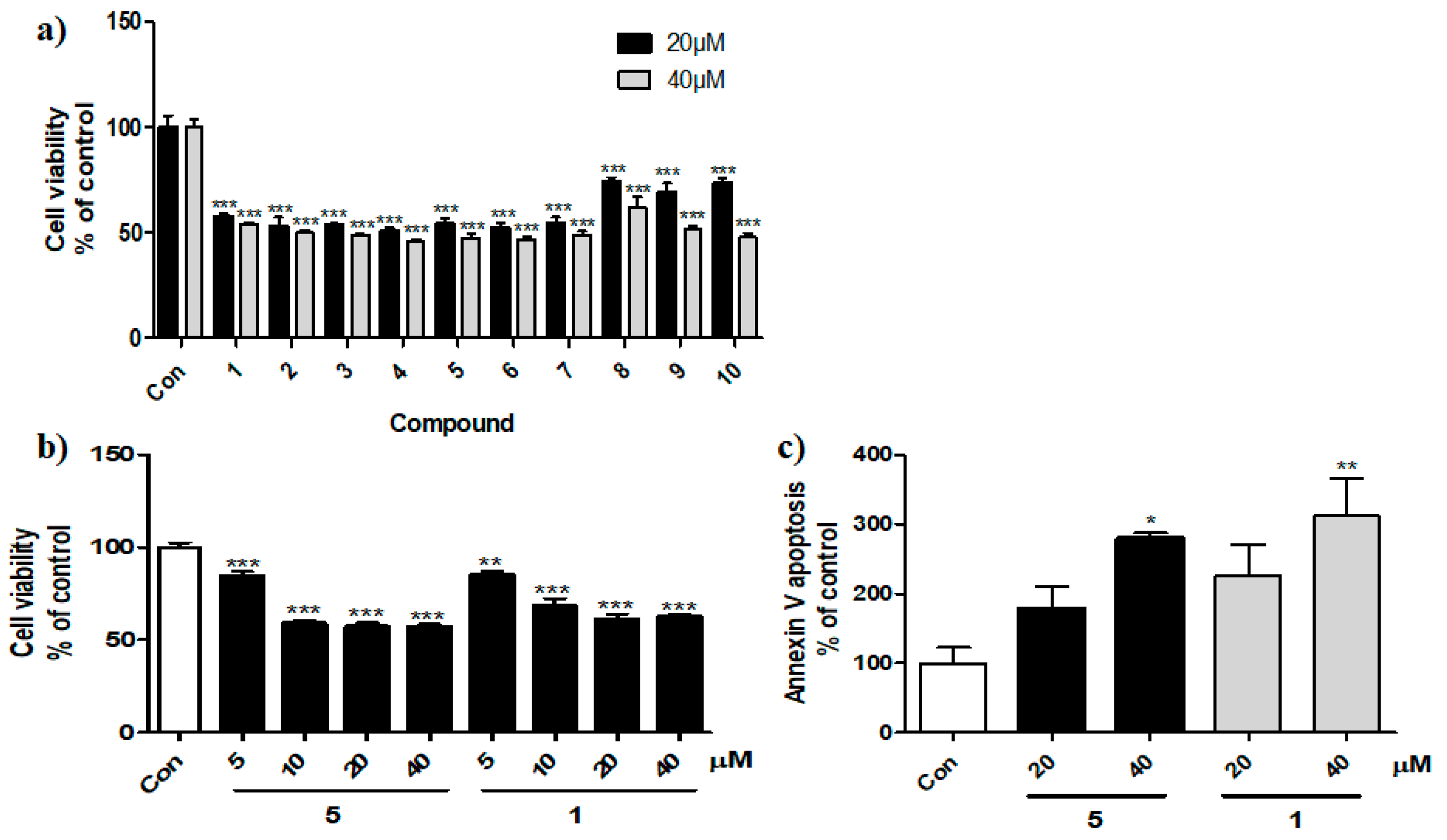
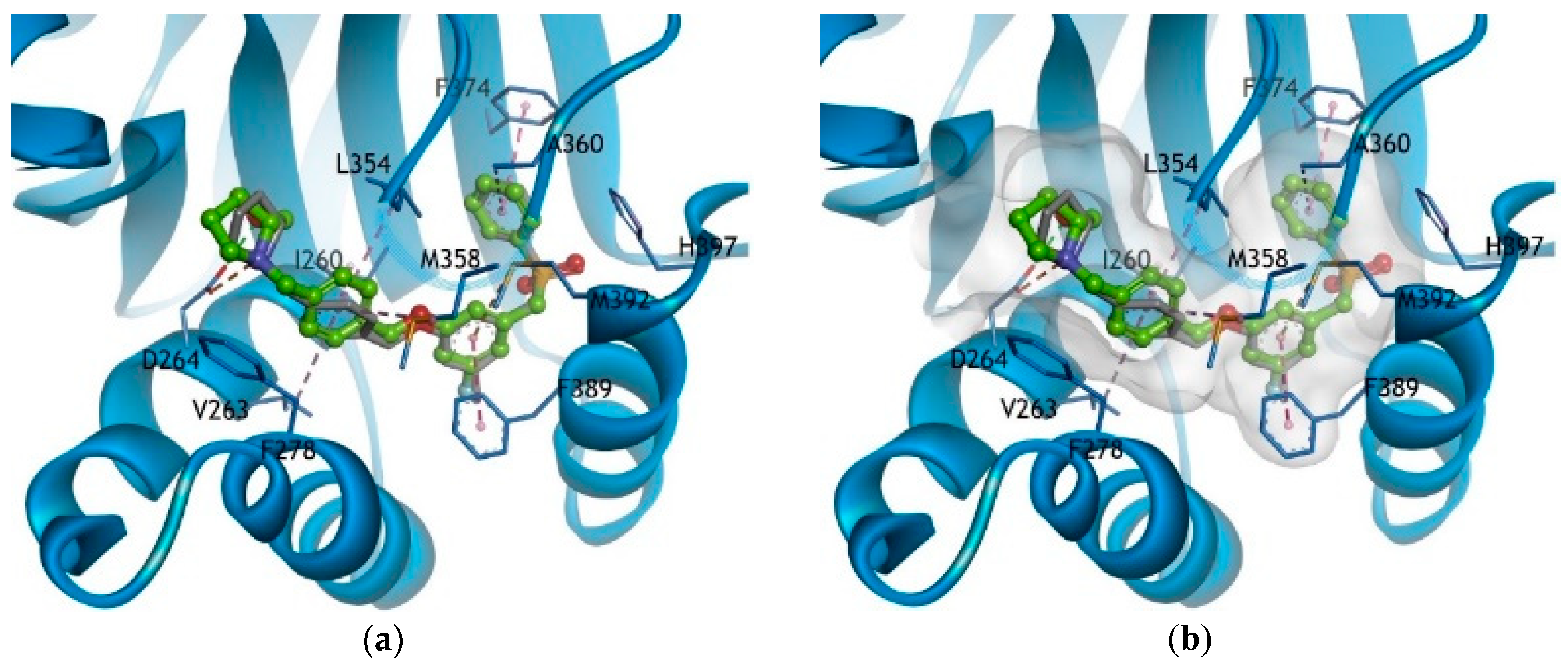
3.2. Synthesis
3.2.1. 3-Fluoro-5-methylphenyl acetate (11)
3-Fluoro-5-methyl phenol (2 g, 0.016 mol) was dissolved in pyridine (80 mL), acetic anhydride (3.0 mL, 0.032 mol) was added, and the mixture was stirred at room temperature for 12 h. Water was added to stop the reaction, and it was concentrated under reduced pressure after EtOAc extraction and MgSO4 drying. The mixture was separated by column chromatography (n-hexane:EtOAc = 20:1) to give compound 11 (2.64 g, 98%): 1H-NMR (500 MHz, CDCl3) δ 6.78–6.74 (m, 1H), 6.69 (ddd, J = 2.7, 1.4, 0.7 Hz, 1H), 6.66–6.62 (m, 1H), 2.33 (s, 3H), 2.27 (s, 3H); 13C-NMR (125 MHz, CDCl3) δ 169.3, 163.8, 161.8, 151.3, 151.2, 141.2, 141.1, 118.1, 113.8, 113.6, 106.9, 106.7, 21.5, 21.2; ESI-HRMS (M + H)+ m/z calcd for C9H10FO2 169.0665, found 169.0632.
3.2.2. 3-(Bromomethyl)-5-fluorophenyl acetate (14)
Compound 11 (1.5 g, 0.0089 mol) was placed in a sealed tube, dissolved in EtOAc (30 mL), and N-bromosuccinimide (1.59 g, 0.0089 mol) was added thereto, followed by stirring at 60 °C for 2 days. The reaction was terminated with water and EtOAc, and the organic layer was dried over MgSO4 and concentrated under reduced pressure. The resulting mixture was separated by column chromatography (n-hexane:EtOAc = 20:1) to give compound 14 (1.03 g, 47%): 1H-NMR (500 MHz, CDCl3) δ 7.00–6.96 (m, 1H), 6.94–6.93 (m, 1H), 6.80 (dt, J = 9.1, 2.2 Hz, 1H), 4.40 (s, 2H), 2.29 (s, 3H); 13C-NMR (125 MHz, CDCl3) δ 168.9, 163.7, 161.7, 151.6, 151.5, 140.6, 118.2, 118.1, 113.7, 113.5, 109.9, 109.7, 31.7, 31.6, 21.2; ESI-HRMS (M + H)+ m/z calcd for C9H9BrFO2 246.9770, found 246.9733.
3.2.3. 5-(Bromomethyl)-1,3-phenylene diacetate (15)
Orcinol (2 g, 0.016 mol) was dissolved in pyridine (80 mL), acetic anhydride (4.57 mL, 0.048 mol) was added thereto, and the mixture was stirred at room temperature for 12 h. Water was added to stop the reaction, and it was concentrated under reduced pressure after EtOAc extraction and MgSO4 drying. The resulting mixture 12 (1.7 g, 0.008 mol) was dissolved in EtOAc (50 mL) without purification, and N-bromosuccinimide (1.89 g, 0.01 mol) was added thereto and stirred at 60 °C for 2 days. The reaction was terminated with water and EtOAc, and the organic layer was dried over MgSO4 and concentrated under reduced pressure. The resulting mixture was separated by column chromatography (n-hexane:EtOAc = 5:1) to give compound 15 (1.19 g, 52%): 1H-NMR (500 MHz, CDCl3) δ 7.02 (d, J = 2.1 Hz, 2H), 6.86 (t, J = 2.1 Hz, 1H), 4.41 (s, 2H), 2.26 (s, 6H); 13C-NMR (125 MHz, CDCl3) δ 168.9, 515.1, 139.9, 119.7, 115.5, 31.9, 21.2; ESI-HRMS (M + H)+ m/z calcd for C11H12BrO4 286.9919, found 286.9947.
3.2.4. 3-Fluoro-5-((phenylsulfonyl)methyl)phenyl acetate (17)
Compound 14 (1.1 g, 0.0045 mol) was placed in a sealed tube, dissolved in THF/DMF (2/1, 30 mL), and benzene sulfinic acid sodium salt (2.2 g, 0.013 mol) was added thereto. The reaction was stirred for 3 days while heating to 80 °C. After the reaction was cooled to room temperature, the reaction was terminated with water and EtOAc, dried over MgSO4 and concentrated under reduced pressure. The mixture was separated by column chromatography (n-hexane:EtOAc = 2:1) to give compound 17 (1.1 g, 80%): 1H-NMR (500 MHz, CDCl3) δ 7.69–7.65 (m, 2H), 7.65–7.60 (m, 1H), 7.51–7.46 (m, 2H), 6.84 (t, J = 2.2 Hz, 0.5H), 6.82 (t, J = 2.2 Hz, 0.5H), 6.71 (t, J = 6.71 Hz, 2H), 6.68 (t, J = 2.1 Hz, 0.5H), 6.66 (t, J = 2.0 Hz, 0.5H), 4.26 (s, 2H), 2.25 (s, 3H); 13C-NMR (125 MHz, CDCl3) δ 168.8, 163.5, 161.5, 151.5. 151.4, 137.5, 134.2, 131.0, 130.9, 129.3, 128.7, 120.1, 115.5, 115.3, 110.5, 110.3, 62.2, 21.1; ESI-HRMS (M + H)+ m/z calcd for C15H14FO4S 309.0597, found 309.0543.
3.2.5. 5-((Phenylsulfonyl)methyl)-1,3-phenylene diacetate (18)
Compound 15 (0.9 g, 0.0031 mol) was dissolved in 25 mL of THF/DMF (2/1) mixed solvent in a sealed tube, benzenesulfinic acid sodium salt (777 mg, 0.0047 mol) was added, and the mixture was stirred at 80 °C for 4 days. The reaction was terminated with water and EtOAc, dried over MgSO4 and concentrated under reduced pressure. The mixture was separated by column chromatography (n-hexane:EtOAc = 3:1) to give compound 18 (744 mg, 68%): 1H-NMR (500 MHz, CDCl3) δ 7.53–7.61 (m, 2H), 7.59–7.56 (m, 1H), 7.44–7.43 (m, 2H), 6.87 (t, J = 2.1 Hz, 1H), 6.73 (d, J = 2.1 Hz, 2H), 4.24 (s, 2H), 2.21 (s, 6H); 13C-NMR (125 MHz, CDCl3) δ 168.5, 150.8, 137.3, 133.7, 128.9, 128.4, 121.2, 115.8, 61.9, 20.8; ESI-HRMS (M + H)+ m/z calcd for C17H17O6S 349.0746, found 349.0721.
3.2.6. 3-((Phenylsulfonyl)methyl)phenol (19)
m-Toly acetate (2 g, 0.013 mol) was dissolved in EtOAc (150 mL), and
N-bromosuccinimide (2.37 g, 0.0133 mol) was added thereto, followed by stirring for 3 days while heating to 70 °C. After cooling to room temperature, water and EtOAc were added thereto and the reaction was completed. The organic layer was separated, dried over MgSO
4, and concentrated to obtain a mixture of compound
13. Compound
13 was placed in a sealed tube and dissolved in 120 mL of THF/DMF (2/1). Benzenesulfinic acid sodium salt (4.27 g, 0.026 mol) was added thereto, followed by stirring for 3 days while heating to 80
oC. The reaction was cooled to room temperature, water and EtOAc were added and the reaction was completed. The organic layer was separated, dried over MgSO
4, and concentrated to obtain mixture compound
18. The reaction mixture was dissolved in 150 mL of MeOH/H
2O (1/1), NaHCO
3 (5 g) was added thereto, and the mixture was stirred for 12 h while heating at 40 °C. The reaction was cooled to room temperature, water and EtOAc were added thereto and the reaction was completed. The organic layer was separated, dried over MgSO
4, and concentrated. The mixture was separated by column chromatogrphy (
n-hexane:EtOAc = 2:1) to obtain compound
19. The chemical spectrum of compound
19 was consistent with that previously reported [
15].
3.2.7. 3-Fluoro-5-((phenylsulfonyl)methyl)phenol (20)
Compound 17 (1.0 g, 0.0032 mol) was dissolved in MeOH/H2O (1/1, 60 mL) and NaHCO3 (2 g) was added thereto. The reaction was stirred for 1 day while heating to 40 °C. The reaction was terminated with water and EtOAc, dried over MgSO4 and concentrated under reduced pressure. The mixture was separated by column chromatography (CH2Cl2:MeOH = 20:1) to give compound 20 (783 mg, 92%): 1H-NMR (500 MHz, CDCl3) δ 7.66 (dd, J = 5.2, 3.4 Hz, 2H), 7.62 – 7.57 (m, 1H), 7.46 (t, J = 7.8 Hz, 2H), 6.53 (t, J = 2.3 Hz, 0.5 H), 6.51 (t, J = 2.2 Hz, 0.5 H), 6.50 (t, J = 1.7 Hz, 1H), 6.25–6.23 (m, 0.5H), 6.23–6.21 (m, 0.5H), 4.21 (s, 2H); 13C-NMR (125 MHz, CDCl3) δ 164.3, 163.4, 162.3, 158.1, 158.0, 137.5, 134.2, 130.4, 130.3, 129.3, 129.2, 128.6, 114.1, 109.5, 109.4, 104.0, 103.8, 62.5; ESI-HRMS (M + H)+ m/z calcd for C13H12FO3S 267.0491, found 267.0466.
3.2.8. 5-((Phenylsulfonyl)methyl)benzene-1,3-diol (21)
Compound 18 (700 mg, 0.002 mol) was dissolved in MeOH (25 mL) and aq. NaHCO3 (10 mL) was added thereto. It was stirred at room temperature for 1 day. The reaction was terminated with water and EtOAc, dried over MgSO4 and concentrated under reduced pressure. The mixture was separated by column chromatography (CH2Cl2:MeOH = 10:1) to give compound 21 (436 mg, 82%): 1H NMR (500 MHz, CDCl3) δ 7.68 (dd, J = 8.4, 1.2 Hz, 2H), 7.62–7.57 (m, 1H), 7.49–7.41 (m, 2H), 6.30 (t, J = 2.2 Hz, 1H), 6.15 (d, J = 2.2 Hz, 2H), 5.28 (s, 2H); 13C-NMR (125 MHz, CDCl3) δ 158.0, 137.7, 133.9, 129.4, 129.0, 128.5, 109.5, 103.2, 62.6; ESI-HRMS (M + H)+ m/z calcd for C13H13O4S 265.0535, found 265.0581.
3.2.9. 4-((3-((Phenylsulfonyl)methyl)phenoxy)methyl)benzaldehyde (22)
Compound
19 (292 mg, 1.176 mmol) was dissolved in MeCN (10 mL), and K
2CO
3 (488 mg, 3.528 mmol) and 4-(bromomethyl)benzaldehyde (257 mg, 1.294 mmol) were added thereto, and the mixture was stirred for 12 h while heating to 60 °C. After cooling the reaction to room temperature, the reaction was terminated with water and EtOAc, dried over MgSO
4 and concentrated under reduced pressure. The reaction was washed with
n-hexane/EtOAc (3/1) to give compound
22. The chemical spectrum of compound
22 was consistent with that previously reported [
15].
3.2.10. 4-((3-Fluoro-5-((phenylsulfonyl)methyl)phenoxy)methyl)benzaldehyde (23)
Compound 20 (0.7 g, 0.0026 mol) was dissolved in MeCN (30 mL), and K2CO3 (1.09 g, 0.079 mol) and 4-(bromomethyl)benzaldehyde (569 mg, 0.0029 mmol) were added thereto. The reaction was stirred at room temperature for 12 h, the reaction was terminated with water and EtOAc, dried over MgSO4 and concentrated under reduced pressure. The mixture was washed with (n-hexane:EtOAc = 3:1) to give compound 23 (730 mg, 73%): 1H-NMR (500 MHz, CDCl3) δ 10.00 (s, 1H), 7.88 (d, J = 8.2 Hz, 2H), 7.66 (dd, J = 8.4, 1.2 Hz, 2H), 7.64–7.59 (m, 1H), 7.52 (d, J = 8.1 Hz, 2H), 7.49–7.43 (m, 2H), 6.63 (t, J = 2.3 Hz, 0.5H), 6.61 (t, J = 2.3 Hz, 0.5H), 6.56 (t, J = 1.7 Hz, 1H), 6.41–6.40 (m, 0.5H), 6.39–6.38 (m, 0.5H), 5.03 (s, 2H), 4.23 (s, 2H); 13C-NMR (125 MHz, CDCl3) δ 191.9, 164.2, 162.3, 159.6, 159.5, 143.0, 137.8, 136.2, 134.1, 131.0, 130.2, 129.2, 128.6, 127.6, 113.1, 110.9, 110.8, 103.5, 103.3, 69.6, 62.5; ESI-HRMS (M + H)+ m/z calcd for C21H18FO4S 385.0910, found 385.0981.
3.2.11. 4-((3-Hydroxy-5-((phenylsulfonyl)methyl)phenoxy)methyl)benzaldehyde (24)
Compound 21 (310 mg, 1.173 mmol) was dissolved in THF (15 mL), and K2CO3 (650 mg, 4.70 mmol) and 4-(bromomethyl)benzaldehyde (210 mg, 1.056 mmol) were added thereto. The reaction was stirred at room temperature for 12 h and terminated with water and EtOAc. Then, it was dried over MgSO4 and concentrated under reduced pressure. The mixture was separated by column chromatography (CH2Cl2:MeOH = 20:1) to give compound 24 (278 mg, 62%): 1H-NMR (500 MHz, CDCl3) δ 9.93 (s, 1H), 7.84 (d, J = 8.0 Hz, 2H), 7.64 – 7.60 (m, 2H), 7.58 (d, J = 7.3 Hz, 1H), 7.50 (d, J = 8.0 Hz, 2H), 7.45 (d, J = 7.7 Hz, 2H), 6.36 (t, J = 2.2 Hz, 1H), 6.17 (t, J = 1.6 Hz, 1H), 6.15 (t, J = 1.6 Hz, 1H), 4.96 (s, 2H), 4.20 (s, 2H); 13C-NMR (125 MHz, CDCl3) δ 192.7, 159.5, 158.3, 144.2, 137.7, 135.8, 133.9, 130.0, 129.0, 128.5, 127.5, 111.2, 108.5, 102.9, 69.1, 62.7; ESI-HRMS (M + H)+ m/z calcd for C21H19O5S 383.0953, found 383.0914.
3.2.12. (R)-(1-(4-((3-((Phenylsulfonyl)methyl)phenoxy)methyl)benzyl)pyrrolidin-2-yl)methanol (2)
Compound
22 (63 mg, 0.17 mmol) was dissolved in 1,2-dicholroethane (5 mL), and (
R)-(-)-prolinol (52 mg, 0.516 mmol) and sodium triacetoxyborohydride (73 mg, 0.34 mmol) were added thereto. The mixture was stirred for 12 h at room temperature. The reaction was terminated with water and EtOAc, dried over MgSO
4 and concentrated under reduced pressure. The mixture was separated by column chromatography (CH
2Cl
2:MeOH = 5:1) to give compound
2 (46 mg, 60%) [
15]:
1H-NMR (500 MHz, CDCl
3) δ 7.63 (dd,
J = 8.4, 1.2 Hz, 2H), 7.61–7.57 (m, 1H), 7.45 (d,
J = 7.6 Hz, 2H), 7.43 (d,
J = 7.6 Hz, 2H), 7.36 (d,
J = 8.1 Hz, 2H), 7.15–7.10 (m, 1H), 6.89 (ddd,
J = 8.3, 2.5, 0.8 Hz, 1H), 6.74–6.70 (m, 1H), 6.62 (d,
J = 7.6 Hz, 1H), 4.93 (s, 2H), 4.25 (s, 2H), 4.14 (d,
J = 13.1 Hz, 1H), 3.70 (dt,
J = 16.6, 8.3 Hz, 2H), 3.58 (dd,
J = 11.8, 3.9 Hz, 1H), 3.18–3.16 (m, 1H), 3.11–3.01 (m, 1H), 2.53 (dd,
J = 16.7, 8.6 Hz, 1H), 1.97 (ddd,
J = 15.8, 9.6, 5.8 Hz, 2H), 1.84 (ddtd,
J = 15.8, 12.3, 8.4, 4.6 Hz, 4H);
13C-NMR (125 MHz, CDCl
3) δ 158.7, 138.0, 133.9, 130.0, 129.7, 129.5, 129.0, 128.7, 127.9, 123.6, 117.0, 115.8, 69.6, 62.9, 61.5, 58.6, 54.3, 27.2, 23.5; ESI-HRMS (M + H)
+ m/z calcd for C
26H
30NO
4S 452.1896, found 452.1877.
3.2.13. (S)-1-(4-((3-((Phenylsulfonyl)methyl)phenoxy)methyl)benzyl)pyrrolidin-3-ol (3)
Using compound 22 (63 mg, 0.17 mmol) and (S)-(-)-3-hydroxy pyrrolidine (45 mg, 0.516 mmol), compound 3 (38 mg, 52%) was obtained by the same method as the synthesis method of compound 2: 1H-NMR (500 MHz, CDCl3) δ 7.63 (dd, J = 8.4, 1.2 Hz, 2H), 7.61–7.57 (m, 1H), 7.46–7.44 (m, 2H), 7.45–7.42 (m, 2H), 7.16–7.09 (m, 1H), 6.92–6.85 (m, 1H), 6.74–6.70 (m, 1H), 6.61 (d, J = 7.6 Hz, 1H), 4.92 (s, 2H), 4.40–4.39 (m, 1H), 4.25 (s, 2H), 3.90 (d, J = 7.9 Hz, 2H), 3.17–3.07 (m, 1H), 2.95 (d, J = 11.1 Hz, 1H), 2.88 (dd, J = 11.1, 5.0 Hz, 1H), 2.72 (dd, J = 15.2, 9.2 Hz, 1H), 2.25–2.13 (m, 1H), 1.95–1.82 (m, 1H); 13C-NMR (125 MHz, CDCl3) δ 158.7, 137.9, 137.0, 133.9, 130.0, 129.7, 129.4, 129.2, 128.7, 127.9, 123.7, 117.1, 115.7, 70.5, 69.6, 62.9, 62.0, 59.5, 52.4, 34.3; ESI-HRMS (M + H)+ m/z calcd for C25H28NO4S 438.1739, found 438.1702.
3.2.14. 1-(4-((3-((Phenylsulfonyl)methyl)phenoxy)methyl)benzyl)piperidin-4-ol (4)
Using compound 22 (63 mg, 0.17 mmol) and 4-hydroxypiperidine (52 mg, 0.516 mmol), compound 4 (53 mg, 69%) was obtained by the same method as the synthesis of compound 2: 1H-NMR (500 MHz, CDCl3) δ 7.63–7.60 (m, 2H), 7.58 (dd, J = 10.6, 4.4 Hz, 1H), 7.43 (dd, J = 8.1, 7.6 Hz, 2H), 7.40 (d, J = 7.9 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.12 (t, J = 7.9 Hz, 1H), 6.94–6.87 (m, 1H), 6.75–6.69 (m, 1H), 6.60 (d, J = 7.6 Hz, 1H), 4.92 (s, 2H), 4.25 (s, 2H), 3.77–3.74 (m, 1H), 3.68 (s, 2H), 2.94–2.82 (m, 1H), 2.57–2.44 (m, 2H), 2.03–1.90 (m, 2H), 1.71–1.57 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ 158.7, 137.9, 136.5, 133.9, 130.2, 129.7, 129.5, 129.1, 128.8, 127.7, 123.6, 117.0, 115.8, 69.7, 62.9, 61.9, 50.7, 33.0; ESI-HRMS (M + H)+ m/z calcd for C26H30NO4S 452.1896, found 452.1890.
3.2.15. (R)-(1-(4-((3-Fluoro-5-((phenylsulfonyl)methyl)phenoxy)methyl)benzyl)pyrrolidin-2-yl)methanol (5)
Using compound 23 (84 mg, 0.22 mmol) and (R)-(-)-prolinol (66 mg, 0.656 mmol), compound 5 (72 mg, 70%) was obtained by the same method as the synthesis of compound 2: 1H-NMR (500 MHz, CDCl3) δ 7.65 (dd, J = 8.3, 1.2 Hz, 2H), 7.64–7.59 (m, 1H), 7.51 (d, J = 8.0 Hz, 2H), 7.49–7.44 (m, 2H), 7.36 (d, J = 8.0 Hz, 2H), 6.60 (dt, J = 10.4, 2.3 Hz, 1H), 6.52–6.51 (m, 1H), 6.40–6.34 (m, 1H), 4.92 (s, 2H), 4.29 (d, J = 13.1 Hz, 1H), 4.21 (s, 2H), 3.90 (d, J = 13.1 Hz, 1H), 3.81–3.66 (m, 2H), 3.33–3.22 (m,2H), 2.74–2.66 (m, 1H), 2.04–1.79 (m, 4H); 13C-NMR (125 MHz, CDCl3) δ 164.2, 162.2, 159.8, 137.8, 137.0, 134.1, 130.8, 129.2, 128.6, 127.9, 113.1, 110.6, 110.5, 103.4, 103.2, 69.8, 67.4, 62.5, 61.3, 58.7, 54.0, 53.6, 50.7, 26.8, 23.4; ESI-HRMS (M + H)+ m/z calcd for C26H29FNO4S 470.1801, found 470.1819.
3.2.16. (S)-1-(4-((3-fluoro-5-((phenylsulfonyl)methyl)phenoxy)methyl)benzyl)pyrrolidin-3-ol (6)
Using compound 23 (65 mg, 0.17 mmol) and (S)-(-)-3-hydroxy pyrrolidine (44 mg, 0.51 mmol), compound 6 (43 mg, 56%) was obtained by the same method as the synthesis of compound 2: 1H-NMR (500 MHz, CDCl3) δ 7.68–7.64 (m, 2H), 7.64–7.60 (m, 1H), 7.50–7.45 (m, 2H), 7.40 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 8.1 Hz, 2H), 6.62 (dt, J = 10.4, 2.3 Hz, 1H), 6.52–6.51 (m, 1H), 6.40–6.35 (m, 1H), 4.92 (s, 2H), 4.22 (s, 2H), 4.08 (d, J = 13.1 Hz, 1H), 3.69 (dd, J = 11.5, 3.3 Hz, 1H), 3.61–3.49 (m, 2H), 3.15–3.06 (m, 1H), 2.97–2. 92 (m, 1H), 2.44 (dt, J = 15.7, 8.0 Hz, 1H), 2.02–1.82 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ 164.2, 162.3, 159.9, 137.8, 134.1, 130.8, 129.8, 129.2, 128.7, 127.8, 113.1, 110.6, 110.4, 103.5, 103.3, 70.1, 62.6, 61.6, 58.5, 54.4, 31.0; ESI-HRMS (M + H)+ m/z calcd for C25H27FNO4S 456.1645, found 456.1697.
3.2.17. 1-(4-((3-Fluoro-5-((phenylsulfonyl)methyl)phenoxy)methyl)benzyl)piperidin-4-ol (7)
Using compound 23 (65 mg, 0.17 mmol) and 4-hydroxypiperidine (51 mg, 0.51 mmol), compound 7 (56 mg, 71%) was obtained by the same method as the synthesis of compound 2: 1H-NMR (500 MHz, CDCl3) δ 7.66 (dd, J = 8.4, 1.2 Hz, 2H), 7.62 (ddt, J = 8.7, 7.4, 1.2 Hz, 1H), 7.49–7.45 (m, 2H), 7.36 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 8.1 Hz, 2H), 6.62 (dt, J = 10.4, 2.3 Hz, 1H), 6.54–6.51 (m, 1H), 6.39–6.34 (m, 1H), 4.91 (s, 2H), 4.22 (s, 2H), 3.79–3.65 (m, 1H), 3.55 (s, 2H), 2.86–2.70 (m, 2H), 2.31–2.17 (m, 2H), 1.94–1.86 (m, 2H), 1.65–1.56 (m, 2H); 13C-NMR (125 MHz, CDCl3) δ 164.2, 162.3, 160.0, 137.8, 134.1, 130.8, 129.7, 129.2, 128.7, 127.6, 113.1, 110.5, 110.3, 103.4, 103.2, 70.2, 62.7, 53.5, 50.9, 34.1, 31.0; ESI-HRMS (M + H)+ m/z calcd for C26H29FNO4S 470.1801, found 470.1840.
3.2.18. (R)-3-((4-((2-(Hydroxymethyl)pyrrolidin-1-yl)methyl)benzyl)oxy)-5-((phenylsulfonyl)methyl)phenol (8)
Compound 24 (58 mg, 0.15 mmol) was dissolved in 1,2-dichloroethane (5 mL), and (R)-(-)-prolinol (46 mg, 0.45 mmol) and sodium triacetoxyborohydride (64 mg, 0.303 mmol) were added. It was stirred at room temperature for 12 h. The reaction was terminated with water and EtOAc, dried over MgSO4 and concentrated under reduced pressure. The mixture was separated by column chromatography (CH2Cl2:MeOH = 5:1) to give compound 8 (41 mg, 58%): 1H-NMR (500 MHz, CDCl3/CD3OD = 3/1) δ 7.52–7.48 (m, 2H), 7.46 (td, J = 2.0, 1 Hz), 7.35–7.30 (m, 2H), 7.21 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 6.23 (t, J = 2.2 Hz. 1H), 6.08–6.02 (m, 1H), 6.01–5.98 (m, 1H), 4.72 (s, 2H), 3.17 (d, J = 1.6 Hz, 2H), 2.92 (dd, J = 5.7, 2.9 Hz, 1H), 2.83 (d, J = 3.3 Hz, 1H), 2.37 (dd, J = 16.7, 9.1 Hz, 1H), 1.91–1.76 (m, 2H), 1.70–1.55 (m, 4H); 13C-NMR (125 MHz, CDCl3/CD3OD = 3/1) δ 159.8, 158.2, 137.7, 133.9, 129.9, 129.7, 129.0, 128.5, 127.7, 111.0, 108.6, 103.0, 69.6, 65.7, 62.8, 62.1, 58.6, 54.3, 29.6, 27.3, 22.8.; ESI-HRMS (M + H)+ m/z calcd for C26H30NO5S 468.1845, found 468.1849.
3.2.19. (S)-1-(4-((3-Hydroxy-5-((phenylsulfonyl)methyl)phenoxy)methyl)benzyl)pyrrolidin-3-ol (9)
Compound 24 (58 mg, 0.15 mmol) was dissolved in 1,2-dichloroethane (5 mL), and then (S)-(-)-3-hydroxypyrrolidine (40 mg, 0.45 mmol) and sodium triacetoxyborohydride (64 mg, 0.455 mmol) were added. It was stirred at room temperature for 12 h. The reaction was terminated with water and EtOAc, dried over MgSO4 and concentrated under reduced pressure. The mixture was separated by column chromatography (CH2Cl2:MeOH = 5:1) to give compound 9 (44 mg, 64%): 1H-NMR (500 MHz, CDCl3/CD3OD = 3/1) δ 7.49 (dd, J = 5.9, 4.7 Hz, 2H), 7.46 (d, J = 7.5 Hz, 1H), 7.32 (t, J = 7.8 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 7.21 (d, J = 8.1 Hz, 2H), 6.23 (t, J = 2.1 Hz, 1H), 6.03 (d, J = 1.6 Hz, 1H), 6.01 (d, J = 1.7 Hz, 1H), 4.72 (s, 2H), 4.36–4.20 (m, 1H), 4.05 (s, 2H), 3.01–2.91 (m, 2H), 2.86–2.78 (m, 1H), 2.73 (d, J = 11.6 Hz, 1H), 2.12 – 1.95 (m, 1H), 1.78–1.64 (m, 1H); 13C-NMR (125 MHz, CDCl3/CD3OD = 3/1) δ 159.7, 158.2, 137.6(2C), 133.9, 130.0, 129.7, 129.0(2C), 128.5, 127.9, 111.0, 108.6, 103.0, 69.7, 69.4, 62.7, 61.4, 59.7, 52.4, 33.7; ESI-HRMS (M + H)+ m/z calcd for C25H28NO5S 454.1688, found 454.1673.
3.2.20. 1-(4-((3-Hydroxy-5-((phenylsulfonyl)methyl)phenoxy)methyl)benzyl)piperidin-4-ol (10)
Compound 24 (58 mg, 0.15 mmol) was dissolved in 1,2-dichloroethane (5 mL), 4-hydroxypiperidine (46 mg, 0.45 mmol) and sodium triacetoxyborohydride (64 mg, 0.455 mmol) were added and stirred at room temperature for 12 h. The reaction was terminated with water and EtOAc, dried over MgSO4 and concentrated under reduced pressure. The mixture was separated by column chromatography (CH2Cl2:MeOH = 5:1) to give compound 10 (48 mg, 68%): 1H-NMR (500 MHz, CDCl3/CD3OD = 3/1) δ 7.54–7.48 (m, 2H), 7.48–7.44 (m, 1H), 7.33 (dd, J = 7.0, 1.2 Hz, 2H), 7.26 (d, J = 8.1 Hz, 2H), 7.21 (d, J = 8.1 Hz, 2H), 6.23 (t, J = 2.2 Hz, 1H), 6.03 (d, J = 1.6 Hz, 1H), 6.01 (d, J = 1.6 Hz, 1H), 4.73 (s, 2H), 4.05 (s, 2H), 3.63–3.65 (m, 1H), 2.85 (ddd, J = 11.2, 7.8, 3.3 Hz, 2H), 2.47 (d, J = 13.7 Hz, 2H), 1.82 (dd, J = 14.8, 6.0 Hz, 2H), 1.67 – 1.62 (m, 2H); 13C NMR (125 MHz, CDCl3/CD3OD = 3/1) δ 159.9, 158.4, 137.8, 134.1, 130.8, 129.8, 129.2, 128.7, 127.9, 111.2, 108.8, 103.2, 69.6, 62.9, 61.4, 53.9, 41.5, 31.8, 29.8; ESI-HRMS (M + H)+ m/z calcd for C26H30NO5S 468.1845, found 468.1829.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}