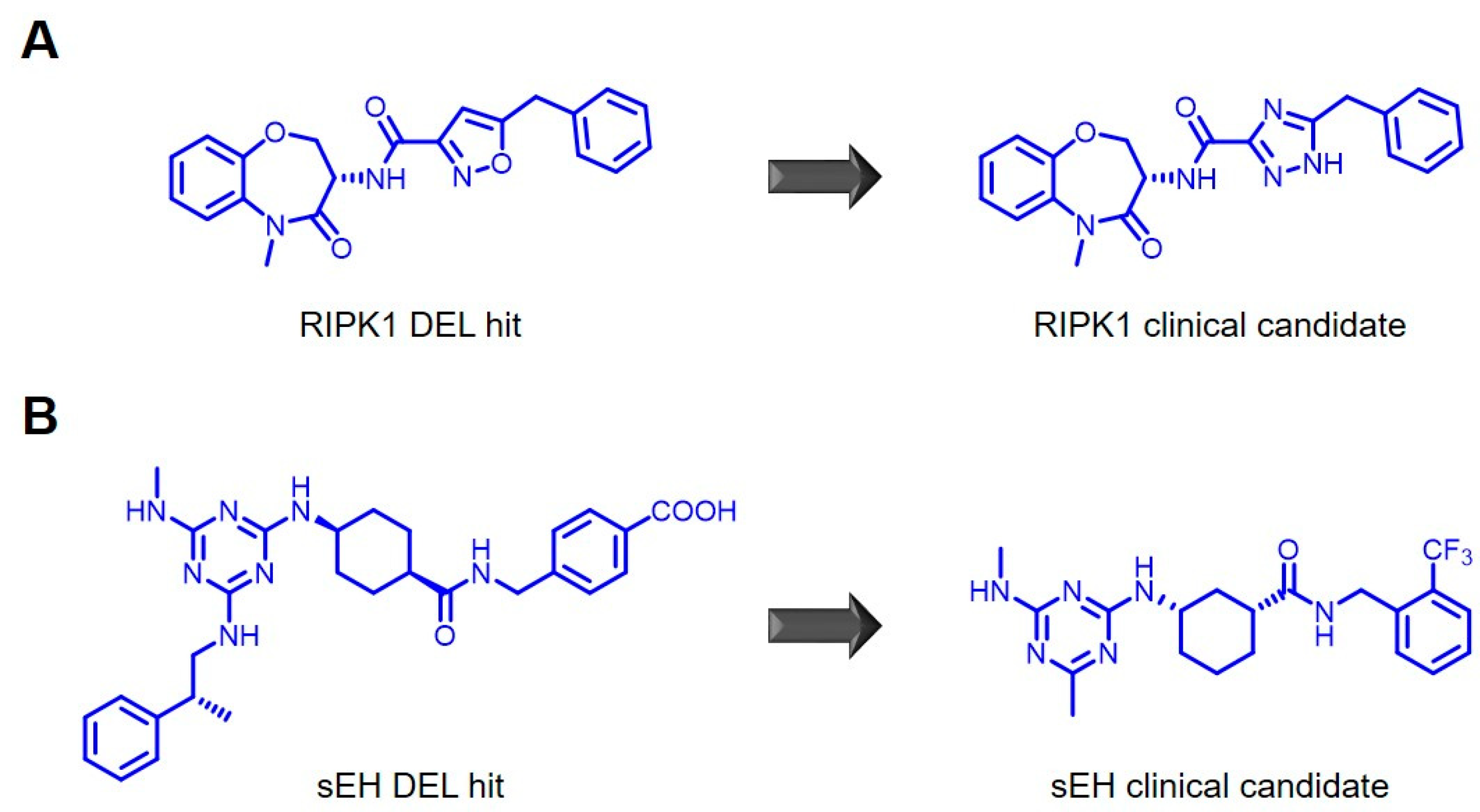
5. Applications of Encoded Library Platforms in Drug Discovery
6. Encoded Library Technologies at Novartis
7. Future Directions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DEL | DNA-encoded library |
| PDP | Peptide Discovery Platform |
| HTS | high-throughput screening |
| POI | protein of interest |
| NGS | Next Generation Sequencing |
| (q)PCR | (quantitative) polymerase chain reaction |
| SAR | structure-activity relationship |
| PDC | peptide-drug conjugate |
| LMW | low molecular weight |
| PPI | protein-protein interaction |
| DNA | deoxyribonucleic acid |
| (t)/ (m)/ (si)RNA | (transfer)/ (messenger)/ (small interfering) ribonucleic acid |
| ARS | amino acyl tRNA synthetases |
| MoA | Mode of action |
| MW | molecular weight |
| (k)Da | (kilo)Dalton |
| RaPID | Random Peptide Integrated Discovery |
| SICLOPPS | Split-Intein Circular Ligation Of Peptides and Proteins |
| PURE | Protein synthesis Using Recombinant Elements |
| IVT | in vitro translation |
| IC50 | half maximal inhibitory concentration |
| GPCR | G protein-coupled receptor |
| RIPK1 | Receptor-interacting serine/threonine-protein kinase |
| sEH | soluble epoxide hydrolase |
| EpCAM | epithelial cell adhesion molecule |
References
- Shalem, O.; Sanjana, N.E.; Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.P.; Zhao, D.X.; Sasik, R.; Luebeck, J.; Birmingham, A.; Bojorquez-Gomez, A.; Licon, K.; Klepper, K.; Pekin, D.; Beckett, A.N.; et al. Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 2017, 14, 573. [Google Scholar] [CrossRef]
- Yamagishi, Y.; Shoji, I.; Miyagawa, S.; Kawakami, T.; Katoh, T.; Goto, Y.; Suga, H. Natural Product-Like Macrocyclic N-Methyl-Peptide Inhibitors against a Ubiquitin Ligase Uncovered from a Ribosome-Expressed De Novo Library. Chem. Biol. 2011, 18, 1562–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenner, S.; Lerner, R.A. Encoded Combinatorial Chemistry. Proc. Natl. Acad. Sci. USA 1992, 89, 5381–5383. [Google Scholar] [CrossRef]
- Castanon, J.; Roman, J.P.; Jessop, T.C.; de Blas, J.; Haro, R. Design and Development of a Technology Platform for DNA-Encoded Library Production and Affinity Selection. Slas Discov. Adv. Life Sci. R&D 2018, 23, 387–396. [Google Scholar]
- Faver, J.C.; Riehle, K.; Lancia, D.R., Jr.; Milbank, J.B.J.; Kollmann, C.S.; Simmons, N.; Yu, Z.; Matzuk, M.M. Quantitative Comparison of Enrichment from DNA-Encoded Chemical Library Selections. ACS Comb. Sci 2019, 21, 75–82. [Google Scholar] [CrossRef]
- Yuen, L.H.; Franzini, R.M. Achievements, Challenges, and Opportunities in DNA-Encoded Library Research: An Academic Point of View. ChemBioChem 2017, 18, 829–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodnow, R.A.; Dumelin, C.E.; Keefe, A.D. DNA-encoded chemistry: Enabling the deeper sampling of chemical space. Nat. Rev. Drug Discov. 2017, 16, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Ledsgaard, L.; Kilstrup, M.; Karatt-Vellatt, A.; McCafferty, J.; Laustsen, A.H. Basics of Antibody Phage Display Technology. Toxins 2018, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Plückthun, A. Ribosome display: A perspective. Methods Mol. Biol. 2012, 805, 3–28. [Google Scholar]
- Kunig, V.; Potowski, M.; Gohla, A.; Brunschweiger, A. DNA-encoded libraries—An efficient small molecule discovery technology for the biomedical sciences. Biol. Chem. 2018, 399, 691–710. [Google Scholar] [CrossRef]
- Neri, D.; Lerner, R.A. DNA-Encoded Chemical Libraries: A Selection System Based on Endowing Organic Compounds with Amplifiable Information. Annu. Rev. Biochem. 2018, 87, 479–502. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.A.; Acharya, R.A.; Arico-Muendel, C.C.; Belyanskaya, S.L.; Benjamin, D.R.; Carlson, N.R.; Centrella, P.A.; Chiu, C.H.; Creaser, S.P.; Cuozzo, J.W.; et al. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat. Chem. Biol. 2009, 5, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Mannocci, L.; Zhang, Y.X.; Scheuermann, J.; Leimbacher, M.; De Bellis, G.; Rizzi, E.; Dumelin, C.; Melkko, S.; Neri, D. High-throughput sequencing allows the identification of binding molecules isolated from DNA-encoded chemical libraries. Proc. Natl. Acad. Sci. USA 2008, 105, 17670–17675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerry, C.J.; Yang, Z.; Stasi, M.; Schreiber, S.L. DNA-Compatible [3 + 2] Nitrone-Olefin Cycloaddition Suitable for DEL Syntheses. Org. Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pels, K.; Dickson, P.; An, H.; Kodadek, T. DNA-Compatible Solid-Phase Combinatorial Synthesis of beta-Cyanoacrylamides and Related Electrophiles. ACS Comb. Sci. 2018, 20, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Phelan, J.P.; Lang, S.B.; Sim, J.; Berritt, S.; Peat, A.J.; Billings, K.; Fan, L.; Molander, G.A. Open-Air Alkylation Reactions in Photoredox-Catalyzed DNA-Encoded Library Synthesis. J. Am. Chem. Soc. 2019. [Google Scholar] [CrossRef]
- Ruff, Y.; Berst, F. Efficient copper-catalyzed amination of DNA-conjugated aryl iodides under mild aqueous conditions. MedChemComm 2018, 9, 1188–1193. [Google Scholar] [CrossRef]
- Franzini, R.M.; Randolph, C. Chemical Space of DNA-Encoded Libraries. J. Med. Chem. 2016, 59, 6629–6644. [Google Scholar] [CrossRef]
- Favalli, N.; Bassi, G.; Scheuermann, J.; Neri, D. DNA-encoded chemical libraries—Achievements and remaining challenges. FEBS Lett. 2018, 592, 2168–2180. [Google Scholar] [CrossRef]
- Chan, A.I.; McGregor, L.M.; Liu, D.R. Novel selection methods for DNA-encoded chemical libraries. Curr. Opin. Chem. Biol. 2015, 26, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satz, A.L.; Hochstrasser, R.; Petersen, A.C. Analysis of Current DNA Encoded Library Screening Data Indicates Higher False Negative Rates for Numerically Larger Libraries. ACS Comb. Sci. 2017, 19, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Decurtins, W.; Wichert, M.; Franzini, R.M.; Buller, F.; Stravs, M.A.; Zhang, Y.X.; Neri, D.; Scheuermann, J. Automated screening for small organic ligands using DNA-encoded chemical libraries. Nat. Protoc. 2016, 11, 764–780. [Google Scholar] [CrossRef] [PubMed]
- Machutta, C.A.; Kollmann, C.S.; Lind, K.E.; Bai, X.P.; Chan, P.F.; Huang, J.Z.; Ballell, L.; Belyanskaya, S.; Besra, G.S.; Barros-Aguirre, D.; et al. Prioritizing multiple therapeutic targets in parallel using automated DNA-encoded library screening. Nat. Commun. 2017, 8, 16081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obexer, R.; Walport, L.J.; Suga, H. Exploring sequence space: Harnessing chemical and biological diversity towards new peptide leads. Curr. Opin. Chem. Biol. 2017, 38, 52–61. [Google Scholar] [CrossRef]
- Huang, Y.; Wiedmann, M.M.; Suga, H. RNA Display Methods for the Discovery of Bioactive Macrocycles. Chem Rev. 2018. [Google Scholar] [CrossRef]
- Lam, K.S.; Lehman, A.L.; Song, A.M.; Doan, N.; Enstrom, A.M.; Maxwell, J.; Liu, R.W. Synthesis and screening of “one-Bead one-compound” combinatorial peptide libraries. Methods Enzymol. 2003, 369, 298–322. [Google Scholar]
- Qian, Z.; Upadhyaya, P.; Pei, D. Synthesis and screening of one-bead-one-compound cyclic peptide libraries. Methods Mol. Biol. 2015, 1248, 39–53. [Google Scholar]
- Vaughan, T.J.; Williams, A.J.; Pritchard, K.; Osbourn, J.K.; Pope, A.R.; Earnshaw, J.C.; McCafferty, J.; Hodits, R.A.; Wilton, J.; Johnson, K.S. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat. Biotechnol. 1996, 14, 309–314. [Google Scholar] [CrossRef]
- Krumpe, L.R.H.; Mori, T. Potential of phage-displayed peptide library technology to identify functional targeting peptides. Expert Opin. Drug Dis. 2007, 2, 525–537. [Google Scholar] [CrossRef] [Green Version]
- Maini, R.; Umemoto, S.; Suga, H. Ribosome-mediated synthesis of natural product-like peptides via cell-free translation. Curr. Opin. Chem. Biol. 2016, 34, 44–52. [Google Scholar] [CrossRef]
- Lennard, K.R.; Tavassoli, A. Peptides Come Round: Using SICLOPPS Libraries for Early Stage Drug Discovery. Chem. Eur. J. 2014, 20, 10608–10614. [Google Scholar] [CrossRef]
- Scott, C.P.; Abel-Santos, E.; Jones, A.D.; Benkovic, S.J. Structural requirements for the biosynthesis of backbone cyclic peptide libraries. Chem. Biol. 2001, 8, 801–815. [Google Scholar] [CrossRef] [Green Version]
- Tavassoli, A. SICLOPPS cyclic peptide libraries in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef]
- Josephson, K.; Ricardo, A.; Szostak, J.W. mRNA display: From basic principles to macrocycle drug discovery. Drug Discov. Today 2014, 19, 388–399. [Google Scholar] [CrossRef]
- Roberts, R.W.; Szostak, J.W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 12297–12302. [Google Scholar] [CrossRef]
- Ishizawa, T.; Kawakami, T.; Reid, P.C.; Murakami, H. TRAP Display: A High-Speed Selection Method for the Generation of Functional Polypeptides. J. Am. Chem. Soc. 2013, 135, 5433–5440. [Google Scholar] [CrossRef]
- Hipolito, C.J.; Suga, H. Ribosomal production and in vitro selection of natural product-like peptidomimetics: The FIT and RaPID systems. Curr. Opin. Chem. Biol. 2012, 16, 196–203. [Google Scholar] [CrossRef]
- Josephson, K.; Hartman, M.C.; Szostak, J.W. Ribosomal synthesis of unnatural peptides. J. Am. Chem. Soc. 2005, 127, 11727–11735. [Google Scholar] [CrossRef]
- Ellman, J.A.; Mendel, D.; Schultz, P.G. Site-Specific Incorporation of Novel Backbone Structures into Proteins. Science 1992, 255, 197–200. [Google Scholar] [CrossRef]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the genetic code of Escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [CrossRef]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef]
- Murakami, H.; Ohta, A.; Ashigai, H.; Suga, H. A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nat. Methods 2006, 3, 357–359. [Google Scholar] [CrossRef]
- Goto, Y.; Suga, H.; Ribozymes; Hartig, J. Methods in Molecular Biology (Methods and Protocols); Humana Press: New York, NY, USA, 2012; Volume 848, pp. 465–478. [Google Scholar]
- Katoh, T.; Iwane, Y.; Suga, H. tRNA engineering for manipulating genetic code. Rna Biol. 2018, 15, 453–460. [Google Scholar] [CrossRef]
- Passioura, T.; Suga, H. A RaPID way to discover nonstandard macrocyclic peptide modulators of drug targets. Chem. Commun. 2017, 53, 1931–1940. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.S.; Cui, H.G. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliver. Rev. 2017, 110, 112–126. [Google Scholar] [CrossRef]
- Lin, W.L.; Reddavide, F.V.; Uzunova, V.; Gur, F.N.; Zhang, Y.X. Characterization of DNA-Conjugated Compounds Using a Regenerable Chip. Anal. Chem. 2015, 87, 864–868. [Google Scholar] [CrossRef]
- Zimmermann, G.; Li, Y.Z.; Rieder, U.; Mattarella, M.; Neri, D.; Scheuermann, J. Hit-Validation Methodologies for Ligands Isolated from DNA-Encoded Chemical Libraries. ChemBioChem 2017, 18, 853–857. [Google Scholar] [CrossRef]
- Skopic, M.K.; Salamon, H.; Bugain, O.; Jung, K.; Gohla, A.; Doetsch, L.J.; dos Santos, D.; Bhat, A.; Wagner, B.; Brunschweiger, A. Acid- and Au(I)-mediated synthesis of hexathymidine-DNA-heterocycle chimeras, an efficient entry to DNA-encoded libraries inspired by drug structures. Chem. Sci. 2017, 8, 3356–3361. [Google Scholar] [CrossRef]
- Fleming, S.R.; Bartges, T.E.; Vinogradov, A.A.; Kirkpatrick, C.L.; Goto, Y.; Suga, H.; Hicks, L.M.; Bowers, A.A. Flexizyme-Enabled Benchtop Biosynthesis of Thiopeptides. J. Am. Chem. Soc. 2019, 141, 758–762. [Google Scholar] [CrossRef]
- Goto, Y.; Ito, Y.; Kato, Y.; Tsunoda, S.; Suga, H. One-Pot Synthesis of Azoline-Containing Peptides in a Cell-free Translation System Integrated with a Posttranslational Cyclodehydratase. Chem. Biol. 2014, 21, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Jalali-Yazdi, F.; Lai, L.H.; Takahashi, T.T.; Roberts, R.W. High-Throughput Measurement of Binding Kinetics by mRNA Display and Next-Generation Sequencing. Angew Chem. Int. Edit. 2016, 55, 4007–4010. [Google Scholar] [CrossRef]
- Atangcho, L.; Navaratna, T.; Thurber, G.M. Hitting Undruggable Targets: Viewing Stabilized Peptide Development through the Lens of Quantitative Systems Pharmacology. Trends Biochem. Sci. 2018. [Google Scholar] [CrossRef]
- Chene, P. Inhibition of the p53-MDM2 interaction: Targeting a protein-protein interface. Mol. Cancer Res. 2004, 2, 20–28. [Google Scholar]
- Straub, C.S. Targeting IAPs as An Approach to Anti-Cancer Therapy. Curr. Top. Med. Chem. 2011, 11, 291–316. [Google Scholar] [CrossRef]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef]
- Belyanskaya, S.L.; Ding, Y.; Callahan, J.F.; Lazaar, A.L.; Israel, D.I. Discovering Drugs with DNA-Encoded Library Technology: From Concept to Clinic with an Inhibitor of Soluble Epoxide Hydrolase. ChemBioChem 2017, 18, 837–842. [Google Scholar] [CrossRef]
- Fernandez-Montalvan, A.E.; Berger, M.; Kuropka, B.; Koo, S.J.; Badock, V.; Weiske, J.; Puetter, V.; Holton, S.J.; Stokigt, D.; ter Laak, A.; et al. Isoform-Selective ATAD2 Chemical Probe with Novel Chemical Structure and Unusual Mode of Action. ACS Chem. Biol. 2017, 12, 2730–2736. [Google Scholar] [CrossRef]
- Arrowsmith, C.H.; Audia, J.E.; Austin, C.; Baell, J.; Bennett, J.; Blagg, J.; Bountra, C.; Brennan, P.E.; Brown, P.J.; Bunnage, M.E.; et al. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541. [Google Scholar] [CrossRef] [Green Version]
- Johannes, J.W.; Bates, S.; Beigie, C.; Belmonte, M.A.; Breen, J.; Cao, S.G.; Centrella, P.A.; Clark, M.A.; Cuozzo, J.W.; Dumelin, C.E.; et al. Structure Based Design of Non-Natural Peptidic Macrocyclic Mcl-1 Inhibitors. ACS Med. Chem. Lett. 2017, 8, 239–244. [Google Scholar] [CrossRef]
- Usanov, D.L.; Chan, A.I.; Maianti, J.P.; Liu, D.R. Second-generation DNA-templated macrocycle libraries for the discovery of bioactive small molecules. Nat. Chem. 2018, 10, 704–714. [Google Scholar] [CrossRef]
- Li, Y.; De Luca, R.; Cazzamalli, S.; Pretto, F.; Bajic, D.; Scheuermann, J.; Neri, D. Versatile protein recognition by the encoded display of multiple chemical elements on a constant macrocyclic scaffold. Nat. Chem. 2018, 10, 441–448. [Google Scholar] [CrossRef]
- Zhu, Z.; Shaginian, A.; Grady, L.C.; O’Keeffe, T.; Shi, X.E.; Davie, C.P.; Simpson, G.L.; Messer, J.A.; Evindar, G.; Bream, R.N.; et al. Design and Application of a DNA-Encoded Macrocyclic Peptide Library. ACS Chem. Biol. 2018, 13, 53–59. [Google Scholar] [CrossRef]
- Brown, D.G.; Brown, G.A.; Centrella, P.; Certel, K.; Cooke, R.M.; Cuozzo, J.W.; Dekker, N.; Dumelin, C.E.; Ferguson, A.; Fiez-Vandal, C.; et al. Agonists and Antagonists of Protease-Activated Receptor 2 Discovered within a DNA-Encoded Chemical Library Using Mutational Stabilization of the Target. Slas. Discov. 2018, 23, 429–436. [Google Scholar] [CrossRef]
- Cheng, R.K.Y.; Fiez-Vandal, C.; Schlenker, O.; Edman, K.; Aggeler, B.; Brown, D.G.; Brown, G.A.; Cooke, R.M.; Dumelin, C.E.; Dore, A.S.; et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 2017, 545, 112. [Google Scholar] [CrossRef]
- Song, X.; Lu, L.Y.; Passioura, T.; Suga, H. Macrocyclic peptide inhibitors for the protein-protein interaction of Zaire Ebola virus protein 24 and karyopherin alpha 5. Org. Biomol. Chem. 2017, 15, 5155–5160. [Google Scholar] [CrossRef]
- Hipolito, C.J.; Tanaka, Y.; Katoh, T.; Nureki, O.; Suga, H. A Macrocyclic Peptide that Serves as a Cocrystallization Ligand and Inhibits the Function of a MATE Family Transporter. Molecules 2013, 18, 10514–10530. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Hipolito, C.J.; Maturana, A.D.; Ito, K.; Kuroda, T.; Higuchi, T.; Katoh, T.; Kato, H.E.; Hattori, M.; Kumazaki, K.; et al. Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature 2013, 496, 247. [Google Scholar] [CrossRef] [PubMed]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Sakiyama, K.; Hipolito, C.J.; Fujioka, A.; Hirokane, R.; Ikeguchi, K.; Watanabe, B.; Hiratake, J.; et al. Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. USA 2014, 111, 4049–4054. [Google Scholar] [CrossRef]
- Kawamura, A.; Munzel, M.; Kojima, T.; Yapp, C.; Bhushan, B.; Goto, Y.; Tumber, A.; Katoh, T.; King, O.N.F.; Passioura, T.; et al. Highly selective inhibition of histone demethylases by de novo macrocyclic peptides. Nat. Commun. 2017, 8, 14773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Hartman, M.C. Ribosome Display and Related Technologies; Douthwaite, J., Jackson, R., Eds.; Methods in Molecular Biology (Methods and Protocols); Springer: New York, NY, USA, 2012; Volume 805, pp. 367–390. [Google Scholar]
- Angelini, A.; Cendron, L.; Chen, S.; Touati, J.; Winter, G.; Zanotti, G.; Heinis, C. Bicyclic peptide inhibitor reveals large contact interface with a protease target. ACS Chem. Biol. 2012, 7, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Urech-Varenne, C.; Radtke, F.; Heinis, C. Phage Selection of Bicyclic Peptide Ligands of the Notch1 Receptor. ChemMedChem 2015, 10, 1754–1761. [Google Scholar] [CrossRef]
- Baeriswyl, V.; Calzavarini, S.; Chen, S.; Zorzi, A.; Bologna, L.; Angelillo-Scherrer, A.; Heinis, C. A Synthetic Factor XIIa Inhibitor Blocks Selectively Intrinsic Coagulation Initiation. ACS Chem. Biol. 2015, 10, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Baeriswyl, V.; Calzavarini, S.; Gerschheimer, C.; Diderich, P.; Angelillo-Scherrer, A.; Heinis, C. Development of a selective peptide macrocycle inhibitor of coagulation factor XII toward the generation of a safe antithrombotic therapy. J. Med. Chem. 2013, 56, 3742–3746. [Google Scholar] [CrossRef] [PubMed]
- Middendorp, S.J.; Wilbs, J.; Quarroz, C.; Calzavarini, S.; Angelillo-Scherrer, A.; Heinis, C. Peptide Macrocycle Inhibitor of Coagulation Factor XII with Subnanomolar Affinity and High Target Selectivity. J. Med. Chem. 2017, 60, 1151–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Krall, N.; Pretto, F.; Decurtins, W.; Bernardes, G.J.; Supuran, C.T.; Neri, D. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem. Int. Ed. Engl. 2014, 53, 4231–4235. [Google Scholar] [CrossRef]
- Krall, N.; Scheuermann, J.; Neri, D. Small targeted cytotoxics: Current state and promises from DNA-encoded chemical libraries. Angew Chem. Int Ed. Engl. 2013, 52, 1384–1402. [Google Scholar] [CrossRef]
- Cazzamalli, S.; Dal Corso, A.; Widmayer, F.; Neri, D. Chemically Defined Antibody- and Small Molecule-Drug Conjugates for in Vivo Tumor Targeting Applications: A Comparative Analysis. J. Am. Chem. Soc. 2018, 140, 1617–1621. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of Lu-177-Dotatate for Midgut Neuroendocrine Tumors. New Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef]
- Iwasaki, K.; Goto, Y.; Katoh, T.; Yamashita, T.; Kaneko, S.; Suga, H. A Fluorescent Imaging Probe Based on a Macrocyclic Scaffold That Binds to Cellular EpCAM. J. Mol. Evol. 2015, 81, 210–217. [Google Scholar] [CrossRef]
- Sakurai, Y.; Mizumura, W.; Murata, M.; Hada, T.; Yamamoto, S.; Ito, K.; Iwasaki, K.; Katoh, T.; Goto, Y.; Takagi, A.; et al. Efficient siRNA Delivery by Lipid Nanoparticles Modified with a Nonstandard Macrocyclic Peptide for EpCAM-Targeting. Mol. Pharm. 2017, 14, 3290–3298. [Google Scholar] [CrossRef]
- Ammala, C.; Drury, W.J., 3rd; Knerr, L.; Ahlstedt, I.; Stillemark-Billton, P.; Wennberg-Huldt, C.; Andersson, E.M.; Valeur, E.; Jansson-Lofmark, R.; Janzen, D.; et al. Targeted delivery of antisense oligonucleotides to pancreatic beta-cells. Sci. Adv. 2018, 4, eaat3386. [Google Scholar] [CrossRef]
- Denton, K.E.; Wang, S.; Gignac, M.C.; Milosevich, N.; Hof, F.; Dykhuizen, E.C.; Krusemark, C.J. Robustness of In Vitro Selection Assays of DNA-Encoded Peptidomimetic Ligands to CBX7 and CBX8. Slas. Discov. 2018, 23, 417–428. [Google Scholar]
- Kollmann, C.S.; Bai, X.P.; Tsai, C.H.; Yang, H.F.; Lind, K.E.; Skinner, S.R.; Zhu, Z.R.; Israel, D.I.; Cuozzo, J.W.; Morgan, B.A.; et al. Application of encoded library technology (ELT) to a protein-protein interaction target: Discovery of a potent class of integrin lymphocyte function-associated antigen 1 (LFA-1) antagonists. Bioorg. Med. Chem. 2014, 22, 2353–2365. [Google Scholar] [CrossRef]
- Ahn, S.; Kahsai, A.W.; Pani, B.; Wang, Q.T.; Zhao, S.; Wall, A.L.; Strachan, R.T.; Staus, D.P.; Wingler, L.M.; Sun, L.D.; et al. Allosteric “beta-blocker” isolated from a DNA-encoded small molecule library. Proc. Natl. Acad. Sci. USA 2017, 114, 1708–1713. [Google Scholar] [CrossRef]
- Ahn, S.; Pani, B.; Kahsai, A.W.; Olsen, E.K.; Husemoen, G.; Vestergaard, M.; Jin, L.; Zhao, S.; Wingler, L.M.; Rambarat, P.K.; et al. Small-Molecule Positive Allosteric Modulators of the beta(2)-Adrenoceptor Isolated from DNA-Encoded Libraries. Mol. Pharm. 2018, 94, 850–861. [Google Scholar] [CrossRef]
- Wu, Z.N.; Graybill, T.L.; Zeng, X.; Platchek, M.; Zhang, J.; Bodmer, V.Q.; Wisnoski, D.D.; Deng, J.H.; Coppo, F.T.; Yao, G.; et al. Cell-Based Selection Expands the Utility of DNA-Encoded Small-Molecule Library Technology to Cell Surface Drug Targets: Identification of Novel Antagonists of the NK3 Tachykinin Receptor. ACS Comb. Sci. 2015, 17, 722–731. [Google Scholar] [CrossRef]
- Schlegel, S.; Hjelm, A.; Baumgarten, T.; Vikstrom, D.; de Gier, J.W. Bacterial-based membrane protein production. Biochim. Biophys. Acta 2014, 1843, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Hollenstein, K.; Kean, J.; Bortolato, A.; Cheng, R.K.; Dore, A.S.; Jazayeri, A.; Cooke, R.M.; Weir, M.; Marshall, F.H. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 2013, 499, 438–443. [Google Scholar] [CrossRef]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Sakai, K.; Suzuki, Y.; Ozawa, N.; Hatta, T.; Natsume, T.; Matsumoto, K.; Suga, H. Artificial human Met agonists based on macrocycle scaffolds. Nat. Commun. 2015, 6, 6373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Grady, L.C.; Ding, Y.; Lind, K.E.; Davie, C.P.; Phelps, C.B.; Evindar, G. Development of a Selection Method for Discovering Irreversible (Covalent) Binders from a DNA-Encoded Library. Slas. Discov. 2019, 24, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.; Rieder, U.; Bajic, D.; Vanetti, S.; Chaikuad, A.; Knapp, S.; Scheuermann, J.; Mattarella, M.; Neri, D. A Specific and Covalent JNK-1 Ligand Selected from an Encoded Self-Assembling Chemical Library. Chemistry 2017, 23, 8152–8155. [Google Scholar] [CrossRef] [PubMed]
- Marzinzik, A.L.; Amstutz, R.; Bold, G.; Bourgier, E.; Cotesta, S.; Glickman, J.F.; Gotte, M.; Henry, C.; Lehmann, S.; Hartwieg, J.C.D.; et al. Discovery of Novel Allosteric Non-Bisphosphonate Inhibitors of Farnesyl Pyrophosphate Synthase by Integrated Lead Finding. ChemMedChem 2015, 10, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Gossert, A.D.; Jahnke, W. NMR in drug discovery: A practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nucl. Mag. Res. Sp. 2016, 97, 82–125. [Google Scholar] [CrossRef] [PubMed]
- Zehender, H.; Le Goff, F.; Lehmann, N.; Filipuzzi, I.; Mayr, L.M. SpeedScreen: The “missing link” between genomics and lead discovery. J. Biomol. Screen 2004, 9, 498–505. [Google Scholar] [CrossRef]
- Miranda, E.; Nordgren, I.K.; Male, A.L.; Lawrence, C.E.; Hoakwie, F.; Cuda, F.; Court, W.; Fox, K.R.; Townsend, P.A.; Packham, G.K.; et al. A Cyclic Peptide Inhibitor of HIF-1 Heterodimerization That Inhibits Hypoxia Signaling in Cancer Cells. J. Am. Chem. Soc. 2013, 135, 10418–10425. [Google Scholar] [CrossRef]
- MacConnell, A.B.; Price, A.K.; Paegel, B.M. An Integrated Microfluidic Processor for DNA-Encoded Combinatorial Library Functional Screening. ACS Comb. Sci 2017, 19, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, W.G.; Malone, M.L.; Dang, V.Q.; Cavett, V.; Satz, A.L.; Paegel, B.M. Activity-Based DNA-Encoded Library Screening. ACS Comb. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Brouzes, E.; Medkova, M.; Savenelli, N.; Marran, D.; Twardowski, M.; Hutchison, J.B.; Rothberg, J.M.; Link, D.R.; Perrimon, N.; Samuels, M.L. Droplet microfluidic technology for single-cell high-throughput screening. Proc. Natl. Acad. Sci. USA 2009, 106, 14195–14200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, F.; Demaree, B.; Ahmed, N.; Abate, A.R. Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nat. Biotechnol. 2017, 35, 640–646. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Advantages | Challenges/Limitations |
|---|---|
|
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottl, J.; Leder, L.; Schaefer, J.V.; Dumelin, C.E. Encoded Library Technologies as Integrated Lead Finding Platforms for Drug Discovery. Molecules 2019, 24, 1629. https://doi.org/10.3390/molecules24081629
Ottl J, Leder L, Schaefer JV, Dumelin CE. Encoded Library Technologies as Integrated Lead Finding Platforms for Drug Discovery. Molecules. 2019; 24(8):1629. https://doi.org/10.3390/molecules24081629
Chicago/Turabian StyleOttl, Johannes, Lukas Leder, Jonas V. Schaefer, and Christoph E. Dumelin. 2019. "Encoded Library Technologies as Integrated Lead Finding Platforms for Drug Discovery" Molecules 24, no. 8: 1629. https://doi.org/10.3390/molecules24081629
APA StyleOttl, J., Leder, L., Schaefer, J. V., & Dumelin, C. E. (2019). Encoded Library Technologies as Integrated Lead Finding Platforms for Drug Discovery. Molecules, 24(8), 1629. https://doi.org/10.3390/molecules24081629