
Peculiar Structural Effects in Pure and Doped Functional Single Crystals of Complex Compositions



Abstract
:1. Introduction
2. Functional Crystals of Complex Compositions: General Information
2.1. Scheelite Family Compounds
2.1.1. PbTO4 (T = Mo,W), Pb(Mo,W)O4, PbMoO4:Nd3+
2.1.2. SrMoO4 and SrMoO4:RE3+
2.1.3. (Na,RE)TO4 (T = Mo, W; RE3+ = La,Gd)
2.2. Eulytin Family Compounds
2.3. Perovskite Family Compounds and Others
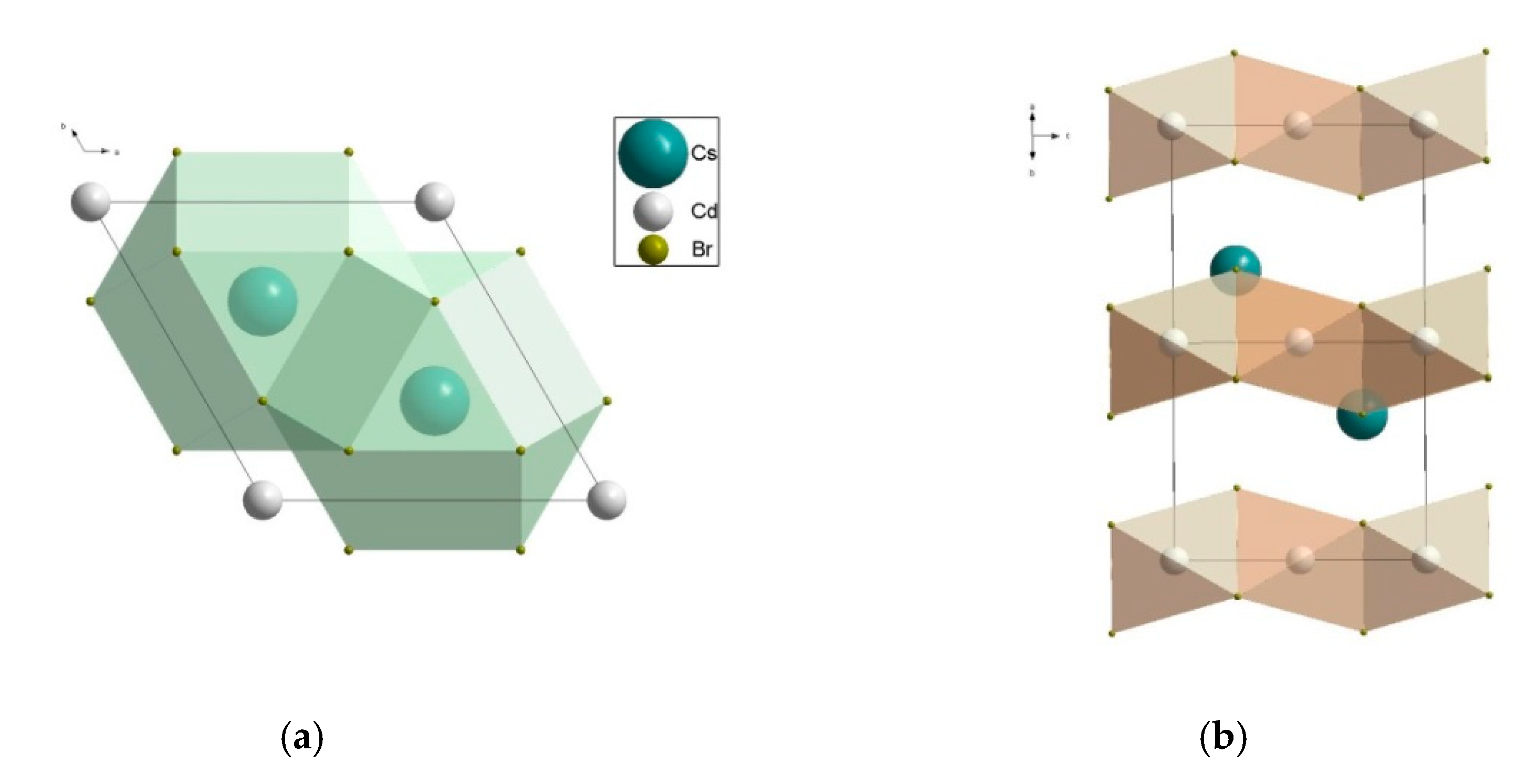
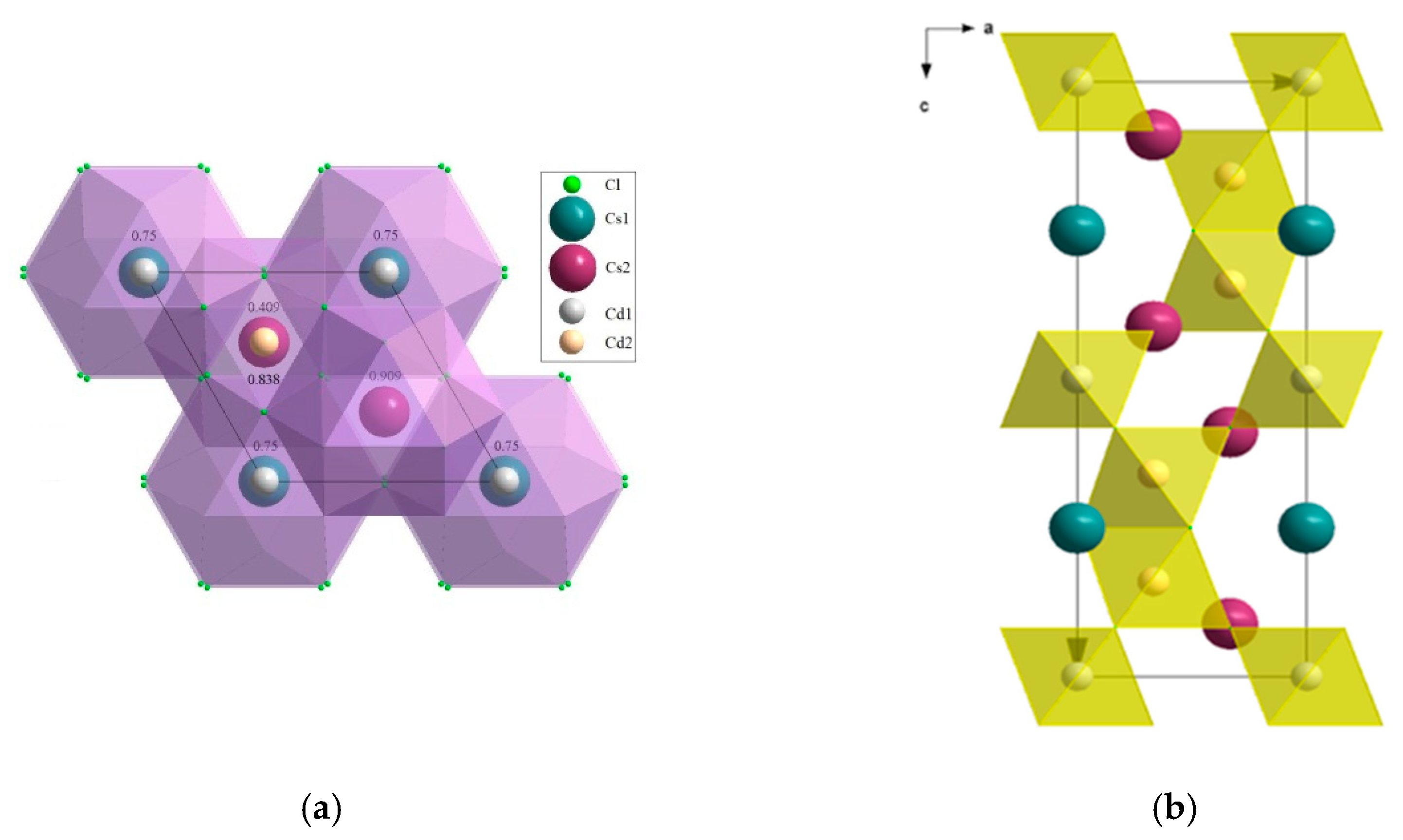
2.3.1. CsCdX3 and CsCdX3:Bi (X = Cl, Br)
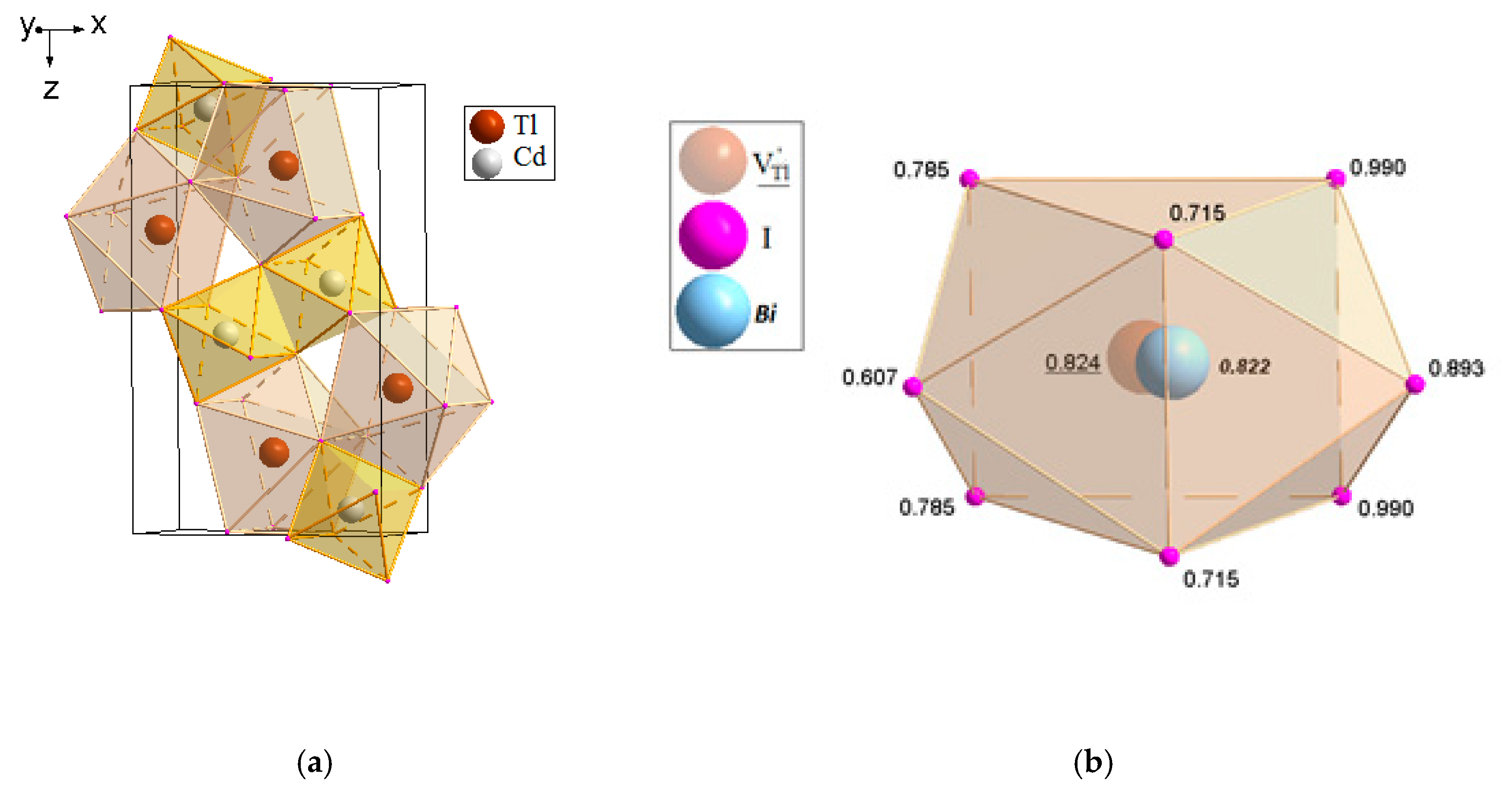
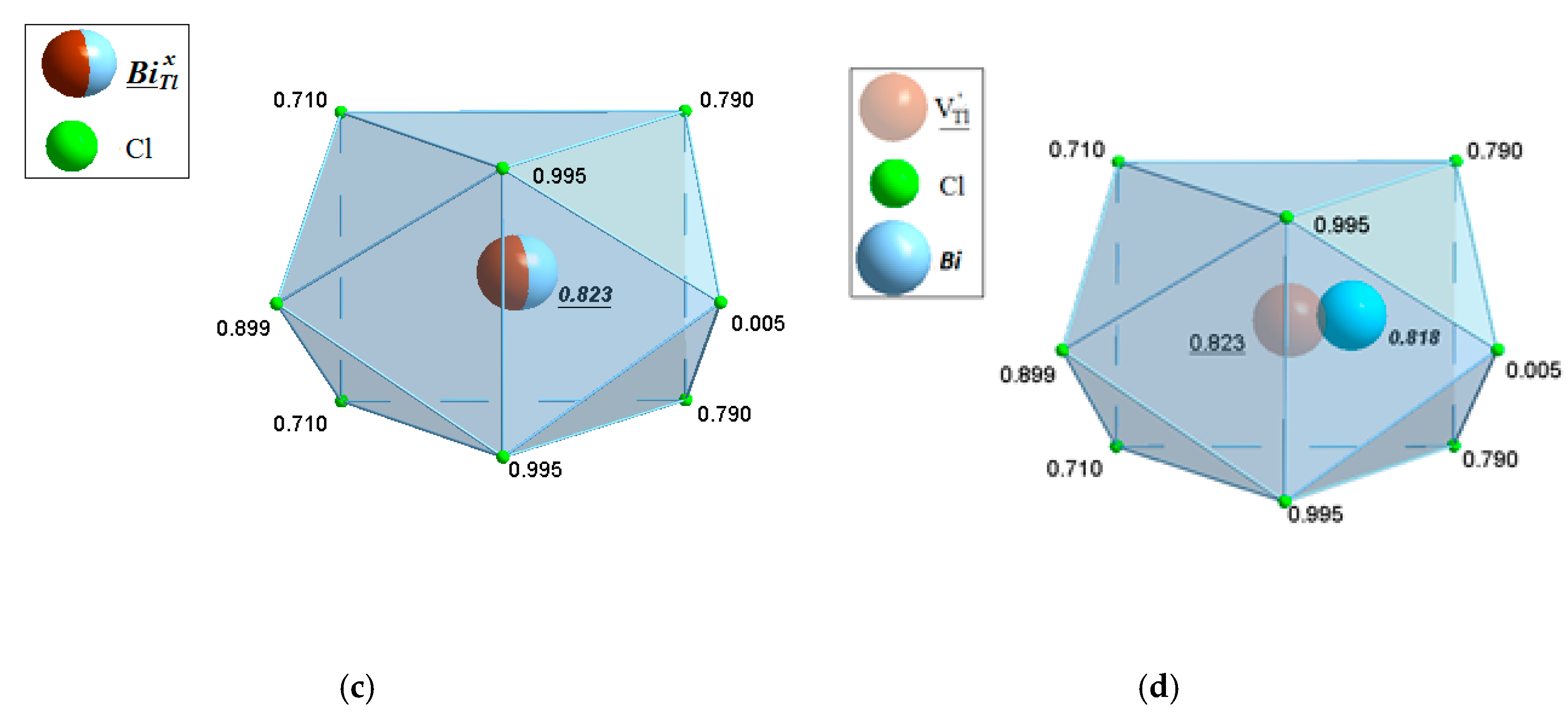
2.3.2. TlCdX3 and TlCdX3:Bi (X = Cl, I)
3. Growth and Investigation Methods
3.1. Crystal Growth
3.1.1. Scheelite Family Compounds
3.1.2. Eulytin Family Compounds
3.1.3. Perovskite Family Compounds and Others
3.2. Characterization Methods
3.2.1. Single-Crystal X-Ray and Neutron Diffraction
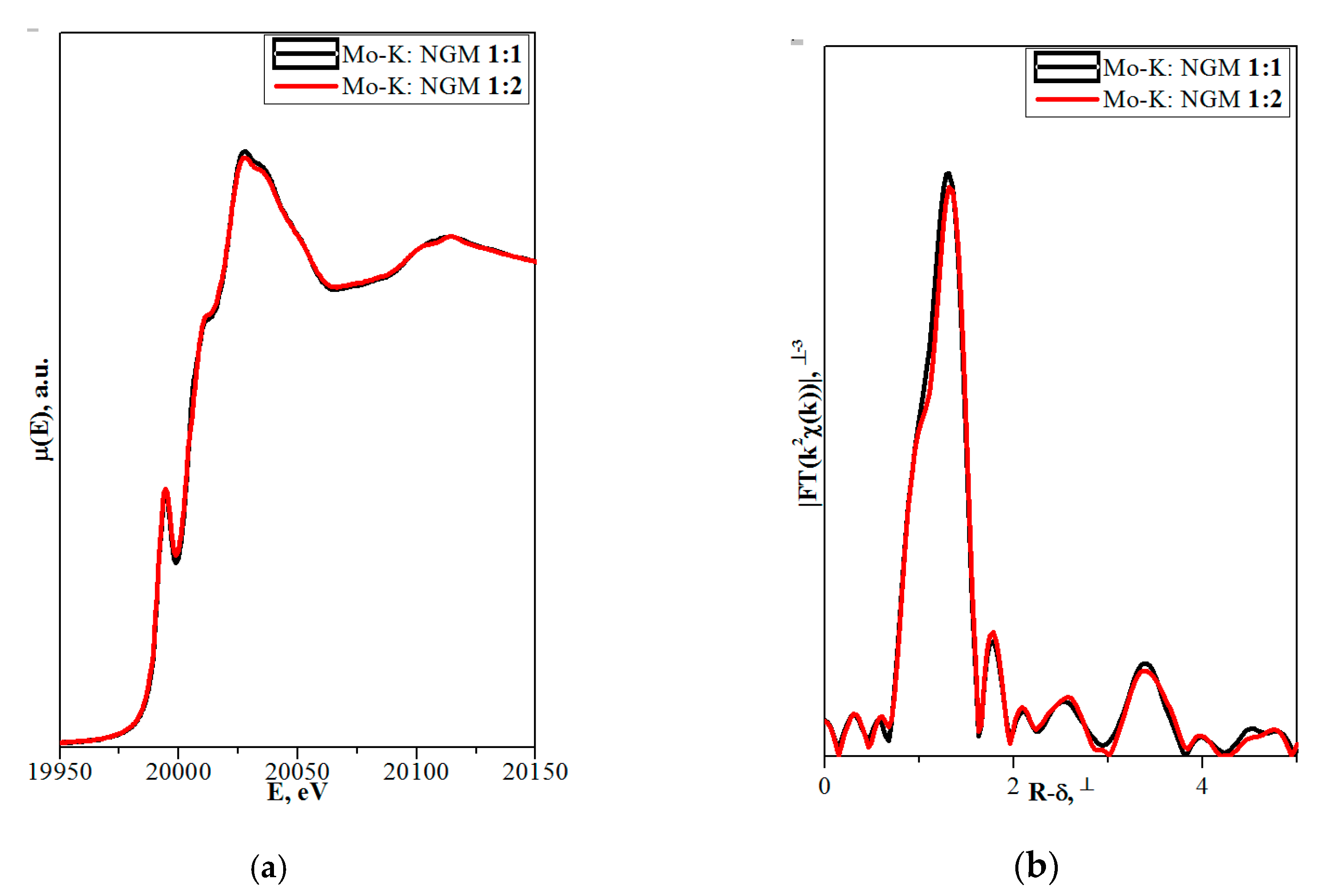
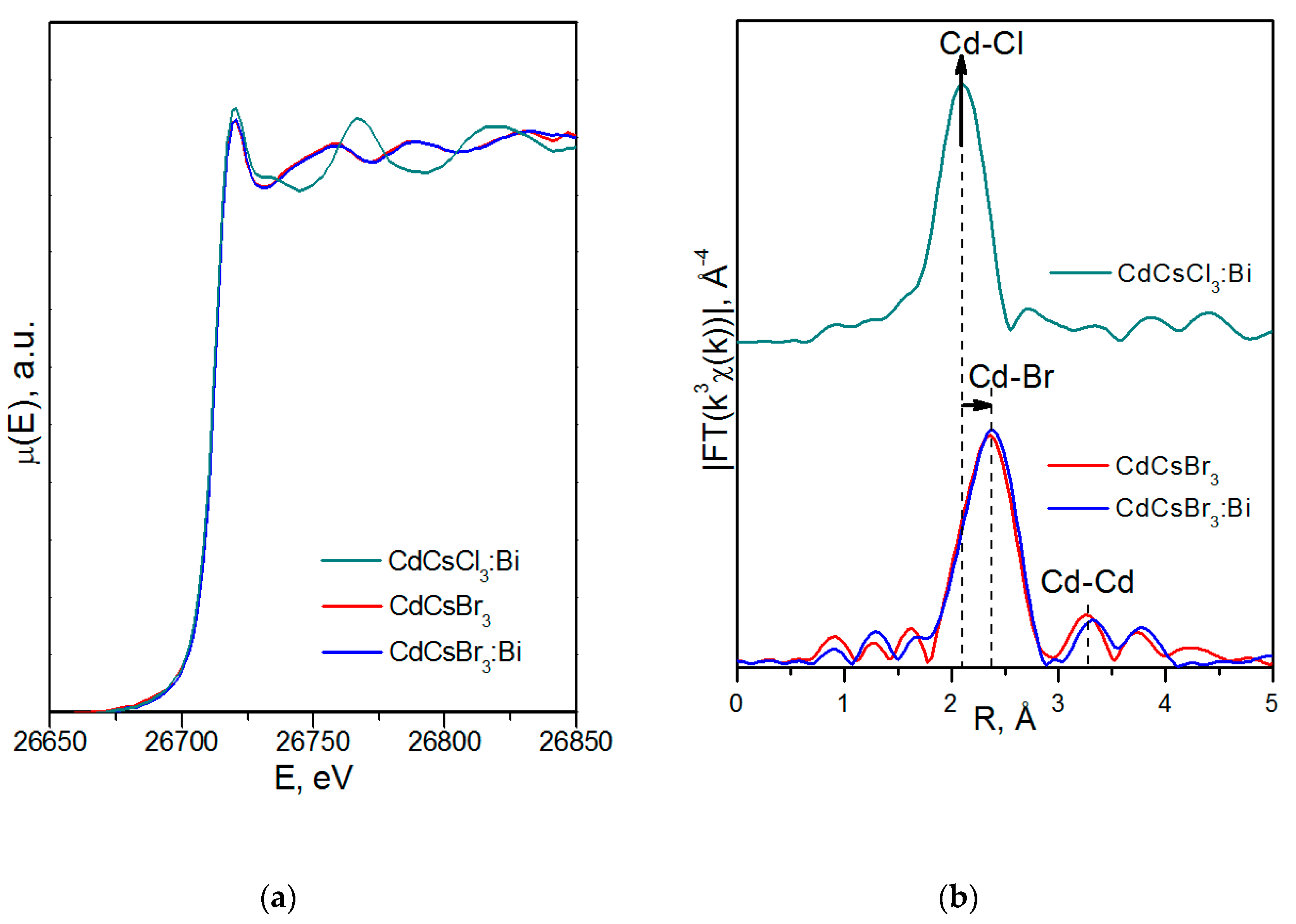
3.2.2. X-Ray Absorption Spectroscopy
3.3. Functional Properties
4. Structural Features in Functional Crystals of Complex Compositions
4.1. Scheelite Family Compounds: PbTO4, SrMoO4, (Na,RE3+)TO4 (T = Mo, W; RE3+ = La,Gd)
4.1.1. PbTO4 (T = Mo,W), Pb(Mo,W)O4, PbMoO4:Nd3+
PbTO4 (T = Mo,W)
Pb(Mo,W)O4 Solid Solutions
PbMoO4:Nd3+
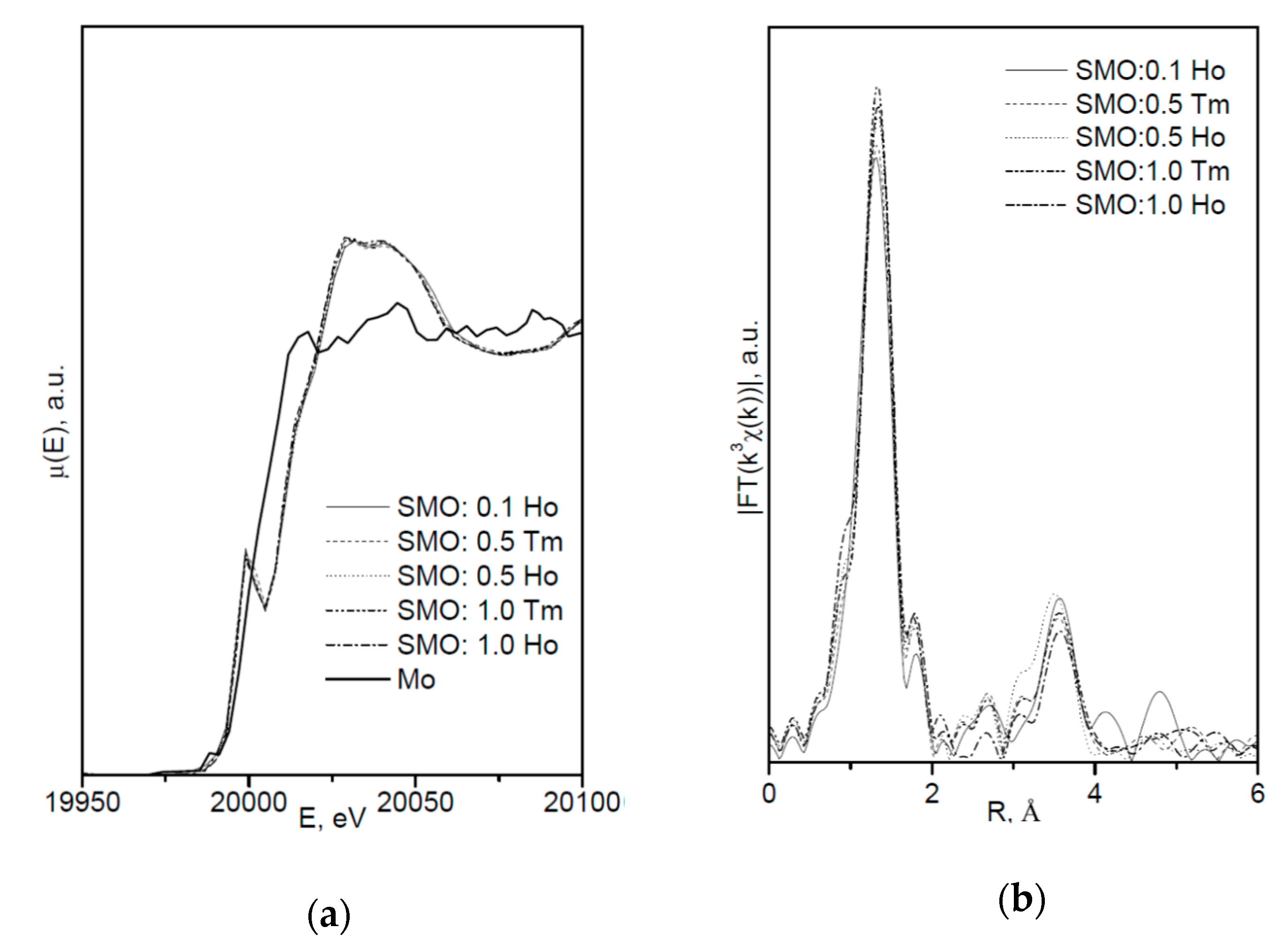
4.1.2. SrMoO4 and SrMoO4:RE (RE3+ = Ho,Tm)
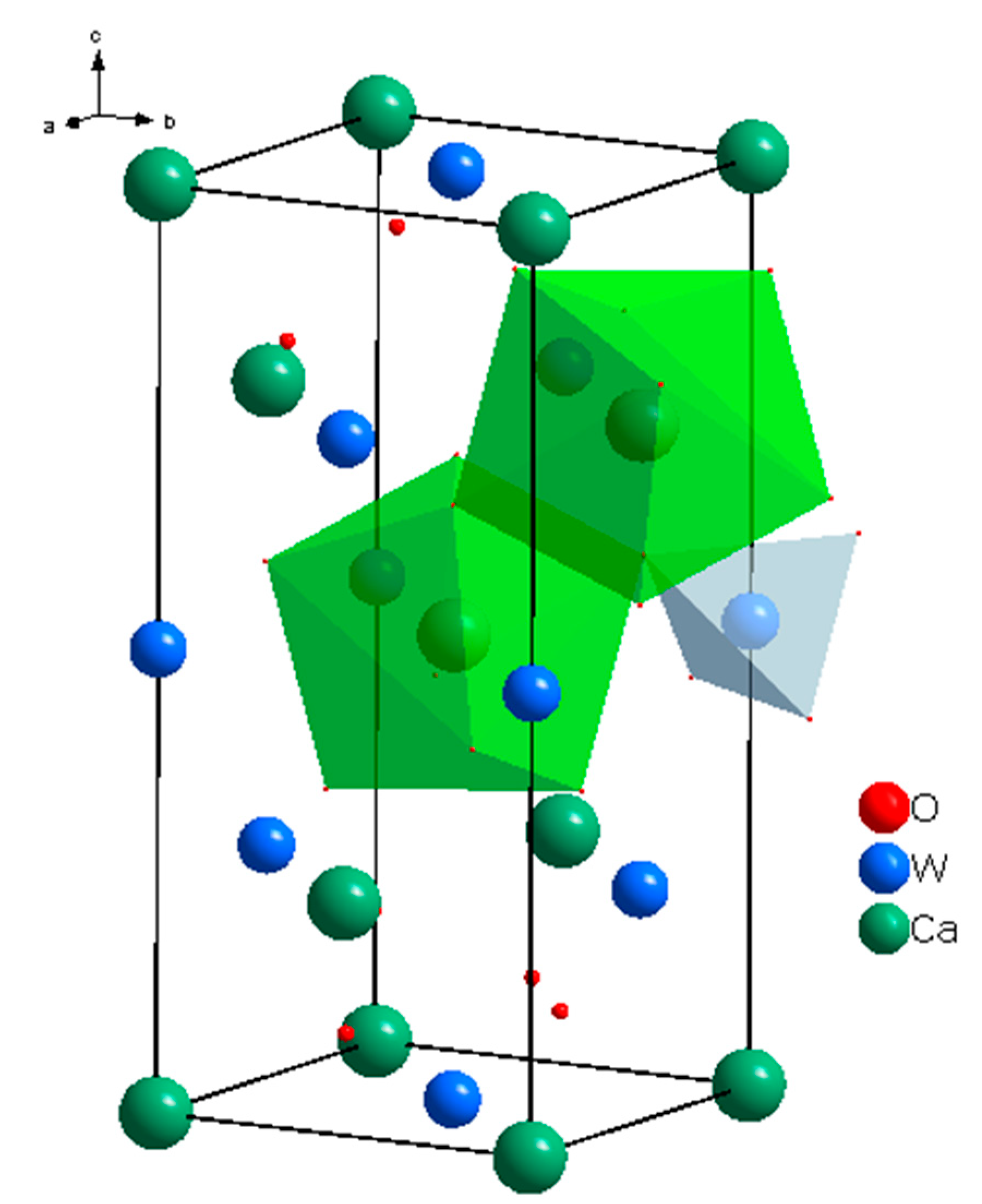
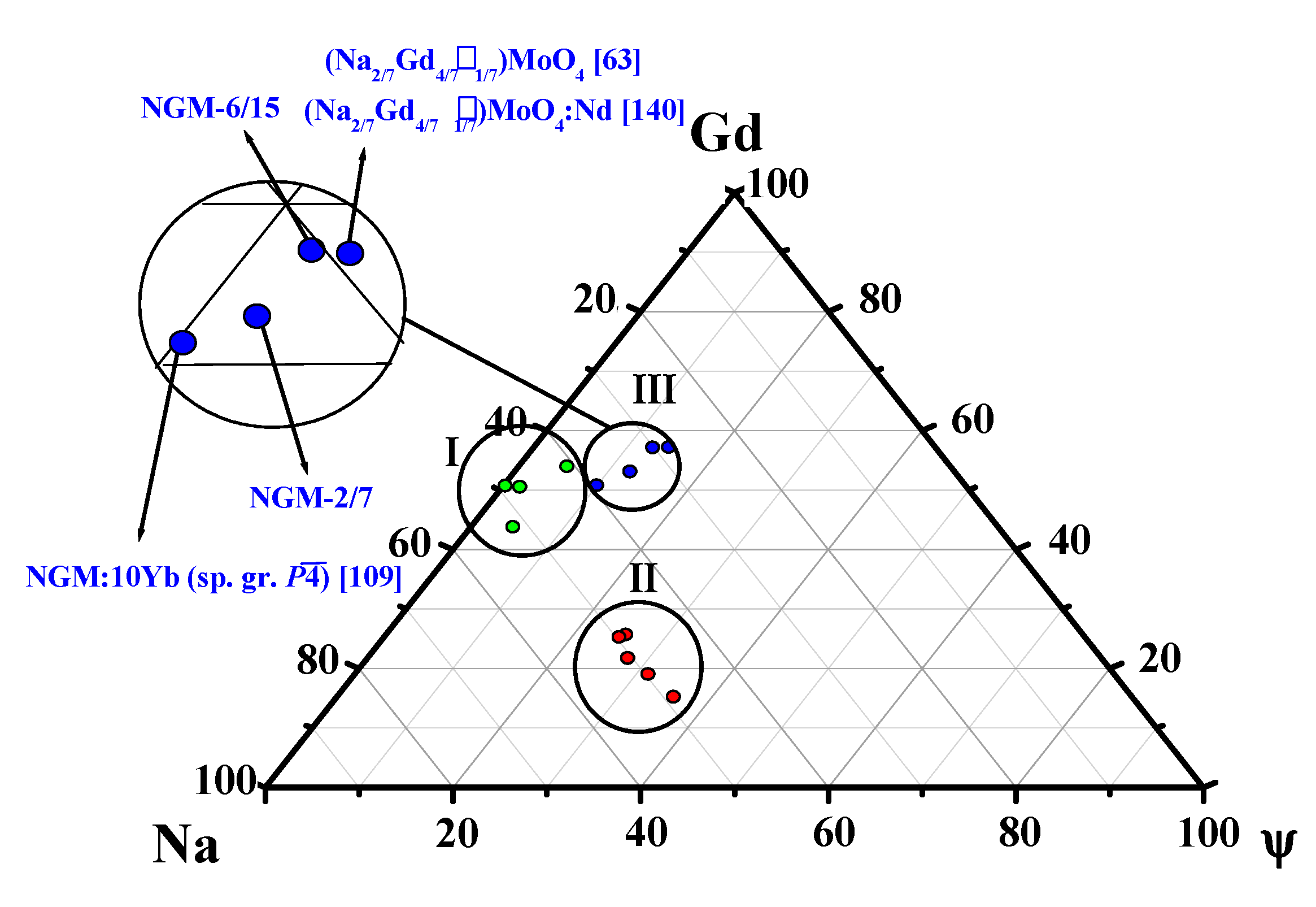
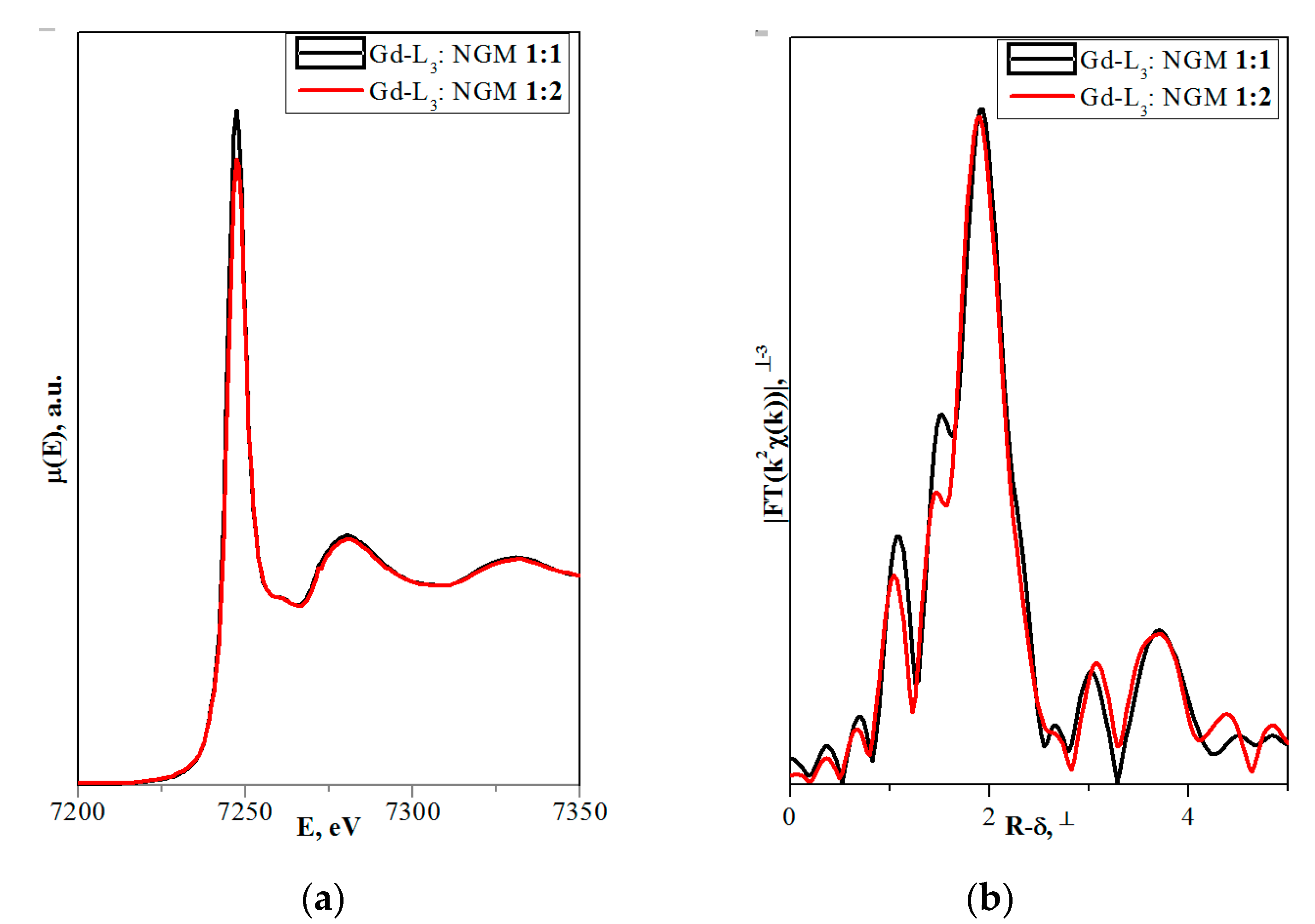
4.1.3. (Na,RE)TO4 (T = Mo, W; RE3+ = La, Gd)
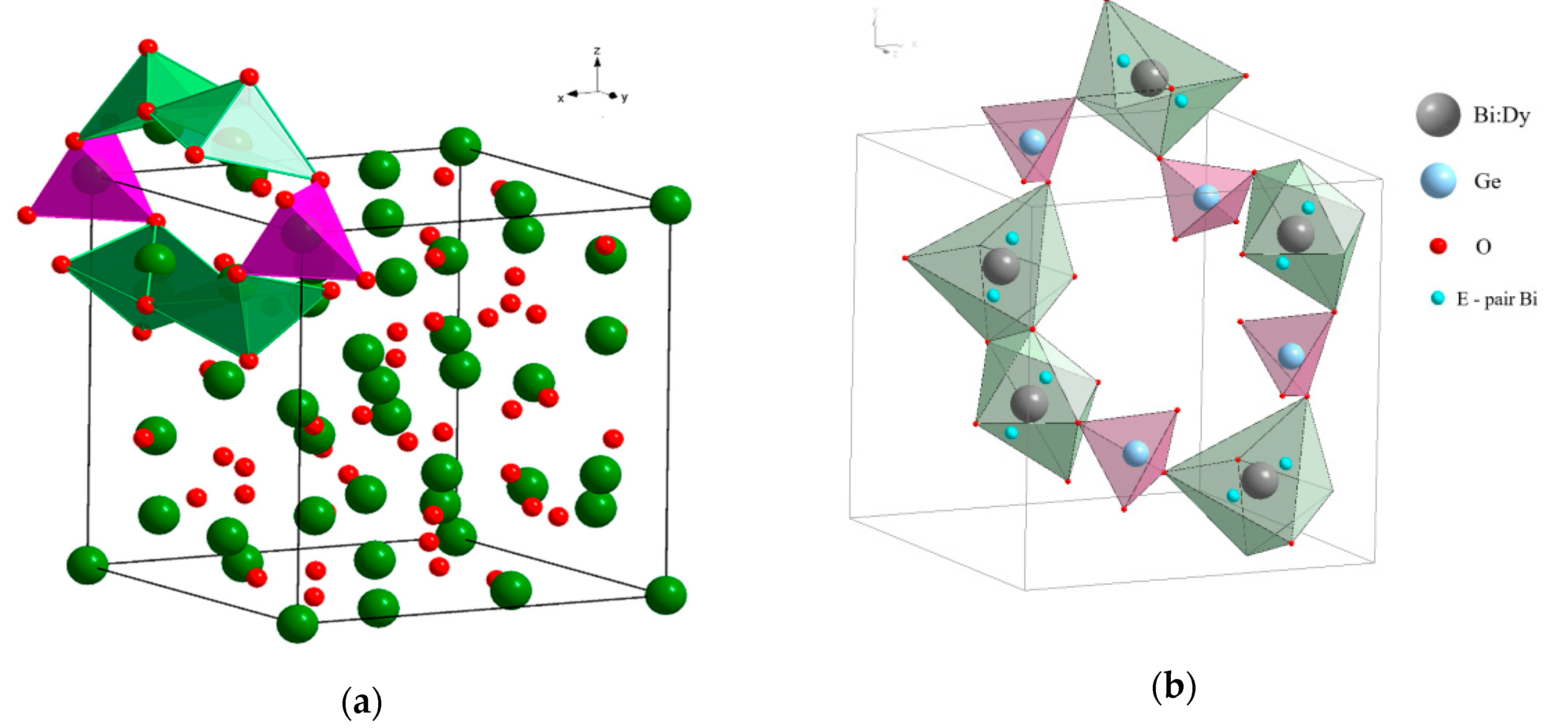
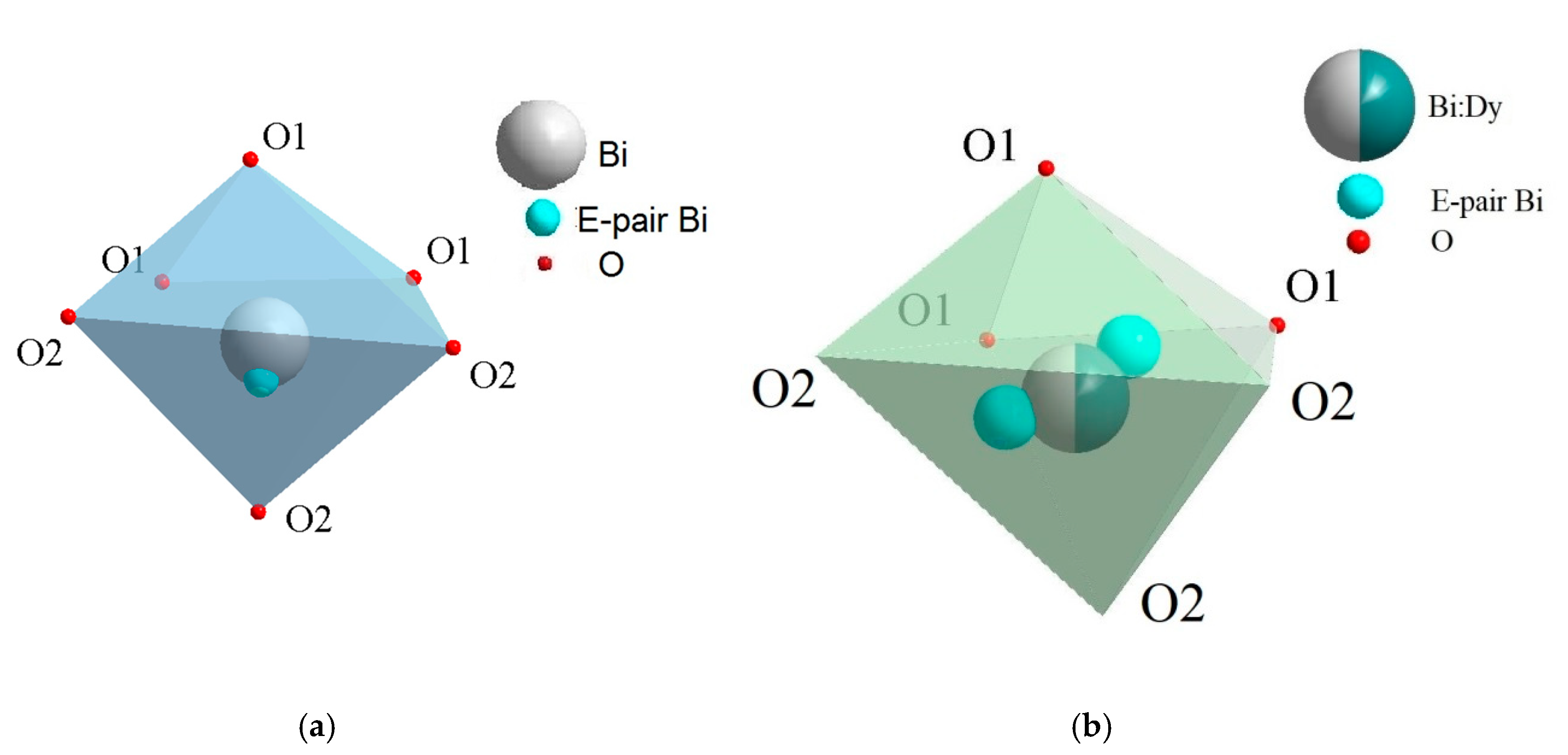
4.2. Eulytin Family Compound Bi4Ge3O12
4.3. Perovskite Family Compound CsCdX3 (X = Cl, Br) as well as TlCdX3 (X = Cl, I)
4.3.1. CsCdX3 (X = Cl, Br)
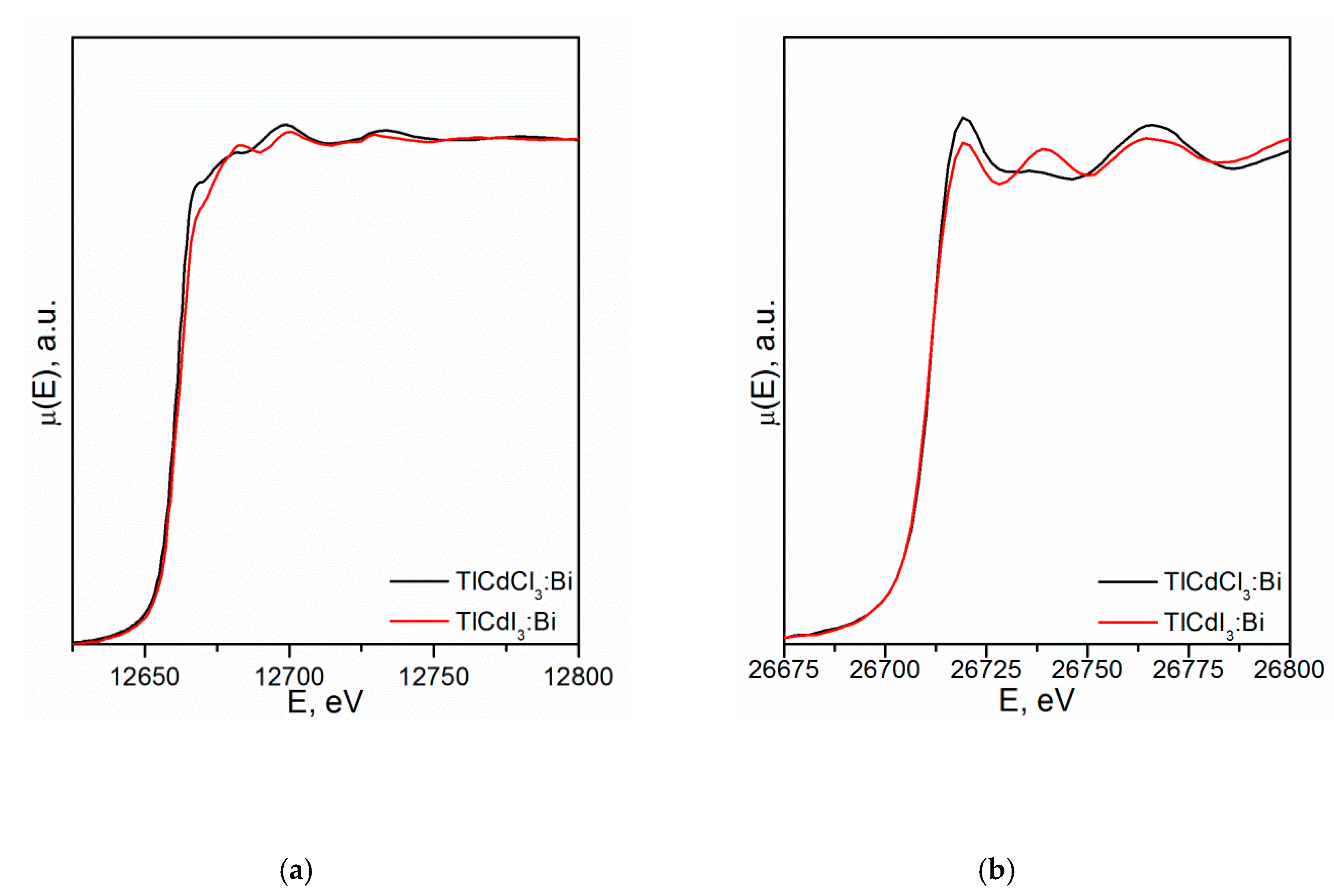
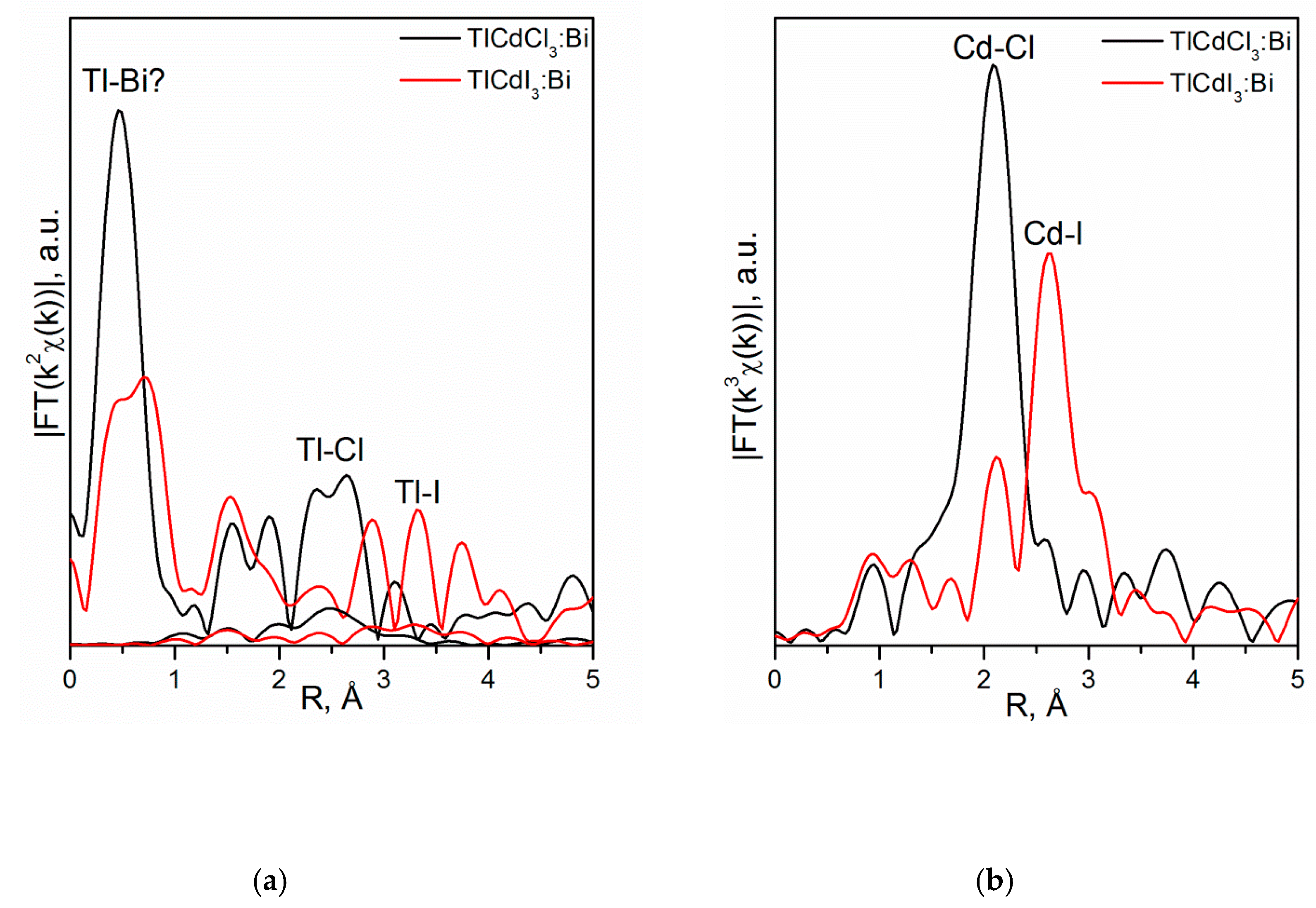
4.3.2. TlCdX3 (X = Cl, I)
5. Summary
- CsCdX3 (X = Cl, Br) (perovskite family, space group P63/mmc) with the refined actual compositions (Cs(1)0.989(1)⎕0.011)Cs(2)(Cd2+(1)0.995(2)Bi3+(1)0.005)(Cd2+(2)0.996(2)Bi3+(2)0.004)Cl6 (a = 7.418(4), c = 18.39(3) Å) and (Cs0.976(10)⎕0.024)(Cd0.967(47)Bi3+0.033)Br3 (a = 7.675(3), c = 6.722(3) Å) (⎕-vacancies);
- TlCdX3 (X = Cl, I) (space group Pnma) with the refined actual compositions Tl1+0.980(12)Bi1+(i)0.014(12)CdI3 and (Tl1+,Bi1+)0.998(1)Bi1+(i)0.005(1)CdCl3.
- (K1+0.994(18)⎕0.006)Ti4+(i)0.006(H0.964(22)⎕0.036)2[(P5+O4)0.946(S6+O4)0.054] (sulfate synthesis of η-TiO2; P),
- (K1+0.963(10)⎕0.037)Ti4+(i)0.037(H0.914(12)⎕0.086)2[(P5+O4)0.939(S6+O4)0.061] (sulfate synthesis of η-TiO2; Pr), described by a quasi-chemical reaction: 0 → VK′ + Tin• + SP• + VH′ (Equation (27));
- (K1+0.994(2)⎕0.004)Ti4+(i)0.002H2(P5+O4) (chloride synthesis of anatase; P),
- (K1+0.990(6)⎕0.010)Ti4+(i)0.003H2(P5+O4) (chloride synthesis of anatase; Pr), described by a quasi-chemical reaction: 0 ® VK′ + Tin• (Equation (28)).
- (Sr2+0.998(2)Ho3+0.002)[(Mo6+,Nb5+)0.998⎕0.002]Nb5+(i)0.002(O3.96(3))(1)(O0.002+x)(2) (Δr/rmin < 2 5%),
- (Sr2+0.992(3)⎕0.008)Tm3+(i)0.008[(Mo6+,Nb5+)0.992⎕0.008]Nb5+(i)0.008(O3.80(4))(1)(O0.008+x)(2) (Δr/rmin > 25%) with new sites for interstitial oxygen–O(2). The greater disordering of the SrMoO4:Tm structure made it possible to propose this crystal as a promising material for tunable (~2 μm) lasers pumped by a diode laser (1700 nm) [95].
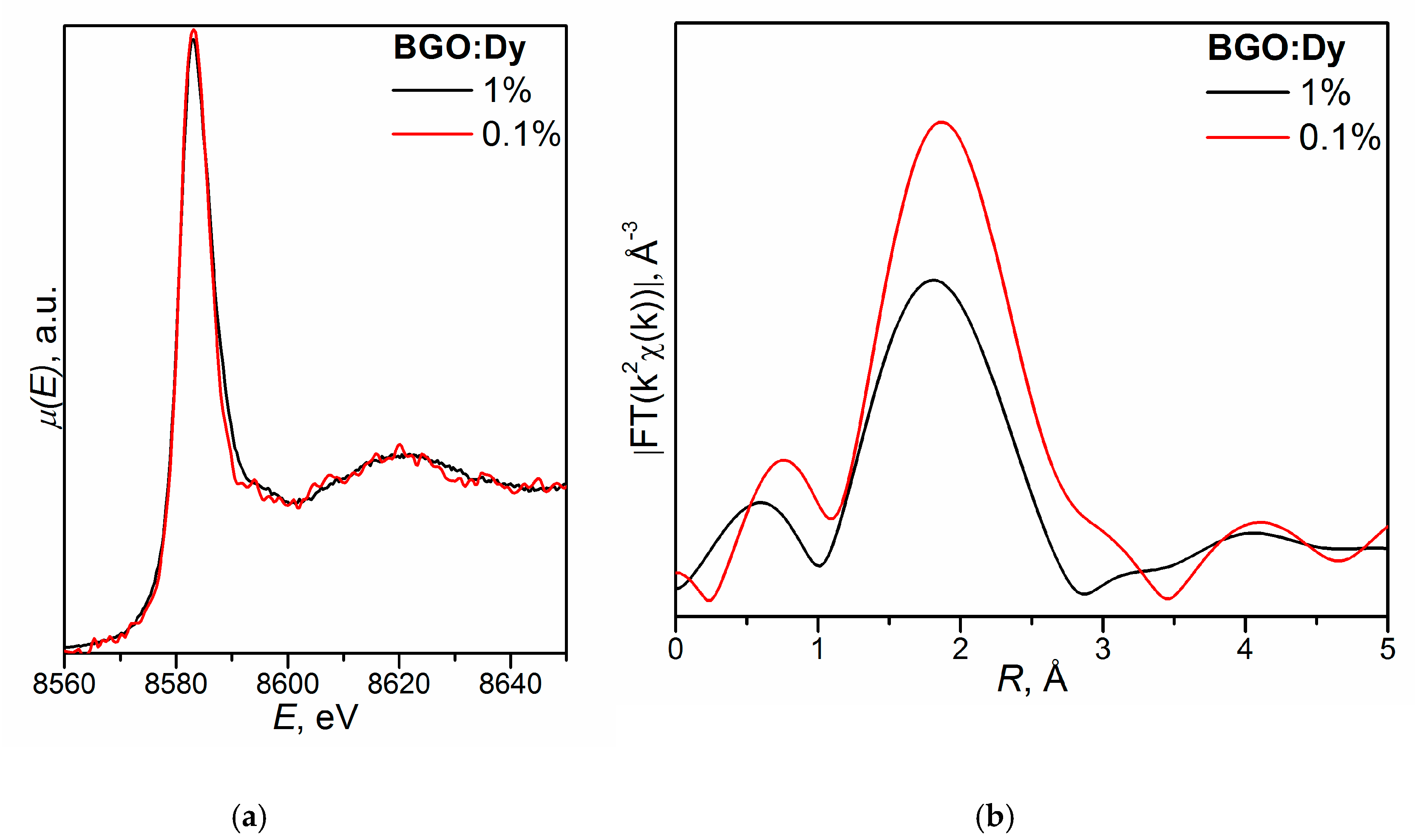
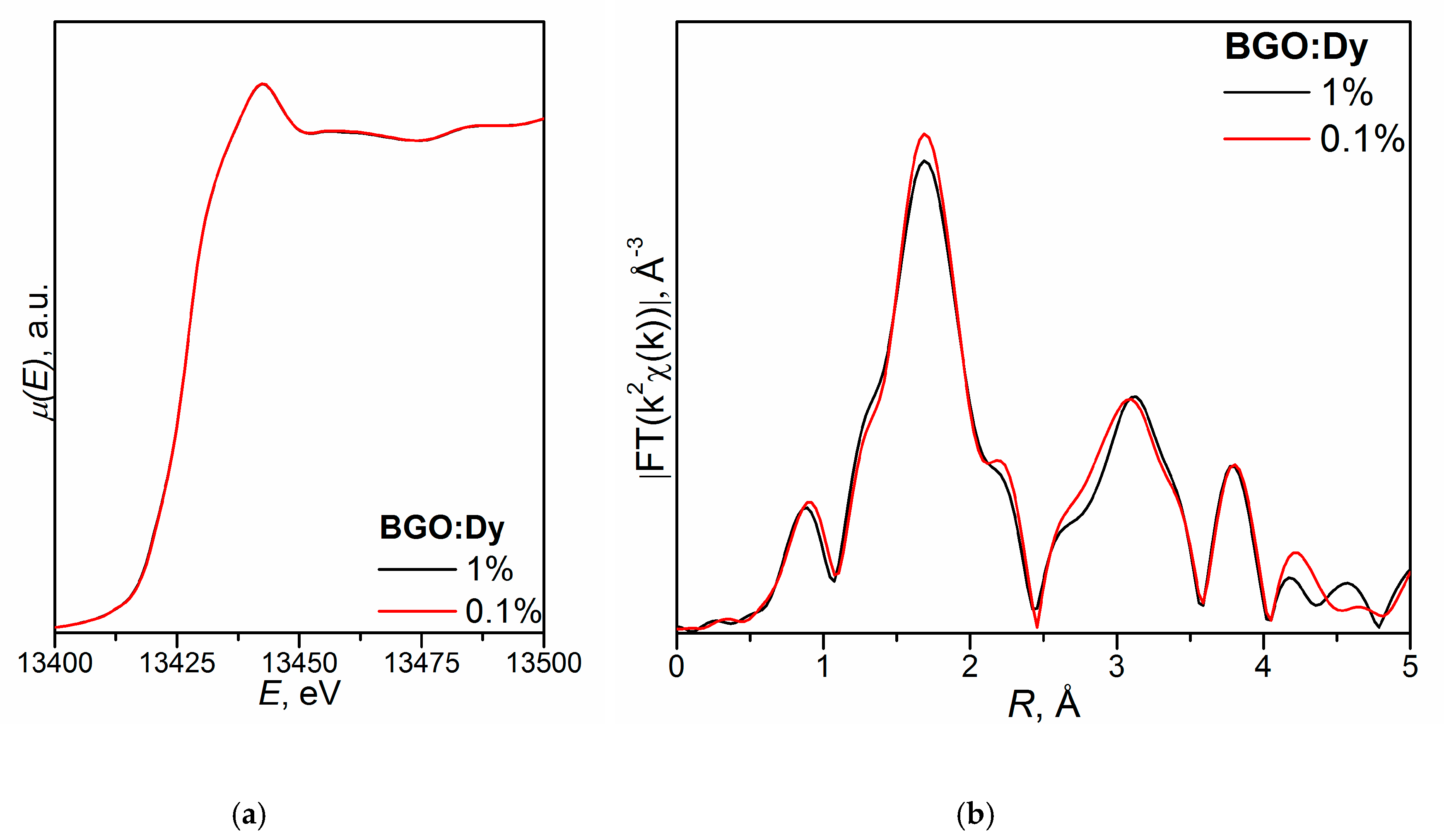
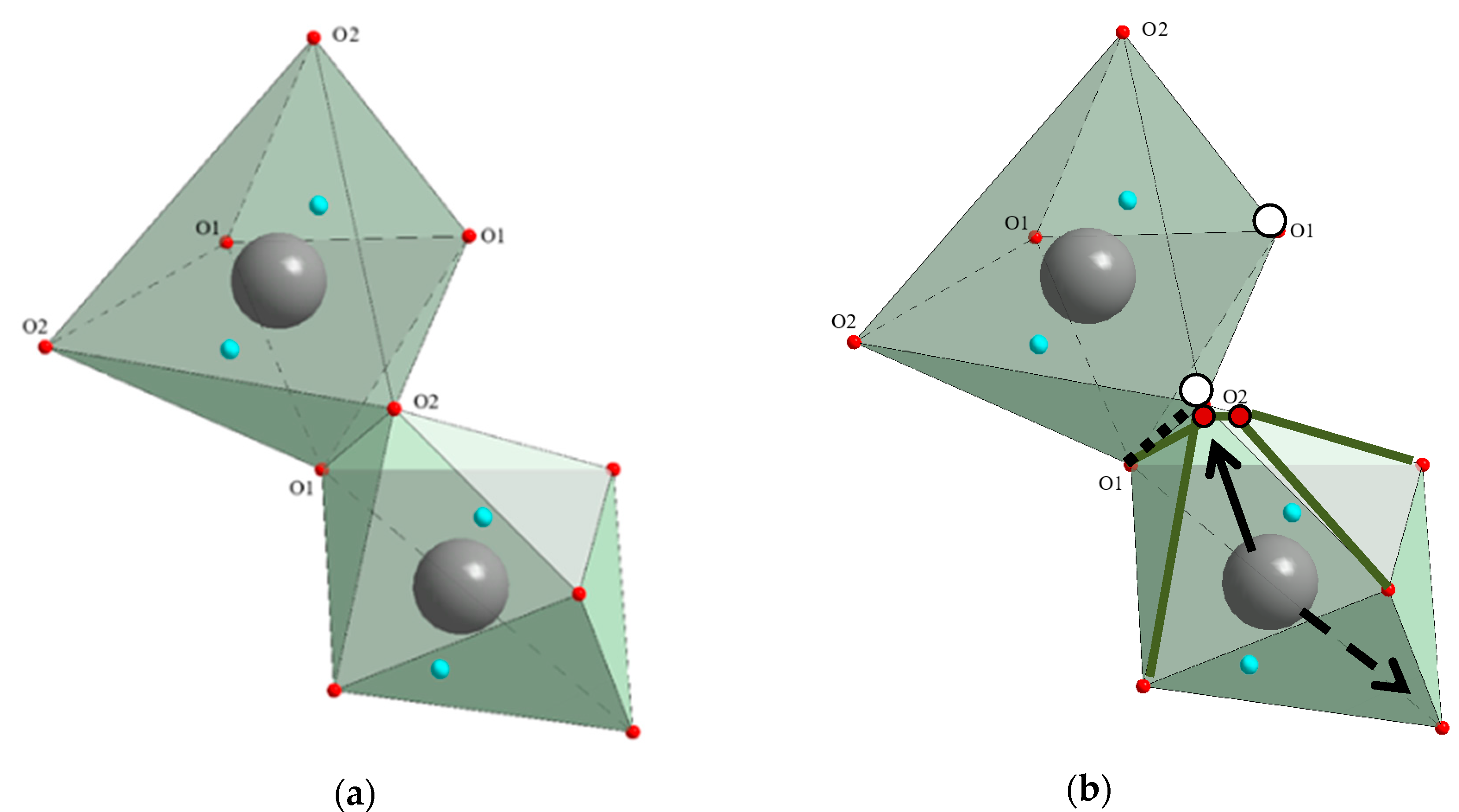
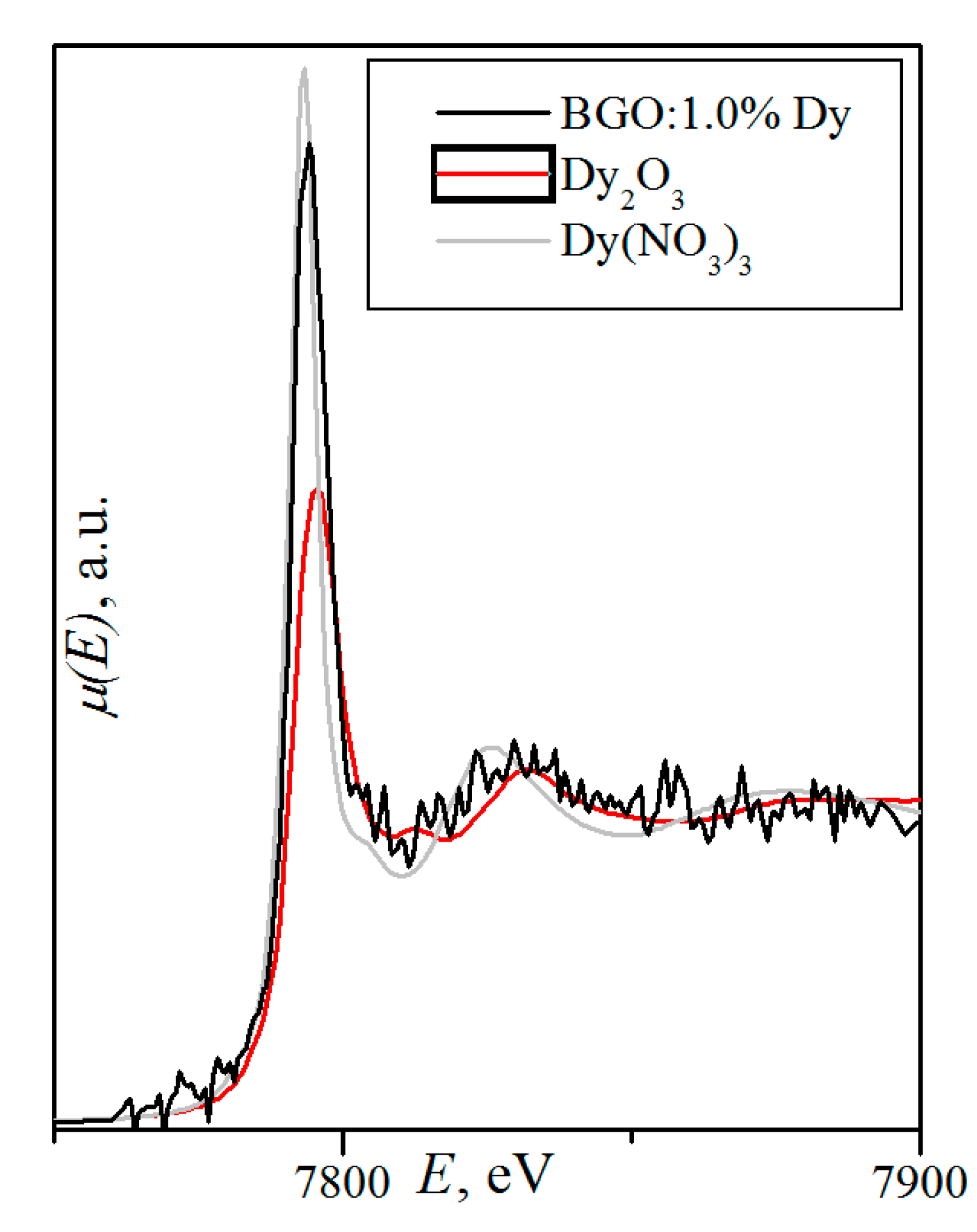
- (Bi3+3.993(5)Dy0.007)Ge3O12 (BGO:0.1% Dy; the DyO7 polyhedron is a derivative of a trigonal prism; the BiO5 polyhedron is a defective octahedron),
- (Bi3+3.972(5)Dy0.028)Ge3O12 (BGO:1.0% Dy3+; the DyO5 polyhedron is a semi-octahedron or polyhedron derived from a trigonal prism; the BiO6 polyhedron is a distorted octahedron) [120].
- SBN:0.5% Ni—Sr(1)0.284(1)Sr(2)0.341(2)Ba(2)0.362(2)Nb(1)(Nb(2)0.975(1)⎕0.025)Ni3+(i)0.025(O5.962⎕0.038);
- SBN:1.0% Ni- Sr(1)0.300(1)Sr(2)0.384(1)Ba(2)0.347(2)Nb(1)(Nb(2)0.972(1)⎕0.028)Ni2+(i)0.028(O5.982(8)⎕0.018).
- (Na,Gd)WO4:Yb—from the space group I41/a to in the local region (up to 70%) of the crystal with increasing Yb content (up to 10%);
6. Conclusions
7. Patents
- Kuzmicheva, G.M.; Podbelsky, V.V.; Chuykin, N.K.; Kaurova, I.A. A software package for studying the dynamics of changes in the structural parameters for compounds with different symmetries. Certificate of state registration of software package No. 2017619941 [Russ.].
- Kuzmicheva, G.M.; Podbelsky, V.V.; Nemashkalo, M.A. A software package for calculating the dynamics of changes in coordination polyhedra during isomorphic substitution in simple scheelites. Certificate of state registration of software package No. 2017610700 [Russ.].
Author Contributions
Funding
Conflicts of Interest
References
- Uda, S. Stoichiometry of oxide crystals. In Handbook of Crystal Growth, 2nd ed.; Nishinaga, T., Ed.; Elsevier: Amsterdam, Netherlands, 2015; pp. 175–214. [Google Scholar]
- Almessiere, M.A.; Slimani, Y.; Tashkandi, N.A.; Baykal, A.; Saraç, M.F.; Trukhanov, A.V.; Ercanb, İ.; Belenlif, İ.; Ozçelik, B. The effect of Nb substitution on magnetic properties of BaFe12O19 nanohexaferrites. Ceram. Int. 2019, 45, 1691–1697. [Google Scholar] [CrossRef]
- Panchuk, O.; Fochuk, P. Doping. In CdTe and Related Compounds; Physics, Defects, Hetero-and Nano-Structures, Crystal Growth, Surfaces and Applications; Triboulet, R., Siffert, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 309–362. [Google Scholar]
- Urusov, V.S. Theory of Isomorphic Mixing Ability; Nauka: Moscow, USSR, 1977. (In Russian) [Google Scholar]
- Rosenberger, F.E. Crystal Growth and Phase Diagrams. In Fundamentals of Crystal Growth I. Springer Series in Solid-State Sciences; Springer: Berlin, Germany, 1979; Volume 5, pp. 145–214. [Google Scholar]
- Kuz’micheva, G.; Svetogorov, R.; Kaurova, I. On symmetry of rare-earth scandium borate RESc3(BO3)4 (RE = Ce, Nd) laser crystals. J. Solid State Chem. 2020, 288, 121393. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M. Some Aspects of the Applied Crystallochemistry; MIREA: Moscow, Russia, 2016. [Google Scholar]
- Kaurova, I.A.; Domoroshchina, E.N.; Kuz’micheva, G.M.; Rybakov, V.B. Evaluation of stability region for scandium-containing rare-earth garnet single crystals and their congruent-melting compositions. J. Cryst. Growth 2017, 468, 452–456. [Google Scholar] [CrossRef]
- Pritula, I.M.; Kolybayeva, M.I.; Salo, V.I.; Puzikov, V.M. Defects of large-size KDP single crystals and their influence on degradation of the optical properties. Opt. Mater. 2007, 30, 98–100. [Google Scholar] [CrossRef]
- Kosinova, A.V.; Kolybaeva, M.I.; Bezkrovnaya, O.N.; Tkachenko, V.F.; Grishina, E.V.; Levchenko, A.N.; Puzikov, V.M.; Pritula, I.M. Structural and mechanical properties of KH2PO4 single crystals with embedded nanoparticles and organic molecules. Cryst. Res. Technol. 2014, 49, 965–974. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Timaeva, O.I.; Kaurova, I.A.; Svetogorov, R.D.; Mühlbauer, M.J. KH2PO4 single crystals activated with the Ti4+ ions in the form of TiO2−x×nH2O nanoparticles: Structural peculiarities, point defects, and dielectric properties. Appl. Phys. Lett. 2018, 112, 121901. [Google Scholar] [CrossRef]
- Krayzman, V.; Levin, I.; Woicik, J.C.; Proffen, T.; Vanderah, T.A.; Tucker, M.G. A combined fit of total scattering and extended X-ray absorption fine structure data for local-structure determination in crystalline materials. J. Appl. Crystallogr. 2009, 42, 867–877. [Google Scholar] [CrossRef]
- Penner-Hahn, J.E. X-ray Absorption Spectroscopy. In Comprehensive Coordination Chemistry II, 2nd ed.; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 2, pp. 159–186. [Google Scholar]
- Aleksandrov, V.B.; Gorbatyii, L.V.; Ilyukhin, V.V. Crystal structure of powellite CaMoO4. Sov. Phys. Crystallogr. 1968, 13, 512–513. [Google Scholar]
- Zhang, Y.; Holzwarth, N.A.W.; Williams, R.T. Electronic band structures of the scheelite materials CaMoO4, CaWO4, PbMoO4, and PbWO4. Phys. Rev. B 1998, 57, 12738. [Google Scholar] [CrossRef]
- Gürmen, E.; Daniels, E.; King, J.S. Crystal structure refinement of SrMoO4, SrWO4, CaMoO4, and BaWO4 by neutron diffraction. J. Chem. Phys. 1971, 55, 1093–1097. [Google Scholar] [CrossRef] [Green Version]
- Kuz’micheva, G.M.; Eremin, A.V.; Rybakov, V.B.; Subbotin, K.A.; Zharikov, E.V. Structural features of phases (Na0.5R0.5)MO4 and (Na0.5R0.5)MO4:R′ (R = Gd, La; R′ = Er, Tm, Yb; M = W, Mo) of the scheelite family. Russ. J. Inorg. Chem. 2009, 54, 854–863. [Google Scholar] [CrossRef]
- Spassky, D.; Ivanov, S.; Kitaeva, I.; Kolobanov, V.; Mikhailin, V.; Ivleva, L.; Voronina, I. Optical and luminescent properties of a series of molybdate single crystals of scheelite crystal structure. Phys. Status Solidi C 2005, 2, 65–68. [Google Scholar] [CrossRef]
- Basiev, T.T.; Doroshenko, M.E.; Ivleva, L.I.; Osiko, V.V.; Kosmyna, M.B.; Sulc, J.; Jelinkova, H. Lasing properties of selectively pumped Raman-active Nd3+-doped molybdate and tungstate crystals. Quantum Electron. 2006, 36, 720. [Google Scholar] [CrossRef]
- Sani, E.; Brugioni, A.; Mercatelli, L.; Parisi, D.; Zharikov, E.V.; Lis, D.A.; Subbotin, K.A. Yb-doped double tungstates for down-conversion applications. Opt. Mater. 2019, 94, 415–422. [Google Scholar] [CrossRef]
- Mikhailik, V.B.; Kraus, H. Cryogenic scintillators in searches for extremely rare events. J. Phys. D Appl. Phys. 2006, 39, 1181. [Google Scholar] [CrossRef] [Green Version]
- Nagornaya, L.L.; Danevich, F.A.; Dubovik, A.M.; Grinyov, B.V.; Henry, S.; Kapustyanyk, V.; Kraus, H.; Poda, D.; Mokina, V.M.; Mikhailik, V.B.; et al. Oxide scintillators to search for dark matter and double beta decay. IEEE Nucl. Sci. Symp. Conf. Rec. 2008, N55-7, 3266–3271. [Google Scholar]
- Danevich, F.A. Radiopure tungstate and molybdate crystal scintillators for double beta decay experiments. Int. J. Mod. Phys. A 2017, 32, 1743008. [Google Scholar] [CrossRef] [Green Version]
- Bain, A.K. Crystal Optics: Properties and Applications, 1st ed.; Wiley-VCH: Weinheim, Germany, 2019; pp. 1–208. [Google Scholar]
- Mantsevich, S.N.; Balakshy, V.I.; Kuznetsov, Y.I. Acousto-optic collinear filter with optoelectronic feedback. Appl. Phys. B 2017, 123, 101. [Google Scholar] [CrossRef]
- Khan, A.; Daniel, D.J.; Kim, H.; Pandey, I.R.; Shlegel, V.; Lee, M.H.; Kim, Y. Luminescence and scintillation characterization of PbMoO4 crystal for neutrinoless double beta decay search. Radiat. Meas. 2019, 123, 34–38. [Google Scholar] [CrossRef]
- Chanu, T.T.T.; Singh, N.R. Multicolor luminescence of PbWO4:RE3+ (Sm3+, Dy3+, Eu3+ and Tb3+) phosphor prepared via hydrothermal method. J. Mater. Sci. Mater. Electron. 2017, 28, 3909–3916. [Google Scholar] [CrossRef]
- Daturi, M.; Busca, G.; Borel, M.M.; Leclaire, A.; Piaggio, P. Vibrational and XRD study of the system CdWO4−CdMoO4. J. Phys. Chem. B 1997, 101, 4358–4369. [Google Scholar] [CrossRef]
- Basiev, T.T.; Doroshenko, M.E.; Smetanin, S.N.; Jelinek, M., Jr.; Kubeček, V.; Jelínková, H.; Shekhovtsov, A.N.; Kosmyna, M.B. Multi-wave SRS oscillation in PbMoO4 and PbMo0.5W0.5O4 crystals under 18 picosecond laser pumping. Laser Phys. Lett. 2012, 9, 853. [Google Scholar] [CrossRef]
- Bochkova, T.M.; Volnyanskii, M.D.; Volnyanskii, D.M.; Shchetinkin, V.S. Color centers in lead molybdate crystals. Phys. Solid State 2003, 45, 244–247. [Google Scholar] [CrossRef]
- Li, W.; Huang, H.; Feng, X. The effects of Nd impurity on the optical, dielectric and electrical properties of PbWO4 single crystals. Phys. Status Solidi A 2005, 202, 2531–2536. [Google Scholar] [CrossRef]
- Huang, H.; Feng, X.; Man, Z.; Tang, T.B.; Dong, M.; Ye, Z.G. Impedance spectroscopy analysis of La-doped PbWO4 single crystals. J. Appl. Phys. 2003, 93, 421–425. [Google Scholar] [CrossRef]
- Gorobets, Y.N.; Kosmyna, M.B.; Luchechko, A.P.; Nazarenko, B.P.; Puzikov, V.M.; Shekhovtsov, A.N.; Sugak, D.Y. Crystal growth of PbWO4:Nd3+ and PbMoO4:Nd3+ crystals and their characterization by means of optical and dielectric relaxation spectroscopy. J. Cryst. Growth 2011, 318, 687–690. [Google Scholar] [CrossRef]
- Zhu, W.L.; Feng, X.Q.; Huang, Y.L.; Lin, Q.S.; Wu, Z.H. Doping mechanism of heavily doped PbWO4:La3+. Phys. Status Solidi A 2002, 193, 211–216. [Google Scholar] [CrossRef]
- Lugli, C.; Medici, L.; Saccardo, D. Natural wulfenite: Structural refinement by single-crystal X-ray diffraction. Neues Jahrb. für Mineral. Monatshefte 1999, 6, 281–288. [Google Scholar]
- Bomio, M.R.D.; Cavalcante, L.S.; Almeida, M.A.P.; Tranquilin, R.L.; Batista, N.C.; Pizani, P.S.; Siu Li, M.; Andres, J.; Longo, E. Structural refinement, growth mechanism, infrared/Raman spectroscopies and photoluminescence properties of PbMoO4 crystals. Polyhedron 2013, 50, 532–545. [Google Scholar] [CrossRef]
- Bi, J.; Wu, L.; Zhang, Y.; Li, Z.; Li, J.; Fu, X. Solvothermal preparation, electronic structure and photocatalytic properties of PbMoO4 and SrMoO4. Appl. Catal. B 2009, 91, 135–143. [Google Scholar] [CrossRef]
- Tanji, K.; Ishii, M.; Usuki, Y.; Kobayashi, M.; Hara, K.; Takano, H.; Senguttuvan, N. Crystal growth of PbWO4 by the vertical Bridgman method: Effect of crucible thickness and melt composition. J. Cryst. Growth 1999, 204, 505–511. [Google Scholar] [CrossRef]
- Arora, S.K.; Chudasama, B. Flux growth and optoelectronic study of PbWO4 single crystals. Cryst. Growth Des. 2007, 7, 296–299. [Google Scholar] [CrossRef]
- Shivakumara, C.; Saraf, R. Eu3+-activated SrMoO4 phosphors for white LEDs applications: Synthesis and structural characterization. Opt. Mater. 2015, 42, 178–186. [Google Scholar] [CrossRef]
- Clarkson, W.A.; Hodgson, N.; Shori, R.K. (Eds.) Solid State Lasers XXVI: Technology and Devices; SPIE: Washington, WA, USA, 2010. [Google Scholar]
- Çiftyürek, E.; Sabolsky, K.; Sabolsky, E.M. Molybdenum and tungsten oxide based gas sensors for high temperature detection of environmentally hazardous sulfur species. Sens. Actuators B Chem. 2016, 237, 262–274. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.Y.; Zheng, G.H.; Dai, Z.X.; Zhao, X.D. Synthesis of Co doping SrMoO4 for enhanced photocatalytic performance via hydrothermal method. Dig. J. Nanomater. Biostruct. 2019, 14, 569–579. [Google Scholar]
- Mikhailik, V.B.; Elyashevskyi, Y.; Kraus, H.; Kim, H.J.; Kapustianyk, V.; Panasyuk, M. Temperature dependence of scintillation properties of SrMoO4. Nucl. Instrum. Methods. Phys. Res. A 2015, 792, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Nie, Z.; Xie, M.; Wang, Y.; Li, D. Excellent optical thermometry based on upconversion emission in SrMoO4:Er3+ phosphor. Opt. Mater. Express 2017, 7, 2404–2410. [Google Scholar] [CrossRef]
- Mao, C.J.; Geng, J.; Wu, X.C.; Zhu, J.J. Selective synthesis and luminescence properties of self-assembled SrMoO4 superstructures via a facile sonochemical route. J. Phys. Chem. C 2010, 114, 1982–1988. [Google Scholar] [CrossRef]
- Park, S.W.; Moon, B.K.; Jeong, J.H.; Bae, J.S.; Kim, J.H. Crystal structure, electronic structure, and photoluminescent properties of SrMoO4:Tb3+ phosphors. Mater. Res. Bull. 2015, 70, 403–411. [Google Scholar] [CrossRef]
- Lin, X.; Qiao, X.; Fan, X. Synthesis and luminescence properties of a novel red SrMoO4:Sm3+,R+ phosphor. Solid State Sci. 2011, 13, 579–583. [Google Scholar] [CrossRef]
- Wu, S.Y.; Dong, H.N.; Wei, W.H. Investigations on the local structures and the EPR parameters for Er3+ in PbMoO4 and SrMoO4. J. Alloys Compd. 2004, 375, 39–43. [Google Scholar] [CrossRef]
- Jiang, X.; Cui, F. SrWO4:Tb3+ nanoparticles: Synthesis, characterization, and luminescence. J. Mater. Sci. Mater. Electron. 2014, 25, 5362–5365. [Google Scholar] [CrossRef]
- Dunaeva, E.E.; Ivleva, L.I.; Doroshenko, M.E.; Zverev, P.G.; Osiko, V.V. SrMoO4:Pr3+ single crystals: Growth and properties. Dokl. Phys. 2015, 60, 122–126. [Google Scholar] [CrossRef]
- Dunaeva, E.E.; Ivleva, L.I.; Doroshenko, M.E.; Zverev, P.G.; Nekhoroshikh, A.V.; Osiko, V.V. Synthesis, characterization, spectroscopy, and laser operation of SrMoO4 crystals co-doped with Tm3+ and Ho3+. J. Cryst. Growth 2015, 432, 1–5. [Google Scholar] [CrossRef]
- Popov, P.A.; Skrobov, S.A.; Matovnikov, A.V.; Ivleva, L.I.; Dunaeva, E.E.; Shekhovtsov, A.N.; Kosmyna, M.B. Investigation of thermophysical characteristics of SrMoO4 crystals, nominally pure and doped with rare earth ions. Crystallogr. Rep. 2015, 60, 915–920. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Zhang, H.; Yu, H.; Pan, Z. Growth and characterization of large SrMoO4 crystals. J. Cryst. Growth 2011, 318, 679–682. [Google Scholar] [CrossRef]
- Ivleva, L.I. Physicochemical and technological peculiarities of multicomponent oxide crystal growth from melt by modified Stepanov technique. Bull. Russ. Acad. Sci. Phys. 2009, 73, 1338. [Google Scholar] [CrossRef]
- Zhao, B.C.; Sun, Y.P.; Zhang, S.B.; Zhu, X.B.; Song, W.H. Flux growth and characterization of SrMo0.93O3 single crystal. J. Cryst. Growth 2006, 290, 292–295. [Google Scholar] [CrossRef]
- Zharikov, E.V.; Zaldo, C.; Diaz, F. Double tungstate and molybdate crystals for laser and nonlinear optical applications. MRS Bull. 2009, 34, 271–276. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, X.; Yan, J.; Lian, H. (Ln3+ = Eu3+/Dy3+/Dy3+, Eu3+) doped NaGd(MoO4)2 phosphors with uniform morphologies: Hydrothermal synthesis, luminescent properties, energy transfer and color tunable emission. J. Lumin. 2018, 195, 170–175. [Google Scholar] [CrossRef]
- Perets, S.; Tseitlin, M.; Shneck, R.Z.; Mogilyanski, D.; Kimmel, G.; Burshtein, Z. Sodium gadolinium tungstate NaGd(WO4)2: Growth, crystallography, and some physical properties. J. Cryst. Growth 2007, 305, 257–264. [Google Scholar] [CrossRef]
- Singh, S.G.; Singh, A.K.; Desai, D.G.; Tiwari, B.; Tyagi, M. Single crystal growth of Yb doped NaGd(WO4)2 and structural and spectroscopic studies. In AIP Conference Proceedings; American Institute of Physics: College Park, MD, USA, 2014; Volume 1591, pp. 1262–1264. [Google Scholar]
- Cascales, C.; Serrano, M.D.; Esteban-Betegón, F.; Zaldo, C.; Peters, R.; Petermann, K. Structural, spectroscopic, and tunable laser properties of Yb3+-doped NaGd(WO4)2. Phys. Rev. B 2006, 74, 174114. [Google Scholar] [CrossRef] [Green Version]
- Cano-Torres, J.M.; Rico, M.; Han, X.; Serrano, M.D.; Cascales, C.; Zaldo, C. Comparative study of crystallographic, spectroscopic, and laser properties of Tm3+ in NaT(WO4)2 (T = La, Gd, Y, and Lu) disordered single crystals. Phys. Rev. B 2011, 84, 174207. [Google Scholar] [CrossRef]
- Morozov, V.; Arakcheeva, A.; Redkin, B.; Sinitsyn, V.; Khasanov, S.; Kudrenko, E.; Raskina, M.; Lebedev, O.; Van Tendeloo, G. Na2/7Gd4/7MoO4: A modulated scheelite-type structure and conductivity properties. Inorg. Chem. 2012, 51, 5313–5324. [Google Scholar] [CrossRef]
- Zhang, J.; Pandey, I.R.; Pan, S.; Kim, H.; Pan, J.; Chen, H. Vertical Bridgman growth and low temperature luminescence properties of NaY(MoO4)2 crystal. J. Lumin. 2019, 212, 184–190. [Google Scholar] [CrossRef]
- Yuan, L.; Ni, H.; Ji, Z.; Chen, J.; Song, G.; Qi, X.; Li, X.; Sun, S.; Wang, S. High Light Response Uniformity in Industrial Growth of 600-mm-Long BGO Crystals for Dark Matter Particle Explorer. IEEE Trans. Nucl. Sci. 2018, 65, 1403–1407. [Google Scholar] [CrossRef]
- Brunner, S.E.; Schaart, D.R. BGO as a hybrid scintillator/Cherenkov radiator for cost-effective time-of-flight PET. Phys. Med. Biol. 2017, 62, 4421. [Google Scholar] [CrossRef]
- Li, L.; Nie, W.; Li, Z.; Romero, C.; Rodriguez-Beltrán, R.I.; de Aldana, J.R.V.; Chen, F. Laser-writing of ring-shaped waveguides in BGO crystal for telecommunication band. Opt. Express 2017, 25, 24236–24241. [Google Scholar] [CrossRef]
- He, R.; Hernández-Palmero, I.; Romero, C.; de Aldana, J.R.V.; Chen, F. Three-dimensional dielectric crystalline waveguide beam splitters in mid-infrared band by direct femtosecond laser writing. Opt. Express 2014, 22, 31293–31298. [Google Scholar] [CrossRef]
- Yu, P.; Su, L.; Zhao, H.; Xu, J. Optical and structural characterization of colored Bi4Ge3O12 crystals. J. Lumin. 2014, 154, 520–524. [Google Scholar] [CrossRef]
- Yu, P.; Su, L.; Zhao, H.; Guo, X.; Li, H.; Feng, X.; Yang, Q.; Xu, J. Spectroscopic properties of red Bi4Ge3O12 by vertical Bridgman method. Opt. Mater. 2011, 33, 831–834. [Google Scholar] [CrossRef]
- Yu, P.; Su, L.; Wang, Q.; Zhao, H.; Guo, X.; Yang, Q.; Xu, J. Near-infrared luminescence of red Bi4Ge3O12 down to a temperature of 8 K. Nucl. Instrum. Methods. Phys. Res. A 2011, 631, 40–43. [Google Scholar] [CrossRef]
- Xiang, D.; Chu, Y.; Xiao, X.; Xu, J.; Zhang, Z.; Liu, Z.; Zhang, Y.; Yang, B. Tunable luminescence and energy transfer of Dy3+-activated Bi4Si3O12-Bi4Ge3O12 pseudo-system phosphor for warm white-emitting. J. Solid State Chem. 2018, 268, 130–135. [Google Scholar] [CrossRef]
- Dunaeva, E.E.; Ivleva, L.I.; Doroshenko, M.E.; Boldyrev, K.N.; Papashvili, A.G. Growth and spectral-luminescence characteristics of modified BGO crystals. J. Cryst. Growth 2019, 525, 125205. [Google Scholar] [CrossRef]
- Yukhin, Y.M.; Mikhailov, Y.I. Chemistry of Bismuth Compounds and Materials; Publishing House of SB RAS: Novosibirsk, Russia, 2001; pp. 1–360. [Google Scholar]
- Bravo, D.; Lopez, F.J. An electron paramagnetic resonance study of Er3+ in Bi4Ge3O12 single crystals. J. Chem. Phys. 1993, 99, 4952–4959. [Google Scholar] [CrossRef]
- Blanzat, B.; Raynal, F.; Parrot, R.; Barthou, C.; Canny, B. Electron paramagnetic resonance of Gd3+ in bismuth germanate. Phys. Status Solidi B 1976, 76, K5–K8. [Google Scholar] [CrossRef]
- Evstratov, I.Y.; Rukolaine, S.; Yuferev, V.S.; Vasiliev, M.G.; Fogelson, A.B.; Mamedov, V.M.; Shlegel, V.N.; Vasiliev, Y.V.; Makarov, Y.N. Global analysis of heat transfer in growing BGO crystals (Bi4Ge3O12) by low-gradient Czochralski method. J. Cryst. Growth 2002, 235, 371–376. [Google Scholar] [CrossRef]
- Kawano, K.; Yoshida, T.; Nakata, R.; Yamada, N.; Sumita, M. Crystal growth of Bi4Ge3O12 and heat transfer analyses of horizontal Bridgman techniques. Jpn. J. Appl. Phys. 1993, 32, 1736. [Google Scholar] [CrossRef]
- Beneventi, P.; Capelletti, R.; Kovács, L.; Péter, A.; Manotti, A.L.; Ugozzoli, F. FTIR spectroscopy of OH stretching modes in BSO, BGO and BTO sillenites. J. Phys. Condens. Matter 1994, 6, 6329. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Saeki, K.; Yanagida, T.; Koshimizu, M.; Asai, K. Luminescence and scintillation properties of TlCdCl3 crystal. Radiat. Meas. 2017, 106, 151–154. [Google Scholar] [CrossRef]
- Sobczyk, M.; Drożdżyński, J. Room temperature near infrared luminescence of an U3+ doped CsCdCl3 potential laser crystal. Mater. Lett. 2007, 61, 2319–2321. [Google Scholar] [CrossRef]
- Romanov, A.N.; Vtyurina, D.N.; Haula, E.V.; Shashkin, D.P.; Pimkin, N.A.; Kuznetsov, M.S.; Lisitsky, I.S.; Korchak, V.N. Broadband infrared photoluminescence of TlCdI3 iodide doped with bismuth. Russ. J. Phys. Chem. B 2017, 11, 83–86. [Google Scholar] [CrossRef]
- Neukum, J.; Bodenschatz, N.; Heber, J. Spectroscopy and upconversion of CsCdBr3:Pr3+. Phys. Rev. B 1994, 50, 3536. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Malkin, B.Z.; Iskhakova, A.I.; Tarasov, V.F.; Shakurov, G.S.; Heber, J.; Altwein, M. Submillimeter EPR spectroscopy of lanthanide compounds: Pair centers of Ho3+ in CsCdBr3. J. Alloys Compd. 1998, 275, 209–213. [Google Scholar] [CrossRef]
- Tarasov, V.F.; Shakurov, G.S.; Malkin, B.Z.; Iskhakova, A.I.; Heber, J.; Altwein, M. Submillimeter electron-nuclear excitation spectra in CsCdBr3:Ln3+(Ln = Tm, Ho) crystals. JETP Lett. 1997, 65, 559–565. [Google Scholar] [CrossRef]
- Mehta, V.; Guillot-Noël, O.; Simons, D.; Gourier, D.; Goldner, P.; Pellé, F. EPR Identification of coupled Yb3+ ion pairs in optically bistable compound CsCdBr3:Yb. J. Alloys Compd. 2001, 323, 308–311. [Google Scholar] [CrossRef]
- Vtyurina, D.N.; Romanov, A.N.; Veber, A.A.; Fattakhova, Z.T.; Antonov, A.A.; Tsvetkov, V.B.; Korchak, V.N. The spectral properties and the NIR photoluminescence of univalent bismuth Bi+ in RbAlCl4, CsAlCl4, RbMgCl3, CsMgCl3, KCdCl3 and RbCdCl3 crystal phases. Russ. J. Phys. Chem. B 2016, 10, 388–393. [Google Scholar] [CrossRef]
- Romanov, A.N.; Veber, A.A.; Fattakhova, Z.T.; Usovich, O.V.; Haula, E.V.; Trusov, L.A.; Kazin, P.E.; Korchak, V.N.; Tsvetkov, V.B.; Sulimov, V.B. Subvalent bismuth monocation Bi+ photoluminescence in ternary halide crystals KAlCl4 and KMgCl3. J. Lumin. 2013, 134, 180–183. [Google Scholar] [CrossRef]
- Romanov, A.N.; Grigoriev, F.V.; Sulimov, V.B. Estimation of Bi+ monocation crystal ionic radius by quantum chemical simulation. Comput. Theor. Chem. 2013, 1017, 159–161. [Google Scholar] [CrossRef]
- Vtyurina, D.N.; Romanov, A.N.; Kuznetsov, M.S.; Fattakhova, Z.T.; Khaula, E.V.; Lisitskii, I.S.; Korchak, V.N. Optical properties of bismuth-doped TlCdCl3 crystal. Russ. J. Phys. Chem. B 2016, 10, 1–4. [Google Scholar] [CrossRef]
- Basiev, T.T.; Baumer, V.N.; Gorobets, Y.N.; Doroshenko, M.E.; Kosmyna, M.B.; Nazarenko, B.P.; Osiko, V.V.; Puzikov, V.M.; Shekhovtsov, A.N. Peculiarities of the growth of PbWO4:Nd3+ and PbMoO4:Nd3+ single crystals. Crystallogr. Rep. 2009, 54, 697–701. [Google Scholar] [CrossRef]
- Baumer, V.N.; Gorobets, Y.N.; Gudzenko, L.V.; Kosmyna, M.B.; Nazarenko, B.P.; Puzikov, V.M.; Shekhovtsov, A.N.; Shtitelman, Z.V. Dependence of PbMoO₄ crystal damage threshold on Nd³⁺ concentration and method of doping. Funct. Mater. 2010, 17, 515–519. [Google Scholar]
- Kosmyna, M.B.; Nazarenko, B.P.; Puzikov, V.M.; Shekhovtsov, A.N. Development of growth technologies for the photonic single crystals by the Czochralski method at Institute for Single Crystals, NAS of Ukraine. Acta Phys. Pol. A 2013, 123, 305–313. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Kaurova, I.A.; Ivleva, L.I.; Eistrikh-Geller, P.A.; Rybakov, V.B.; Khramov, E.V. New structural effects in SrMoO4:Tm3+/Ho3+ crystals. CrystEngComm 2017, 19, 295–303. [Google Scholar] [CrossRef]
- Dunaeva, E.E.; Zverev, P.G.; Doroshenko, M.E.; Nekhoroshikh, A.V.; Ivleva, L.I.; Osiko, V.V. Growth and spectral-luminescent study of SrMoO4 crystals doped with Tm3+ ions. Dokl. Phys. 2016, 61, 119–123. [Google Scholar] [CrossRef]
- Zharikov, E.V.; Lis, D.A.; Subbotin, K.A.; Dudnikova, V.B.; Zaitseva, O.N. Growth of oxide laser crystals by Czochralski method. Acta Phys. Pol. A 2013, 124, 274–285. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Lis, D.A.; Subbotin, K.A.; Rybakov, V.B.; Zharikov, E.V. Growth and structural X-ray investigations of scheelite-like single crystals Er,Ce:NaLa(MoO4)2 and Yb:NaGd(WO4)2. J. Cryst. Growth 2005, 275, 1835–1842. [Google Scholar] [CrossRef]
- Voron’ko, Y.K.; Subbotin, K.A.; Shukshin, V.E.; Lis, D.A.; Ushakov, S.N.; Popov, A.V.; Zharikov, E.V. Growth and spectroscopic investigations of Yb3+-doped NaGd(MoO4)2 and NaLa(MoO4)2—New promising laser crystals. Opt. Mater. 2006, 29, 246–252. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Rybakov, V.B.; Zharikov, E.V.; Lis, D.A.; Subbotin, K.A. Unusual structural properties of (Na0.5La0.5)MoO4:Er,Ce crystals. Inorg. Mater. 2006, 42, 303–309. [Google Scholar] [CrossRef]
- Romanov, A.N.; Veber, A.A.; Fattakhova, Z.T.; Vtyurina, D.N.; Kouznetsov, M.S.; Zaramenskikh, K.S.; Lisitsky, I.S.; Korchak, V.N.; Tsvetkov, V.B.; Sulimov, V.B. Spectral properties and NIR photoluminescence of Bi+ impurity in CsCdCl3 ternary chloride. J. Lumin. 2014, 149, 292–296. [Google Scholar] [CrossRef]
- Romanov, A.N.; Veber, A.A.; Vtyurina, D.N.; Kouznetsov, M.S.; Zaramenskikh, K.S.; Lisitsky, I.S.; Fattakhova, Z.T.; Haula, E.V.; Loiko, P.A.; Yumashev, K.V.; et al. NIR photoluminescence of bismuth-doped CsCdBr3–The first ternary bromide phase with a univalent bismuth impurity center. J. Lumin. 2015, 167, 371–375. [Google Scholar] [CrossRef]
- Gorobets, Y.N.; Kaurova, I.A.; Kuz’micheva, G.M.; Shekhovtsov, A.N.; Rybakov, V.B.; Cousson, A. Influence of the dopant type on point defects in PbMoO4 crystals. J. Surf. Investig.: X-Ray Synchrotron Neutron Tech. 2014, 8, 734–744. [Google Scholar] [CrossRef]
- Kaurova, I.A.; Kuz’micheva, G.M.; Brykovskiy, A.A.; Rybakov, V.B.; Gorobets, Y.N.; Shekhovtsov, A.N.; Cousson, A. Influence of growth conditions on structural parameters of scheelite PbTO4 (T = Mo, W) crystals. Mater. Des. 2016, 97, 56–63. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Kaurova, I.A.; Brykovskiy, A.A.; Rybakov, V.B.; Gorobets, Y.N.; Shekhovtsov, A.N.; Cousson, A. Structural investigation of Pb(MoxW1−x)O4 solid solutions via X-ray and neutron diffraction. Mater. Res. Bull. 2016, 78, 134–140. [Google Scholar] [CrossRef]
- Fulle, K.; McMillen, C.D.; Sanjeewa, L.D.; Kolis, J.W. Hydrothermal Chemistry and Growth of Fergusonite-type RENbO4 (RE = La–Lu, Y) Single Crystals and New Niobate Hydroxides. Cryst. Growth Des. 2016, 16, 4910–4917. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Zharikov, E.V.; Subbotin, K.A.; Lis, D.A.; Rybakov, V.B. X-Ray diffraction study of the structure and defect system of nominally pure and Er3+- and Ce3+-activated (Na0.5La0.5)MoO4 crystals. Inorg. Mater. 2005, 41, 998–1009. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Kaurova, I.A.; Rybakov, V.B.; Eistrikh-Geller, P.A.; Zharikov, E.V.; Lis, D.A.; Subbotin, K.A. Influence of initial charge composition and growth/annealing atmospheres on the structural parameters of Czochralski-grown (NaxGd1−x)MoO4 crystals. CrystEngComm 2016, 18, 2921–2928. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Kaurova, I.A.; Zagorul’ko, E.A.; Bolotina, N.B.; Rybakov, V.B.; Brykovskiy, A.A.; Zharikov, E.V.; Lis, D.A.; Subbotin, K.A. Structural perfection of (Na0.5Gd0.5)MoO4:Yb laser crystals. Acta Mater. 2015, 87, 25–33. [Google Scholar]
- Kuz’micheva, G.M.; Kaurova, I.A.; Ivleva, L.I.; Khramov, E.V.; Eistrikh-Geller, P.A.; Rybakov, V.B.; Chukhlovina, T.V.; Firstov, S.V. Structure and composition peculiarities and spectral-luminescent properties of colorless and pink Bi4Ge3O12 scintillation crystals. Arab. J. Chem. 2018, 11, 1270–1280. [Google Scholar] [CrossRef]
- Vtyurina, D.N.; Kaurova, I.A.; Kuz’micheva, G.M.; Rybakov, V.B.; Chernyshov, D.Y.; Khramov, E.V.; Firstov, S.V.; Korchak, V.N. Structural peculiarities, point defects and luminescence in Bi-doped CsCdX3 (X = Cl, Br) single crystals. J. Alloys Compd. 2019, 803, 912–921. [Google Scholar] [CrossRef]
- Vtyurina, D.N.; Eistrikh-Geller, P.A.; Kuz’micheva, G.M.; Rybakov, V.B.; Khramov, E.V.; Kaurova, I.A.; Chernyshov, D.Y.; Korchak, V.N. Influence of monovalent Bi+ doping on real composition, point defects, and photoluminescence in TlCdCl3 and TlCdI3 single crystals. Sci. China Mater. 2017, 60, 1253–1263. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- North, A.C.T.; Phillips, D.T.; Mathews, F.S. A semi-empirical method of absorption correction. Acta Crystallogr. A 1968, 24, 351–359. [Google Scholar] [CrossRef]
- Walker, N.T.; Stuart, D. An empirical method for correcting diffractometer data for absorption effects. Acta Crystallogr. A 1983, 39, 158–166. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Ivleva, L.I.; Kaurova, I.A.; Rybakov, V.B.; Cousson, A. Structure and real composition of undoped and Cr-and Ni-doped Sr0.61Ba0.39Nb2O6 single crystals. Struct. Chem. 2016, 27, 1623–1634. [Google Scholar] [CrossRef]
- Trofimova, N.N.; Veligzhanin, A.A.; Murzin, V.Y.; Chernyshov, A.A.; Khramov, E.V.; Zabluda, V.N.; Edel’man, I.S.; Slovokhotov, Y.L.; Zubavichus, Y.V. Structural diagnostics of functional nanomaterials with the use of X-ray synchrotron radiation. Nanotechnol. Russ. 2013, 8, 396–401. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 2005, 12, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Kuz’micheva, G.M.; Ivleva, L.I.; Kaurova, I.A.; Khramov, E.V.; Dunaeva, E.E.; Svetogorov, R.D.; Rybakov, V.B. Statistical and Local Structure of Dy-doped Bi4Ge3O12 Crystals: A Role of Activator Concentration in Structural Effects. CrystEngComm 2020. (under review). [Google Scholar]
- Zalkin, A.; Templeton, D.H. X-Ray Diffraction Refinement of the Calcium Tungstate Structure. J. Chem. Phys. 1964, 40, 501–504. [Google Scholar] [CrossRef] [Green Version]
- Grzechnik, A.; Crichton, W.A.; Hanfland, M.; van Smaalen, S. Scheelite CaWO4 at high pressures. J. Phys. Cond. Matter 2003, 15, 7261. [Google Scholar] [CrossRef]
- Kröger, F.A.; Vink, H.J. Relations between the concentrations of imperfections in crystalline solids. Solid State Phys. 1956, 3, 307–435. [Google Scholar]
- Kroger, F.A. Defect chemistry in crystalline solids. Annual Rev. Mater. Sci. 1977, 7, 449–475. [Google Scholar] [CrossRef]
- Morozov, V.A.; Bertha, A.; Meert, K.W.; Van Rompaey, S.; Batuk, D.; Martinez, G.T.; van Aert, S.; Smet, P.F.; Raskina, M.V.; Poelman, D.; et al. Incommensurate Modulation and Luminescence in the CaGd2(1–x)Eu2x(MoO4)4(1–y)(WO4)4y (0 ≤ x ≤ 1, 0 ≤ y ≤ 1) Red Phosphors. Chem. Mater. 2013, 25, 4387–4395. [Google Scholar] [CrossRef]
- Lecog, P.; Annenkov, A.; Gektin, A.; Korzhik, M.; Pedrini, C. Inorganic Scintillators for Detector Systems: Physical Principle and Crystal Engineering; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Oeder, R.; Scharmann, A.; Schwabe, D.; Vitt, B. Growth and properties of PbWO4 and Pb(WO4)1−x(MoO4)x mixed crystals. J. Crys. Growth 1978, 43, 537–540. [Google Scholar] [CrossRef]
- Adya, V.C.; Mohapatra, M.; Bhide, M.K.; Natarajan, V. Observation of “cool daylight” white light emission from Eu and Tb co-doped SrMoO4 nano ceramics. Mater. Sci. Eng. B 2016, 203, 35–40. [Google Scholar] [CrossRef]
- Cao, J.; Wang, Y.; Ma, X.; Li, J.; Zhu, Z.; You, Z.; Yang, F.; Sun, C.; Cao, T.; Ji, Y.; et al. Spectroscopic properties of Pr3+: SrMoO4 crystal. J. Aalloys Compd. 2011, 509, 185–189. [Google Scholar] [CrossRef]
- Ma, X.; Li, J.; Zhu, Z.; You, Z.; Wang, Y.; Tu, C. Optical properties of Er3+:SrMoO4 single crystal. J. Phys. Chem. Solids 2008, 69, 2411–2415. [Google Scholar] [CrossRef]
- Ma, X.; You, Z.; Zhu, Z.; Li, J.; Wu, B.; Wang, Y.; Tu, C. Thermal and optical properties of Tm3+: SrMoO4 crystal. J. Alloys Compd. 2008, 465, 406–411. [Google Scholar] [CrossRef]
- Ma, X.; Zhu, Z.; Li, J.; You, Z.; Wang, Y.; Tu, C. Optical properties of Ho3+:SrMoO4 single crystal. Mater. Res. Bull. 2009, 44, 571–575. [Google Scholar] [CrossRef]
- Niu, N.; Yang, P.; Wang, W.; He, F.; Gai, S.; Wang, D.; Lin, J. Solvothermal synthesis of SrMoO4:Ln (Ln = Eu3+, Tb3+, Dy3+) nanoparticles and its photoluminescence properties at room temperature. Mater. Res. Bull. 2011, 46, 333–339. [Google Scholar] [CrossRef]
- Thongtem, T.; Phuruangrat, A.; Thongtem, S. Microwave-assisted synthesis and characterization of SrMoO4 and SrWO4 nanocrystals. J. Nanopart. Res. 2010, 12, 2287–2294. [Google Scholar] [CrossRef]
- Doroshenko, M.E.; Papashvili, A.G.; Dunaeva, E.E.; Ivleva, L.I.; Osiko, V.V.; Jelinkova, H.; Sulc, J.; Nemec, M. Spectroscopic and laser properties of SrMoO4:Tm3+ crystal under 1700-nm laser diode pumping. Opt. Mater. 2016, 60, 119–122. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Rybakov, V.B.; Panyutin, V.L.; Zharikov, E.V.; Subbotin, K.A. Symmetry of (Na0.5R0.5)MO4 Crystals (R = Gd, La; M = W, Mo). Russ. J. Inorg. Chem. 2010, 55, 1448–1453. [Google Scholar] [CrossRef]
- Zharikov, E.V.; Kuz’micheva, G.M.; Lis, D.A.; Papin, Y.M.; Rybakov, V.B.; Smirnov, B.A.; Subbotin, K.A. X-ray diffraction study of the structure and defect system of Yb-activated (Na0.5Gd0.5)WO4 crystals. Inorg. Mater. 2003, 39, 151–160. [Google Scholar] [CrossRef]
- Sokolenko, E.V.; Zhukovskii, V.M.; Buyanova, E.S.; Krasnobaev, Y.A. Luminescent Properties of Oxygen-disordered Scheelite-Structure Tungstates: I. Steady-State Luminescence. Neorg. Mater. 1998, 34, 499–502. [Google Scholar]
- Kuzmicheva, G.M.; Rybakov, V.B.; Subbotin, K.A.; Zharikov, E.V.; Lis, D.A.; Zaharko, O.; Nikolaev, D.A.; Senin, V.G. Colors of mixed-substituted double molybdate single crystals having scheelite structure. Russ. J. Inorg. Chem. 2012, 57, 1128–1133. [Google Scholar] [CrossRef]
- Zhao, W.; Lin, Z.; Zhang, L.; Wang, G. Growth and spectroscopic properties of Nd3+-doped Na2Gd4(MoO4)7 crystal. J. Alloys Compd. 2011, 509, 2815–2818. [Google Scholar] [CrossRef]
- Bolschikov, F.A.; Kuz’micheva, G.M.; Lis, D.A.; Papin, Y.M.; Popov, A.V.; Ryabochkina, P.A.; Rybakov, V.B.; Senin, V.G.; Smirnov, V.A.; Subbotin, K.A.; et al. Growth, refined structural and spectroscopic characteristics of Tm3+-doped NaGd(WO4)2 single crystals. J. Cryst. Growth 2009, 311, 4171–4178. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Zagorul’ko, E.A.; Bolotina, N.B.; Rybakov, V.B.; Zharikov, E.V.; Lis, D.A.; Subbotin, K.A. Structure-property relationship for complex-substituted molybdates and tungstates of the scheelite family. Crystallogr. Rep. 2014, 59, 22–29. [Google Scholar] [CrossRef]
- Putnis, A.; McConnell, J.D.C. Principles of Mineral Behaviour.; Blackwell: Oxford, UK; London, UK, 1980. [Google Scholar]
- Shtukenberg, A.G.; Punin, Y.O.; Frank-Kamenetskaya, O.V. The kinetic ordering and growth dissymmetrisation in crystalline solid solutions. Russ. Chem. Rev. 2006, 75, 1083. [Google Scholar] [CrossRef]
- Takai, S.; Touda, S.; Oikawa, K.; Mori, K.; Torii, S.; Kamiyama, T.; Esaka, T. Powder neutron diffraction study of Ln-substituted PbWO4 oxide ion conductors. Solid State Ionics 2002, 148, 123–133. [Google Scholar] [CrossRef]
- Lin, Q.; Feng, X.; Chen, J. The structure of a PWO:La3+ crystal. J. Alloys Compd. 2000, 307, 245–248. [Google Scholar] [CrossRef]
- Dardenne, K.; Bosbach, D.; Denecke, M.A.; Brendebach, B. EXAFS investigation of the NaLn(MoO₄)₂-Ca₂(MoO₄)₂ solid solution series local structure. In Proceedings of the 4th Workshop on Speciation, Techniques, and Facilities for Radioactive Materials at Synchrotron Light Sources (Actinide-XAS-2006), Karlsruhe, Germany, 18–20 September 2006; p. 193. [Google Scholar]
- Suvorova, E.I.; Kuz’micheva, G.M.; Morozkin, A.V.; Zharikov, E.V.; Lis, D.A.; Subbotin, K.A. Microstructure of (Na0.5La0.5)MoO4 crystals coactivated with cerium and erbium ions. Inorg. Mater. 2007, 43, 287–291. [Google Scholar] [CrossRef]
- Mel’nikova, T.I.; Kuz’micheva, G.M.; Bolotina, N.B.; Rybakov, V.B.; Zubavichus, Y.V.; Sadovskaya, N.V.; Mar’ina, E.A. Structural features of compounds of the sillenite family. Crystallogr. Rep. 2014, 59, 353–361. [Google Scholar] [CrossRef]
- Mel’nikova, T.I.; Kuz’micheva, G.M.; Bolotina, N.B.; Sadovskaya, N.V. Development of methods of X-ray diffraction analysis for determining the composition and structure of sillenite-family crystals. Crystallogr. Rep. 2014, 59, 155–159. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Mel’nikova, T.I.; Kaurova, I.A.; Zubavichus, Y.V.; Nikolaychik, V.I. Isomorphous substitutions in sillenite-family single-crystal Bi24(M2−xMn4+x)O40 solid solutions (M = Al3+, Fe3+, Ge4+, Ti4+, Cr4+, V5+). J. Cryst. Growth 2019, 507, 413–420. [Google Scholar] [CrossRef]
- Segal, D.J.; Santoro, Β.; Newnham, R.E. Neutron-diffraction study of Bi4Si3O12. Z. Kristallogr. Cryst. Mater. 1966, 123, 73–76. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Kozlikin, S.N.; Zharikov, E.V.; Kalitin, S.P.; Osiko, V.V. Point defects in gadolinium-gallium garnet. Zhurnal Neorganicheskoj Khimii 1988, 33, 2200–2204. [Google Scholar]
- Kuz’micheva, G.M.; Mel’nikova, T.I.; Kaurova, I.A.; Bolotina, N.B. Structure peculiarities and point defects in new sillenite-type (Bi,Sm)24V2O40 crystals. Mater. Lett. 2017, 199, 75–78. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Kaurova, I.A.; Rybakov, V.B.; Podbel’skiy, V.V. Crystallochemical Design of Huntite-Family Compounds. Crystals 2019, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, N.; Kumar, S.; Ghosh, A.K.; Chatterjee, S.; Jha, S.N.; Bhattacharyya, D. Structural investigations of (Mn,Dy) co-doped ZnO nanocrystals using X-ray absorption studies. RSC Adv. 2017, 7, 56662–56675. [Google Scholar] [CrossRef] [Green Version]
- Kuz’micheva, G.M.; Kaurova, I.A.; Khramov, E.V.; Ivleva, L.I.; Chernyshev, V.V.; Rybakov, V.B. A comprehensive structural analysis of relaxor ferroelectric Cr-and Ni-doped Sr0.61Ba0.39Nb2O6 crystals. J. Alloys Compd. 2017, 724, 879–888. [Google Scholar] [CrossRef]
- Berastegui, P.; Hull, S.; Eriksson, S.G. A low-temperature structural phase transition in CsPbF3. J. Phys. Cond. Matter 2001, 13, 5077. [Google Scholar] [CrossRef]
- Siegel, S.; Gebert, E. The structures of hexagonal CsCdCl3 and tetragonal Cs2CdCl4. Acta Crystallogr. 1964, 17, 790. [Google Scholar] [CrossRef]
- McPherson, G.L.; McPherson, A.M.; Atwood, J.L. Structures of CsMgBr3, CsCdBr3 and CsMgI3 - diamagnetic linear chain lattices. J. Phys. Chem. Solids 1980, 41, 495–499. [Google Scholar] [CrossRef]
- Aleksandrov, K.S.; Beznosikov, V.V. Hierarchies of perovskite-like crystals. Phys. Solid State 1997, 39, 695–715. [Google Scholar] [CrossRef]
- Bogdanova, A.V.; Zaslavskaya, N.P.; Sinichka, E.V.; Fedyna, M.F.; Mokra, I.R.; Gasinets, S.M. Synthesis and Crystal Structure of TlCdCl3 and TlCdBr3. Chem. Inform 1993, 24. [Google Scholar] [CrossRef]
- Zandbergen, H.W.; Verschoor, G.C.; Ijdo, D.J.W. The structures of thallium cadmium triiodide and dirubidium iron tetraiodide. Acta Crystallogr. B 1979, 35, 1425–1427. [Google Scholar] [CrossRef]
- Veber, A.A.; Romanov, A.N.; Usovich, O.V.; Fattakhova, Z.T.; Haula, E.V.; Korchak, V.N.; Trusov, L.A.; Kazin, P.E.; Sulimov, V.B.; Tsvetkov, V.B. Luminescent properties of Bi-doped polycrystalline KAlCl4. Appl. Phys. B 2012, 108, 733–736. [Google Scholar] [CrossRef] [Green Version]
- Veber, A.A.; Romanov, A.N.; Usovich, O.V.; Fattakhova, Z.T.; Haula, E.V.; Korchak, V.N.; Trusov, L.A.; Kazin, P.E.; Sulimov, V.B.; Tsvetkov, V.B. Optical properties of the Bi+ center in KAlCl4. J. Lumin. 2014, 151, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Romanov, A.N.; Veber, A.A.; Vtyurina, D.N.; Fattakhova, Z.T.; Haula, E.V.; Shashkin, D.P.; Sulimov, V.B.; Tsvetkov, V.B.; Korchak, V.N. Near infrared photoluminescence of the univalent bismuth impurity center in leucite and pollucite crystal hosts. J. Mater. Chem. C 2015, 3, 3592–3598. [Google Scholar] [CrossRef] [Green Version]
- Okhrimchuk, A.G.; Butvina, L.N.; Dianov, E.M.; Lichkova, N.V.; Zagorodnev, V.N.; Boldyrev, K.N. Near-infrared luminescence of RbPb2Cl5:Bi crystals. Opt. Lett. 2008, 33, 2182–2184. [Google Scholar] [CrossRef]
- Kuz’micheva, G.M.; Timaeva, O.I.; Rybakov, V.B.; Kaurova, I.A.; Kosinova, A.V.; Grebenev, V.V. Growth, structure peculiarities, and dielectric properties of ferroelectric KDP/TiO2 single crystals. J. Mater. Sci. 2016, 51, 3045–3055. [Google Scholar] [CrossRef]
- Antonio, M.R.; Brazdil, G.F.; Glaeser, L.C.; Mehicic, M.; Teller, R.G. Structural Environments and Oxidation States of Metal Cations in Bismuth Cerium Molybdate Solid Solutions by X-ray Absorption Spectroscopy. J. Phys. Chem. 1988, 92, 2338–2345. [Google Scholar] [CrossRef]
- Goryhnov, A.V.; Kuz’micheva, G.M.; Mukhin, B.V.; Zharikov, E.V.; Ageev, A.Y.; Kutovoj, S.A.; Kuz’min, O.V. An X-ray diffraction study of LaSc3(BO3)4 crystals activated with chromium and neodymium ions. Russ. J. Inorg. Chem. 1996, 41, 1531–1536. [Google Scholar]
- Kaurova, I.A.; Gorshkov, D.M.; Kuz’micheva, G.M.; Rybakov, V.B. Composition and Structure of the Huntite-Family Compounds. Fine Chem. Technol. 2018, 13, 42–51. [Google Scholar] [CrossRef]
- Ivleva, L.I.; Dunaeva, E.E.; Voronina, I.S.; Doroshenko, M.E.; Papashvili, A.G. Ca3(VO4)2:Tm3+—A new crystalline medium for 2-μm lasers. J. Cryst. Growth 2018, 501, 18–21. [Google Scholar] [CrossRef]
- Doroshenko, M.E.; Papashvili, A.G.; Voronina, I.S.; Ivleva, L.I.; Sulc, J.; Kratochvíl, J.; Jelinkova, H. Spectroscopic and laser properties of Tm3+ ions in Ca3(VO4)2 crystal. J. Lumin. 2019, 205, 482–486. [Google Scholar] [CrossRef]
- Ivleva, L.I.; Dunaeva, E.E.; Voronina, I.S.; Doroshenko, M.E.; Papashvili, A.G.; Sulc, J.; Kratochvíl, J.; Jelinkova, H. Impact of Tm3+/Ho3+ co-doping on spectroscopic and laser properties of Ca3(VO4)2 single crystal. J. Cryst. Growth 2019, 513, 10–14. [Google Scholar] [CrossRef]
- Belik, A.A.; Morozov, V.A.; Grechkin, S.V.; Khasanov, S.S.; Lazoryak, B.I. Crystal structures of double vanadates Ca9R(VO4)7. III. R = Nd, Sm, Gd, or Ce. Crystallogr. Rep. 2000, 45, 728–733. [Google Scholar] [CrossRef]
- Belik, A.A.; Morozov, V.A.; Kotov, R.N.; Khasanov, S.S.; Lazoryak, B.I. Crystal structures of double vanadates Ca9R(VO4)7. II. R = Tb, Dy, Ho, and Y. Crystallogr. Rep. 2000, 45, 389–394. [Google Scholar]
- Pogosova, M.A.; Azarmi, F.; Eliseev, A.A.; Kazin, P.E. Eu and Cu co-substituted calcium vanadate—The crystal structure, luminescence and color. Dyes Pigm. 2018, 148, 219–223. [Google Scholar] [CrossRef]
- Mel’nikova, T.I.; Kuz’micheva, G.M.; Rybakov, V.B.; Bolotina, N.B.; Dubovsky, A.B. Synthesis, Composition, and Structure of Sillenite-Type Solid Solutions in the Bi2O3−SiO2−MnO2 System. Inorg. Chem. 2011, 50, 2002–2009. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds studied are available from the authors. |




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Designation | Growth Atmosphere | Color | Space Group | Refined Composition 1 |
|---|---|---|---|---|---|
| PbMoO4 | PMO-A | Air | Yellow | I41/a | (Pb0.976(1)⎕0.024)MoO4.00(3) |
| PbWO4 | PWO-A | PbW(O3.982(3)⎕0.018) PbW(O3.94(4)⎕0.06) 2 | |||
| PbMoO4 | PMO-N2 | Nitrogen | I41/a | (Pb0.960(1)⎕0.040)MoO4.00(4) | |
| PbWO4 | PWO-N2 | Pb(W0.996(1)⎕0.004)(O3.855(3)⎕0.145) Pb0.999(1)W(O3.93(4)⎕0.07) 2 |
| Sample | Designation | Color | Unit Cell Parameters (a, c, Å) and Cell Volume (V, Å3) | Refined Composition 1 |
|---|---|---|---|---|
| PbMo0.2W0.8O4 | PMWO-1 | Colorless | 5.4411(4) 12.0261(12) 356.04 5.4468(2) 2 12.0679(4) 2 358.03 2 | (Pb2+0.980(5)Y3+0.020)(Mo6+0.300(5)W6+0.700)(O3.990(8)⎕0.010) or (Pb2+0.980Y0.020)[(Mo6+,W6+)0.960Nb5+0.040](O3.990(8)⎕0.010) (Pb2+0.972Y3+0.028(4))(Mo6+0.490W0.482(8)Nb5+0.028)O4 2 |
| PbMo0.5W0.5O4 | PMWO-2 | 5.4350(5) 12.0926(15) 357.21 5.4406(3) 2 12.0700(3) 2 357.27 2 | (Pb2+0.975(8)Y3+0.025)(Mo6+0.536(5)W6+0.464)(O3.985(10)⎕0.015) or (Pb2+0.975Y0.025)[(Mo6+,W6+)0.945 Nb5+0.055](O3.985(10)⎕0.015) (Pb2+0.960Y3+0.040(4))(Mo6+0.519W0.441(8)Nb5+0.040)O4 2 | |
| PbMo0.8W0.2O4 | PMWO-3 | 5.4398(3) 12.0534(9) 356.68 5.4296(3) 2 12.0629(4) 2 355.62 2 | (Pb2+0.930(10)Y3+0.070)(Mo6+0.770(8)W0.230)(O3.960(20) ⎕0.040) or (Pb2+0.930(10)Y3+0.070)[(Mo6+,W6+)0.850Nb0.150](O3.960(20) ⎕0.060) (Pb2+0.950Y3+0.050(8))[(Mo6+0.719W0.183(8)Nb5+0.100](O3.980(10)⎕0.020) 2 |
| Number | Sample | Color | Refined Composition 1 |
|---|---|---|---|
| 1 | PbMoO4 | Yellowish transparent | Pb2+(Mo0.992(4)⎕0.008)O4 Pb2+ Mo1.000(16)O3.960(44) 2 |
| 2 | PbMoO4:Nd2O3 | Lilac yellow with predominant yellow | (Pb2+0.970(4)Nd3+0.030)Mo6+O4 (Pb2+0.865(45)Nd3+0.085⎕0.050)Mo6+O4 2 |
| 3 | PbMoO4:NdNbO4 | Lilac | (Pb2+0.980(6)Nd3+0.020)(Mo6+0.980Nb5+0.020)O4 (Pb2+0.968(16)Nd3+0.032)(Mo6+0.970(20)Nb5+0.030)O4 2 |
| 4 | PbMoO4:Nd2(MoO4)3 | Lilac yellow with predominant yellow | (Pb2+0.980(4)Nd3+0.020)Mo6+O4 (Pb2+0.964(15)Nd3+0.025⎕0.011)Mo6+O4 2 |
| 5 | PbMoO4:(Na0.5Nd0.5)MoO4 | Deep lilac | (Pb2+0.960(6)Nd3+0.020(8)Na+10.015)Mo6+O4 (Pb2+0.935(12)Nd3+0.033Na+10.033)Mo6+O4 2 |
| Sample | Designation | Refined Composition (XRD) | Refined Composition 1 (XRD + EXAFS/XANES) |
|---|---|---|---|
| SrMoO4 | SMO | SrMoO4 | - |
| SrMoO4:HoNbO4 (0.1 wt%) | SMO:0.1Ho | (Sr0.996(4)Ho0.004)MoO4 | - |
| SrMoO4:HoNbO4 (0.5 wt%) | SMO:0.5Ho | (Sr0.992(3)Ho0.008)MoO4 | - |
| SrMoO4:HoNbO4 (1.0 wt%) | SMO:1.0Ho | (Sr0.998(2)Ho0.002) [(Mo0.998(2)⎕0.002)(Nb0.002)i]O3.96(3) | (Sr2+0.998(2)Ho3+0.002) [(Mo6+,Nb5+)0.998⎕0.002][(Nb5+0.002)i] [(O3.96(3))(1)(O0.002+x)(2)] |
| SrMoO4:TmNbO4 (0.5 wt%) | SMO:0.5Tm | [Sr0.996(2)⎕0.004)(Tm0.004)i] [Mo0.996(3)⎕0.004)(Nb0.004)i]O4 | - |
| SrMoO4:TmNbO4 (1.0 wt%) | SMO:1.0Tm | [(Sr0.992(3)⎕0.008)(Tm0.008)i)] [(Mo0.992(3)⎕0.002)(Nb0.008)i)]O3.80(4) | [(Sr2+0.992(3)⎕0.008)(Tm3+0.008i)] [(Mo6+,Nb5+)0.992⎕0.008][(Nb5+0.008)i] [(O3.80(4))(1)(O0.008+x)(2)] |
| Sample | CN Mo | dEXAFS, Å |
|---|---|---|
| SMO | 4 | 1.768(5) |
| SMO:0.1Ho | 4.2(7) | 1.792(5) |
| SMO:0.5Ho | 4.1(7) | 1.794(6) |
| SMO:1.0Ho | 3.8(3) | 1.799(4) |
| SMO:0.5Tm | 4.1(7) | 1.791(7) |
| SMO:1.0Tm | 3.9(3) | 1.787(5) |
| Sample | Designation | Color | Growth/Annealing Atmosphere | Refined Composition 1 |
|---|---|---|---|---|
| (Na0.5Gd0.5)MoO4 | NGM-I 2 | Dark gray | N2 + 2 vol% O2/– | (Na0.493(3)Gd0.507)Mo(O3.920(5)⎕0.080) |
| (Na0.5Gd0.5)MoO4 | NGM-A 2 | Slightly yellow | N2 + 2 vol% O2/Air | (Na0.495(3)Gd0.505)Mo(O3.996(8)⎕0.004) |
| (Na0.5Gd0.5)MoO4 | NGM-Ar(1) 3 NGM-Ar(2) 2 (NGM 1:1) | Dark gray Almost colorless | Ar/– | (Na0.489(4)Gd0.511)(Mo0.995(3)⎕0.005)(O3.915(10)⎕0.085) (Na0.498(2)Gd0.502)(Mo0.999(4)⎕0.001)O4 |
| (Na2/7Gd4/7)MoO4 | NGM-2/7 2 (NGM 1:2) | Yellow | Air/– | (Na0.348(8)Gd0.528⎕0.124)(Mo6+0.996(3)⎕0.004)O4 |
| (Na6/15Gd8/15)MoO4 | NGM-6/15 3 | Dark gray | Air/– | (Na0.300(8)Gd0.576⎕0.124)Mo(O3.880(10)⎕0.120) |
| CaMoO4 [14] | Nominal Compositions and their Designation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| (Na1/2Gd1/2)MoO4 (NGM 1:1) | (Na2/7Gd4/7⎕1/7)MoO4 (NGM 1:2) | ||||||||
| CN | d, Å | CN for Scattering Paths | d, Å/σ2, Å2 | CN for Scattering Paths | d, Å/σ2, Å2 | ||||
| The parameters of local structure obtained from Gd L3-edge EXAFS modeling | |||||||||
 |  |  |  |  |  | ||||
| The parameters of local structure obtained from Mo K-edge EXAFS modeling | |||||||||
 |  |  |  |  |  | ||||
| Initial Charge Composition | Designation | Color | Refined Composition 1 |
|---|---|---|---|
| Bi4Ge3O12 | BGO(C) | colorless | (Bi3+3.994(40)⎕0.006)[Ge2.980(30)Bi5+0.020)O12.00 |
| Bi4Ge3O12 | BGO(P) | pink | (Bi3+3.987(45)⎕0.013)(Ge2.988(35)Bi5+0.012)(O11.98(5)⎕0.02) |
| Bi4Ge3O12:1.0% Dy | BGO:1.0Dy | colorless | (Bi3+3.952(47)Dy0.048)Ge3O12 |
| Bi4Ge3O12:0.1% Dy | BGO:0.1Dy | colorless | (Bi3+3.996(11)Dy0.004)Ge3O12 |
| Initial Charge Composition | Coordination Number (CN) | Interatomic Distance, Å | |
|---|---|---|---|
| Bi4Ge3O12:0.1Dy | Dy-O | 7.6 | 2.21 |
| Bi-3O | 2.5 | 2.13 | |
| Bi-3O | 2.5 | 2.58 | |
| Bi4Ge3O12:1.0Dy | Dy-O | 4.7 | 2.31 |
| Bi-3O | 2.9 | 2.13 | |
| Bi-3O | 2.9 | 2.57 | |
| Sample | Scattering Path | CN | dEXAFS, Å | σ2, Å2 | Rf,% |
|---|---|---|---|---|---|
| CsCdCl3:Bi | Cd1–Cl1 | 2 | 2.60 | 0.0040 | 1.4 |
| Cd2–Cl1 | 2 | 2.56 | 0.0022 | ||
| Cd2–Cl2 | 2 | 2.72 | 0.0019 | ||
| CsCdBr3 | Cd–Br | 6.0 | 2.73 | 0.0091 | 2.1 |
| Cd–Cd | 1.8 | 3.38 | 0.0078 | ||
| CsCdBr3:Bi | Cd–Br | 6.0 | 2.74 | 0.0094 | 0.8 |
| Cd–Cd | 2.3 | 3.36 | 0.0119 |
| Initial Charge Composition | Coordination Number (CN) | Interatomic Distance, Å | |
|---|---|---|---|
| TlCdI3:Bi1+ | Tl–I1 | 1.5 | 3.86 |
| Tl–I2 | 0.7 | 4.02 | |
| Tl–I3 | 3.0 | 4.09 | |
| TlCdCl3:Bi1+ | Tl–Cl1 | 2.0 | 2.53 |
| Tl–Cl2 | 2.0 | 2.73 | |
| Tl–Cl3 | 2.0 | 3.17 | |
| TlCdI3:Bi1+ | Cd–Cl1 | 1.3 | 2.58 |
| Cd–Cl2 | 4.4 | 2.71 | |
| TlCdCl3:Bi1+ | Cd–I1 | 0.7 | 2.87 |
| Cd–I2 | 3.8 | 3.00 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuz’micheva, G.; Kaurova, I. Peculiar Structural Effects in Pure and Doped Functional Single Crystals of Complex Compositions. Molecules 2020, 25, 2451. https://doi.org/10.3390/molecules25102451
Kuz’micheva G, Kaurova I. Peculiar Structural Effects in Pure and Doped Functional Single Crystals of Complex Compositions. Molecules. 2020; 25(10):2451. https://doi.org/10.3390/molecules25102451
Chicago/Turabian StyleKuz’micheva, Galina, and Irina Kaurova. 2020. "Peculiar Structural Effects in Pure and Doped Functional Single Crystals of Complex Compositions" Molecules 25, no. 10: 2451. https://doi.org/10.3390/molecules25102451
APA StyleKuz’micheva, G., & Kaurova, I. (2020). Peculiar Structural Effects in Pure and Doped Functional Single Crystals of Complex Compositions. Molecules, 25(10), 2451. https://doi.org/10.3390/molecules25102451