Different adaptive NO-dependent Mechanisms in Normal and Hypertensive Conditions



Abstract
:1. Introduction
2. Results
2.1. Blood Pressure, Body Weight, Cardiac and Kidney Weight, Cytokine Levels
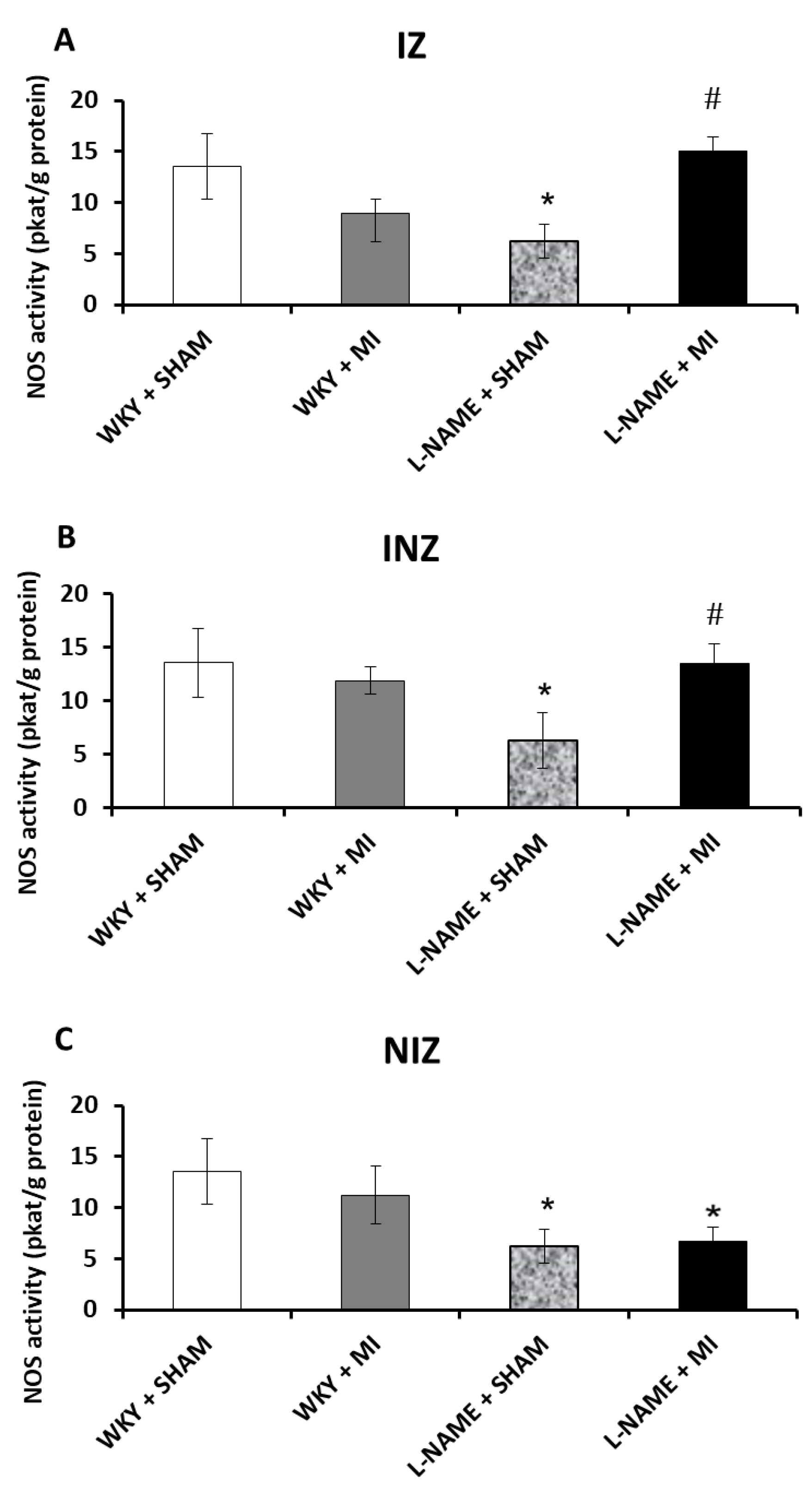
2.2. Total NOS Activity
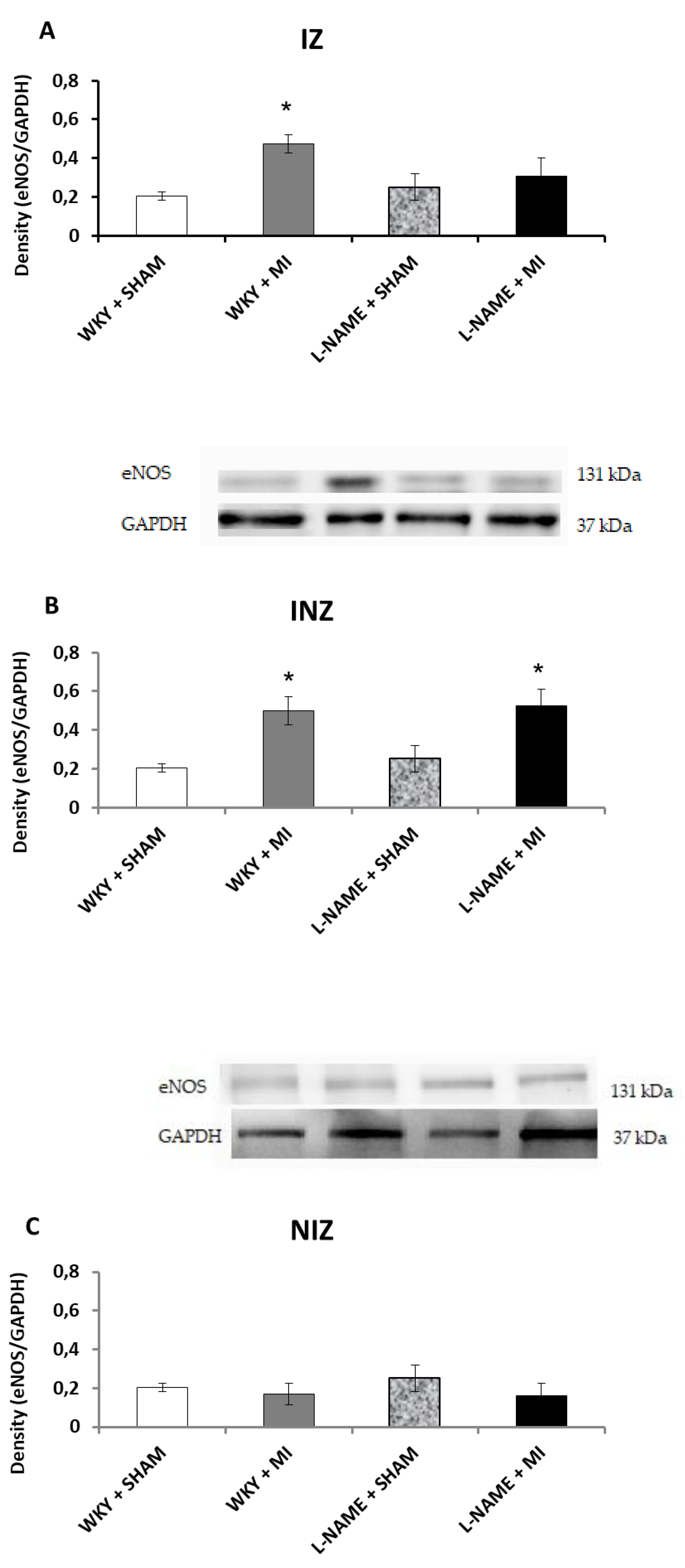
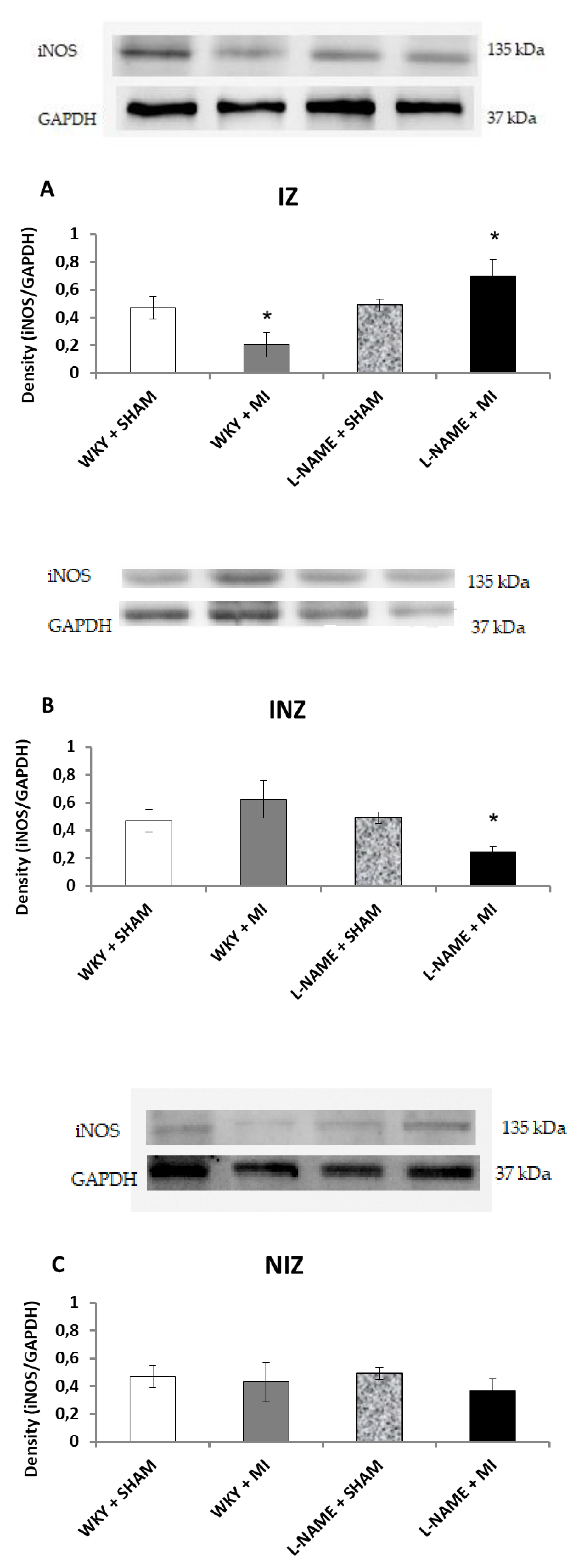
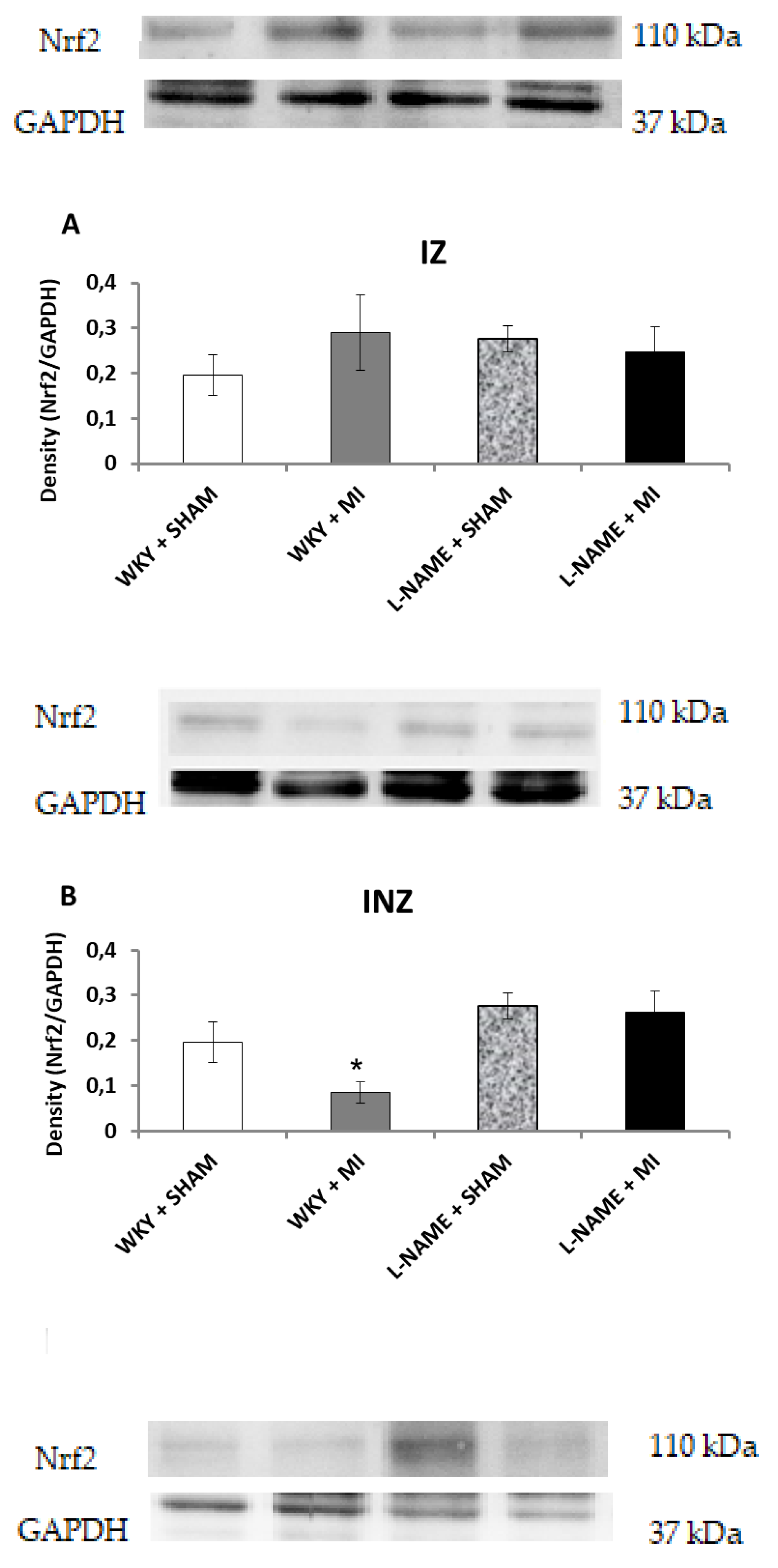
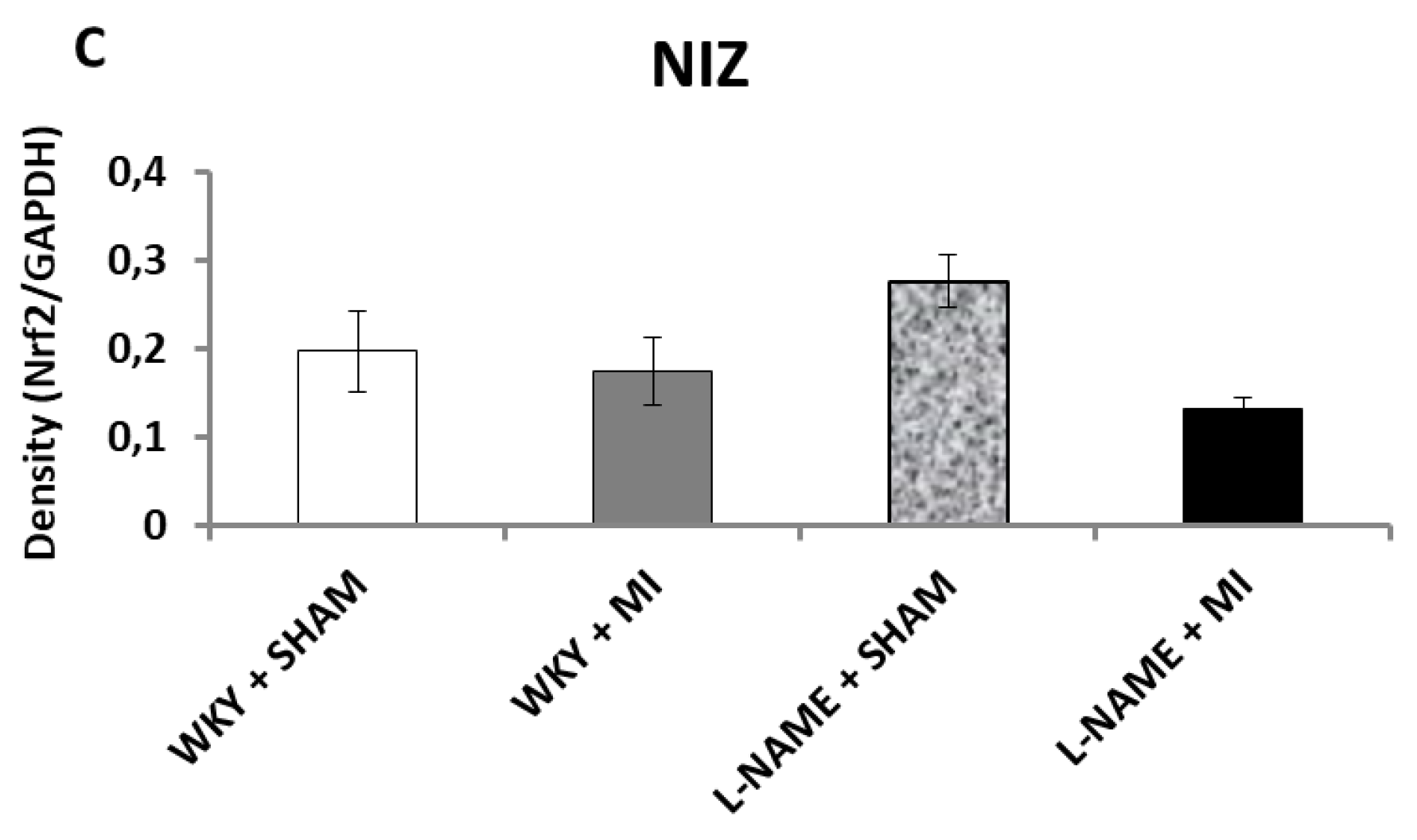
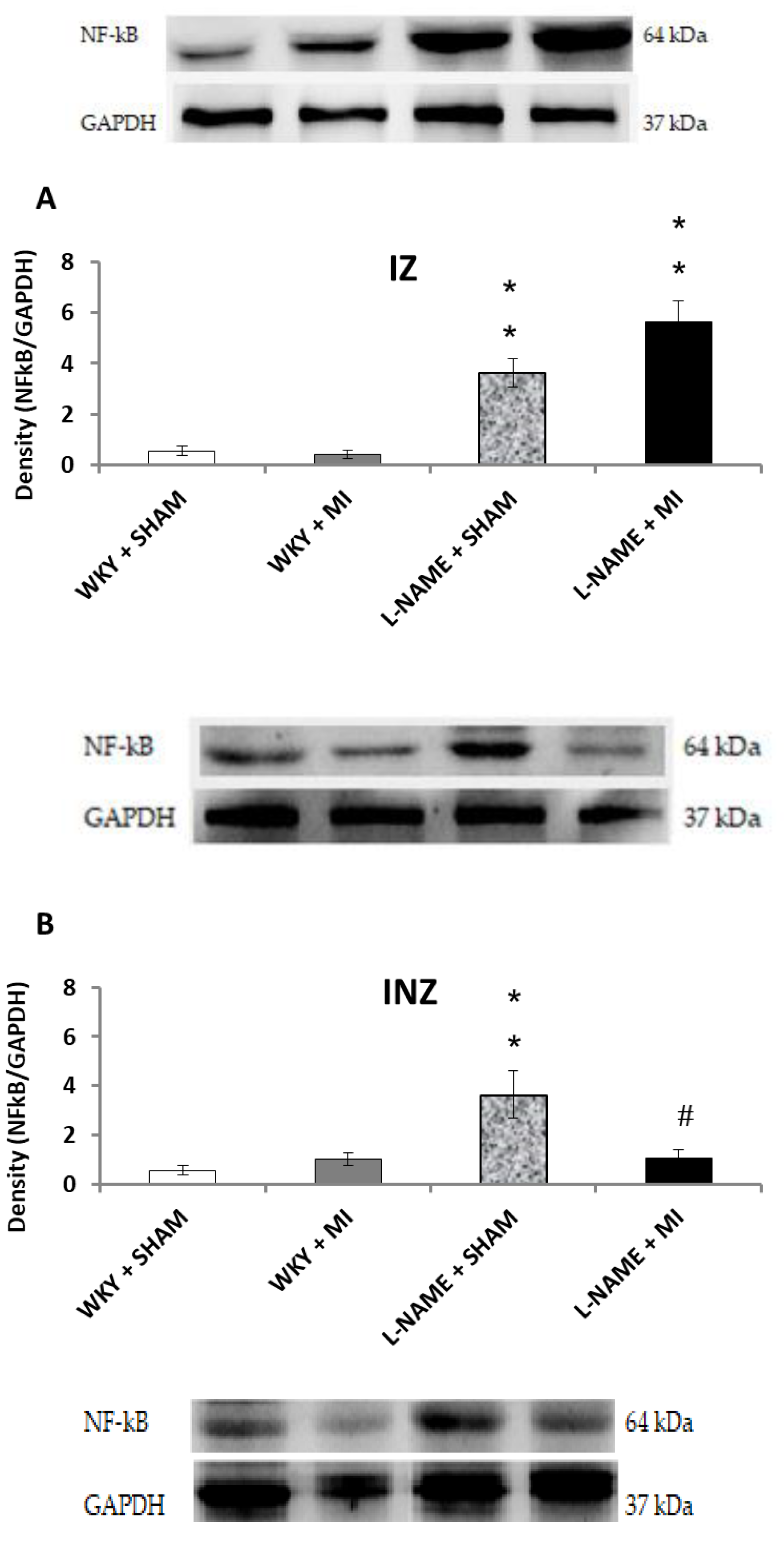
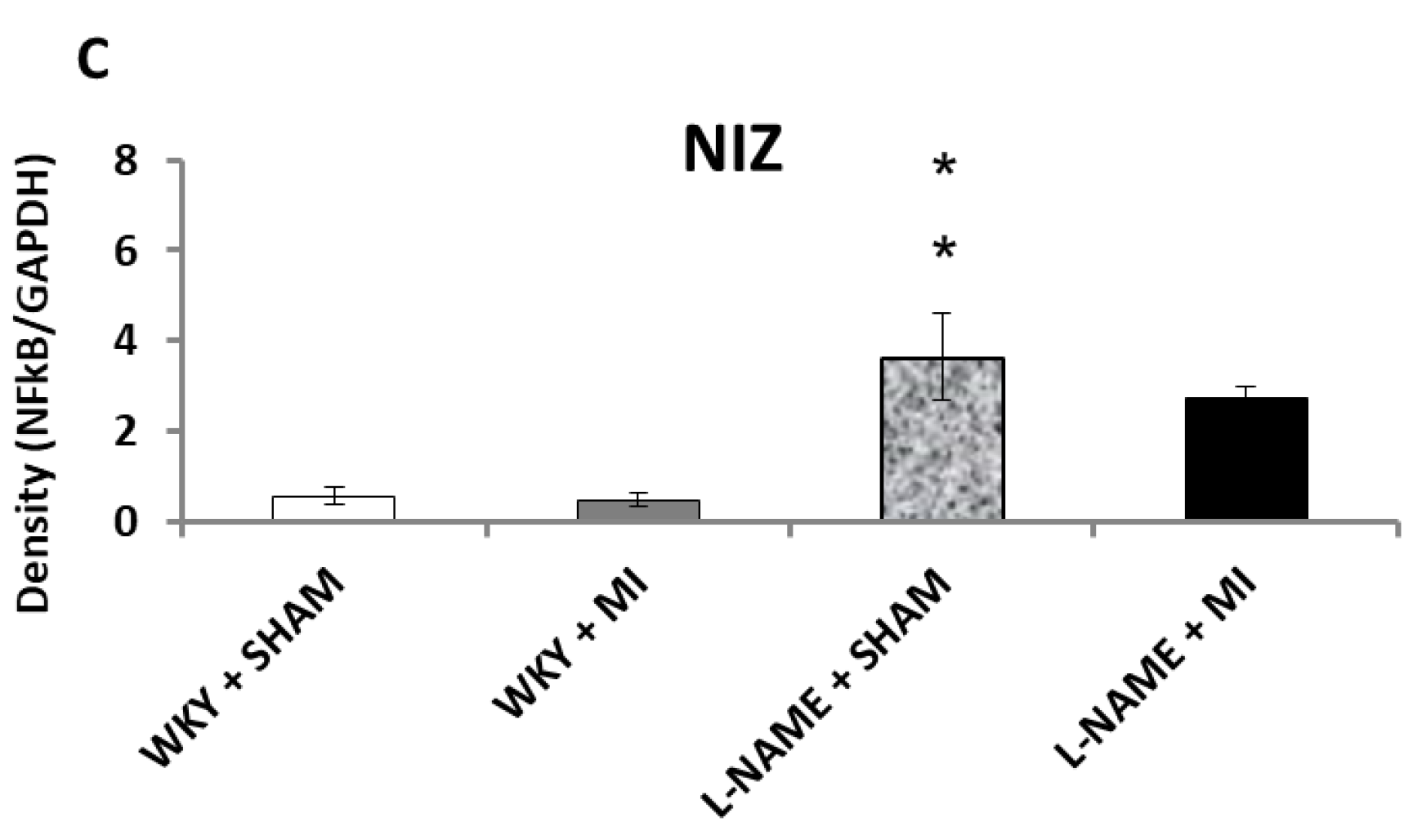
2.3. Protein Expression Levels
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. Experimentally Induced Myocardial Infarction
4.3. Total NOS Activity
4.4. Western Blot Analysis
4.5. Statistical Analysis
5. Conclusion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2017 Update: A Report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef]
- World Health Organization Cardiovascular diseases (CVDs) [Fact sheet]. Available online: http://www.who.int/mediacentre/factsheets/fs317/en/ (accessed on 20 January 2018).
- Anzai, T. Post-infarction inflammation and left ventricular remodeling: A double-edged sword. Circ. J. 2013, 77, 580–587. [Google Scholar] [CrossRef]
- Bhatt, A.S.; Ambrosy, A.P.; Velazquez, E.J. Adverse remodeling and reverse remodeling after myocardial infarction. Curr. Cardiol. Rep. 2017, 19, 71. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Corvera, J.; Halkos, M.; Kerendi, F.; Wang, N.; Guyton, R.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K.; Gounder, S.S.; Davidson, C.J.; Boeheme, C.; Hoidal, J.R.; Wang, L.; Rajasekaran, N.S. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 2012, 52, 366–376. [Google Scholar] [CrossRef]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–H1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.Y.; Reddy, S.P.; Yamamoto, M.; Kleeberger, S.R. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004, 18, 1258–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Santulli, G.; Guo, X.; Gao, M.; Chen, B.X.; Marks, A.R. Imaging atrial arrhythmic intracellular calcium in intact heart. J. Mol. Cell Cardiol. 2013, 64, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G.; Boengler, K.; Schulz, R. Inhibition of mitochondrial permeability transition pore opening: The Holy Grail of cardioprotection. Basic Res. Cardiol. 2010, 105, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Pechanova, O.; Varga, Z.V.; Cebova, M.; Giricz, Z.; Pacher, P.; Ferdinandy, P. Cardiac NO signalling in the metabolic syndrome. Br. J. Pharmacol. 2014, 172, 1415–1433. [Google Scholar] [CrossRef] [Green Version]
- Cebova, M.; Kosutova, M.; Pechanova, O. Cardiovascular effects of gasotransmitter donors. Physiol. Res. 2016, 65 (Suppl. 3), S291–S307. [Google Scholar]
- Aragón, J.P.; Condit, M.E.; Bhushan, S.; Predmore, B.L.; Pate, S.S.; Grinsfelder, D.B.; Gundewar, S.; Jha, S.; Calvert, J.W.; Barouch, L.A.; Lavu, M.; et al. Beta3-adrenoreceptor stimulation ameliorates myocardial ischemia-reperfusion injury via endothelial nitric oxide synthase and neuronal nitric oxide synthase activation. J. Am. Coll. Cardiol. 2011, 58, 2683–2691. [Google Scholar] [CrossRef]
- Luedike, P.; Hendgen-Cotta, U.B.; Sobierajski, J.; Totzeck, M.; Reeh, M.; Dewor, M.; Lue, H.; Krisp, C.; Wolters, D.; Kelm, M.; et al. Cardioprotection through S-nitros(yl)ation of macrophage migration inhibitory factor. Circulation 2012, 125, 1880–1889. [Google Scholar] [CrossRef]
- Pechanova, O.; Bernatova, I.; Pelouch, V.; Babal, P. L-NAME-induced protein remodeling and fibrosis in the rat heart. Physiol. Res. 1999, 48, 353–362. [Google Scholar]
- González-Montero, J.; Brito, R.; Gajardo, A.I.; Rodrigo, R. Myocardial reperfusion injury and oxidative stress: Therapeutic opportunities. World J Cardiol. 2018, 10, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cheng, Z.Y.; Chen, X.J.; Xue, H.Z. Ulinastatin protects rats with myocardial infarction by activating Nrf2/NOS pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8990–8998. [Google Scholar] [CrossRef]
- Liang, J.; Li, L.; Sun, Y.; He, W.; Wang, X.; Su, Q. The protective effect of activating Nrf2 / HO-1 signaling pathway on cardiomyocyte apoptosis after coronary microembolization in rats. BMC Cardiovasc. Disord. 2017, 17, 272. [Google Scholar] [CrossRef] [PubMed]
- Babal, P.; Pechanova, O.; Bernatova, I.; Stvrtina, S. Chronic inhibition of NO synthesis produces myocardial fibrosis and arterial media hyperplasia. Histol Histopathol. 1997, 12, 623–629. [Google Scholar]
- Pechanova, O.; Matuskova, J.; Capikova, D.; Jendekova, L.; Paulis, L.; Simko, F. Effect of spironolactone and captopril on nitric oxide and S-nitrosothiol formation in kidney of L-NAME-treated rats. Kidney Int. 2006, 7, 170–176. [Google Scholar] [CrossRef]
- Cebova, M.; Kristek, F. Age-dependent ultrastructural changes of coronary artery in spontaneously hypertensive rats. Gen Physiol Biophys. 2011, 30, 364–372. [Google Scholar] [CrossRef]
- Guzmán-Hernández, E.A.; Villalobos-Molina, R.; Sánchez-Mendoza, M.A.; Del Valle-Mondragón, L.; Pastelín-Hernández, G.; Ibarra-Barajas, M. Early co-expression of cyclooxygenase-2 and renin in the rat kidney cortex contributes to the development of N(G)-nitro-L-arginine methyl ester induced hypertension. Can J Physiol Pharmacol. 2015, 93, 299–308. [Google Scholar] [CrossRef]
- Cebova, M.; Klimentova, J.; Janega, P.; Pechanova, O. Effect of bioactive compound of Aronia melanocarpa on cardiovascular system in experimental hypertension. Oxid. Med. Cell Longev. 2017, 2017, 8156594. [Google Scholar] [CrossRef]
- Simko, F.; Baka, T.; Krajcirovicova, K.; Repova, K.; Aziriova, S.; Zorad, S.; Poglitsch, M.; Adamcova, M.; Reiter, R.J.; Paulis, L. Effect of Melatonin on the Renin-Angiotensin-Aldosterone System in l-NAME-Induced Hypertension. Molecules 2018, 23, 265. [Google Scholar] [CrossRef]
- Bauersachs, J.; Bouloumié, A.; Fraccarollo, D.; Hu, K.; Busse, R.; Ertl, G. Endothelial dysfunction in chronic myocardial infarction despite increased vascular endothelial nitric oxide synthase and soluble guanylate cyclase expression: Role of enhanced vascular superoxide production. Circulation 1999, 100, 292–298. [Google Scholar] [CrossRef]
- Chen, H.; Xu, Y.; Wang, J.; Zhao, W.; Ruan, H. Baicalin ameliorates isoproterenol-induced acute myocardial infarction through iNOS, inflammation and oxidative stress in rat. Int. J. Clin. Exp. Pathol. 2015, 8, 10139–10147. [Google Scholar]
- Grumbach, I.M.; Chen, W.; Mertens, S.A.; Harrison, D.G. A negative feedback mechanism involving nitric oxide and nuclear factor Kappa-B modulates endothelial nitric oxide synthase transcription. J. Mol. Cell Cardiol. 2005, 39, 595–603. [Google Scholar] [CrossRef]
- Malik, S.; Sharma, A.K.; Bharti, S.; Nepal, S.; Bhatia, J.; Nag, T.C.; Narang, R.; Arya, D.S. In vivo cardioprotection by pitavastatin from ischemic-reperfusion injury through suppression of IKK/NF-κB and upregulation of pAkt-e-NOS. J. Cardiovasc. Pharmacol. 2011, 58, 199–206. [Google Scholar] [CrossRef]
- Hall, G.; Singh, I.S.; Hester, L.; Hasday, J.D.; Rogers, T.B. Inhibitor-kappaB kinase-beta regulates LPS-induced TNF-alpha production in cardiac myocytes through modulation of NF-kappaB p65 subunit phosphorylation. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2103–H2111. [Google Scholar] [CrossRef]
- Ghigo, A.; Franco, I.; Morello, F.; Hirsch, E. Myocyte signalling in leucocyte recruitment to the heart. Cardiovasc Res. 2014, 102, 270–280. [Google Scholar] [CrossRef]
- Shahzad, S.; Mateen, S.; Hasan, A.; Moin, S. GRACE score of myocardial infarction patients correlates with oxidative stress index, hsCRP and inflammation. Immunobiology 2019, 18, 30212-2. [Google Scholar] [CrossRef]
- Wang, X.; Guo, Z.; Ding, Z.; Mehta, J.L. Inflammation, Autophagy, and Apoptosis after Myocardial Infarction. J. Am. Heart Assoc. 2018, 7, e008024. [Google Scholar] [CrossRef]
- Oleksowicz, L.; Mrowiec, Z.; Isaacs, R.; Dutcher, J.P.; Puszkin, E. Morphologic and ultrastructural evidence for interleukin-6 induced platelet activation. Am. J. Hematol. 1995, 48, 92–99. [Google Scholar] [CrossRef]
- Yang, S.; Chou, G.; Li, Q. Cardioprotective role of azafrin in against myocardial injury in rats via activation of the Nrf2-ARE pathway. Phytomedicine 2018, 47, 12–22. [Google Scholar] [CrossRef]
- Pechanova, O.; Bernatova, I.; Pelouch, V.; Simko, F. Protein remodelling of the heart in NO-deficient hypertension: The effect of captopril. J. Mol. Cell Cardiol. 1997, 29, 3365–3374. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Week 0 (mmHg) | Week 4 (mmHg) | Week 5 (mmHg) | |
|---|---|---|---|
| WKY + Sham | 128 ± 3 | 119 ± 5 | 123 ± 4 |
| WKY + MI | 128 ± 3 | 133 ± 8 | 130 ± 8 |
| L-NAME + Sham | 128 ± 3 | 158 ± 5 ** | 161 ± 5 ** |
| L-NAME + MI | 128 ± 3 | 160 ± 3** | 163 ± 4 ** |
| BW (g) | HW (mmHg) | LKW (g) | |
|---|---|---|---|
| WKY + Sham | 304.20 ± 8.96 | 1.183 ± 0.24 | 1.065 ± 0.03 |
| WKY + MI | 301.67 ± 7.57 | 1.097 ± 0.08 | 1.186 ± 0.05 |
| L-NAME + Sham | 297.00 ± 4.94 | 1.37 ± 0.03 | 1.211 ± 0.06 |
| L-NAME + MI | 295.00 ± 5.87 | 1.059 ± 0.01 | 1.132 ± 0.05 |
| TNF/α [pg/mL] | IL-6 [pg/mL] | |
|---|---|---|
| WKY + Sham | 12.11 ± 2.41 | 22.41 ± 3.78 |
| WKY + MI | 27.72 ± 3.2 ** | 40.03 ± 2.51 ** |
| L-NAME + Sham | 18.19 ± 1.38 | 36.43 ± 3.08 |
| L-NAME + MI | 41.92 ± 2.71 ## | 53.17 ± 3.24 ## |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosutova, M.; Pechanova, O.; Barta, A.; Franova, S.; Cebova, M. Different adaptive NO-dependent Mechanisms in Normal and Hypertensive Conditions. Molecules 2019, 24, 1682. https://doi.org/10.3390/molecules24091682
Kosutova M, Pechanova O, Barta A, Franova S, Cebova M. Different adaptive NO-dependent Mechanisms in Normal and Hypertensive Conditions. Molecules. 2019; 24(9):1682. https://doi.org/10.3390/molecules24091682
Chicago/Turabian StyleKosutova, Michaela, Olga Pechanova, Andrej Barta, Sona Franova, and Martina Cebova. 2019. "Different adaptive NO-dependent Mechanisms in Normal and Hypertensive Conditions" Molecules 24, no. 9: 1682. https://doi.org/10.3390/molecules24091682
APA StyleKosutova, M., Pechanova, O., Barta, A., Franova, S., & Cebova, M. (2019). Different adaptive NO-dependent Mechanisms in Normal and Hypertensive Conditions. Molecules, 24(9), 1682. https://doi.org/10.3390/molecules24091682