
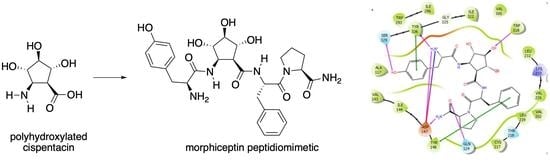
New Morphiceptin Peptidomimetic Incorporating (1S,2R,3S,4S,5R)-2-Amino-3,4,5-trihydroxycyclopen-tane-1-carboxylic acid: Synthesis and Structural Study

 ,
,  ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
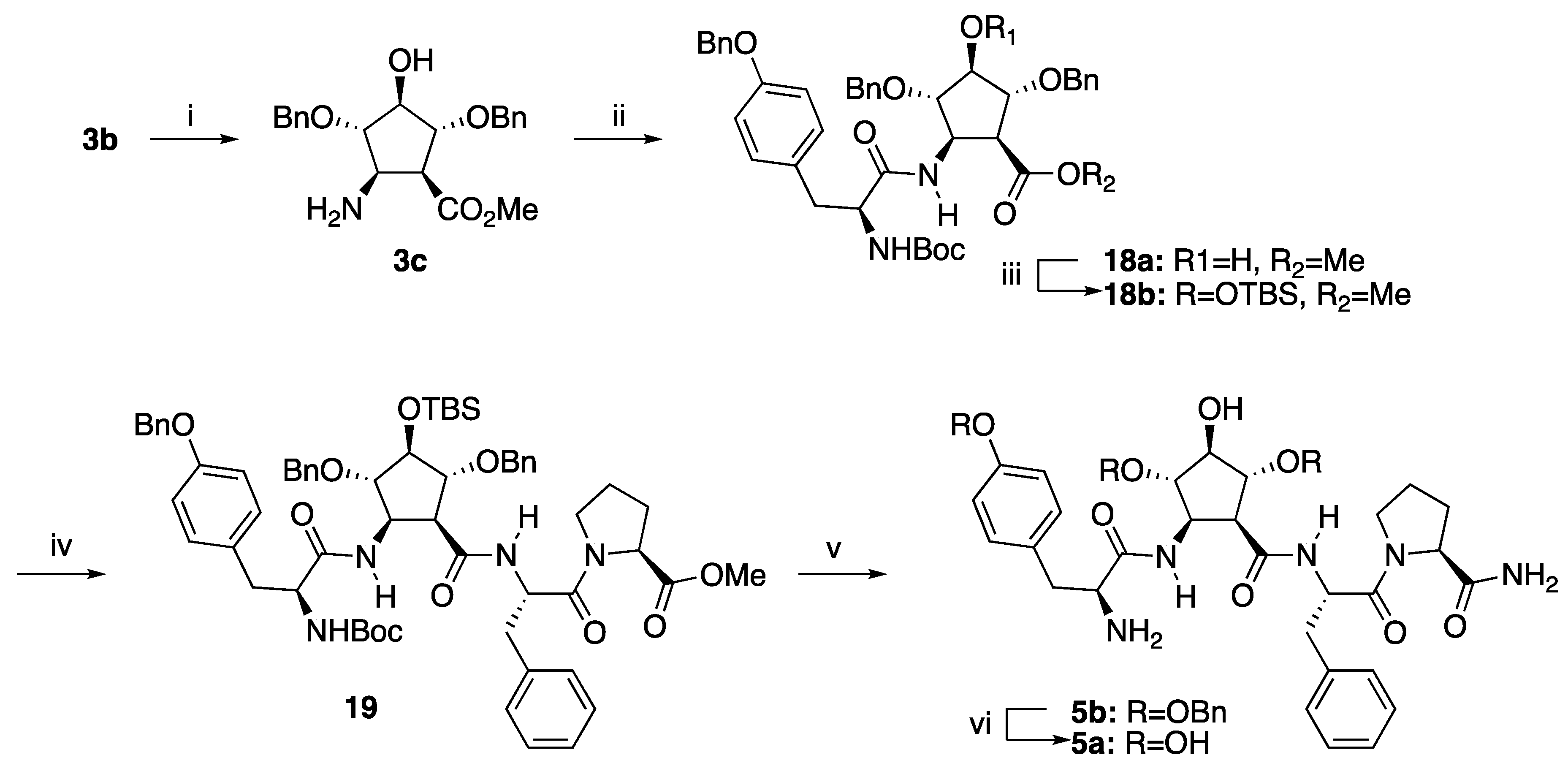
2.1. Synthesis of the Polyhydroxylated 2-Aminocyclopentanecarboxylic acid (3b)
2.2. Preparation of the Morphiceptin Peptidomimetic 5a
2.3. Docking Studies
2.4. Conformation of the Morphiceptin Peptidomimetic 5a in Solution
3. Materials and Methods
3.1. Chemistry
3.1.1. (1S,4S,5R,6S,7R)-6,7-Dibenzyloxy-5-nitro-2-oxabicyclo-[2.2.1]heptan-3-one (8)
3.1.2. (1S,4S,5R,6S,7R)-6,7-Dibenzyloxy-5-t-butoxycarbonylamino-2-oxabicyclo-[2.2.1]heptan-3-one (9)
3.1.3. Methyl (1S,2R,3S,4S,5R)-2,4-Dibenzyloxy-5-t-butoxycarbonylamino-3-hydroxycyclo-pentanecarboxylate (Pcp) (3b)
3.1.4. (1S3S,4S,5R,6S,7R)-6,7-Dibenzyloxy-3-methoxy-5-nitro-2-oxabicycle[2.2.1]heptane (12) and (1S,3R,4S,5R,6S,7R)-6,7-Dibenzyloxy-3-methoxy-5-nitro-2-oxabicycle[2.2.1]heptane (13)
3.1.5. Methyl (1S,2R,3S,4S,5R)-5-(N-tert-butoxycarbonyl-O-benzyloxycarbonyl-l-tyrosylamino)-2,4-dibenzyloxy-3-hydroxy-cyclopentanoate (18a)
3.1.6. Methyl (1S,2R,3S,4S,5R)-5-(N-tert-butoxycarbonyl-O-benzyloxycarbonyl-l-tyrosylamino)-2,4-dibenzyloxy-3-tert-butyldimethylsilyloxy-cyclopentanoate (18b)
3.1.7. (1S,2R,3S,4S,5R)-5-[(N-tert-butoxycarbonyl-O-benzyloxycarbonyl)-l-tyrosylamino]-2,4-dibenzyloxy-3-tert-butyldimethylsilyloxy-1-(metoxy-L-prolyl-L-phenylalanylcarbonyl) Cyclopentane (19)
3.1.8. (1S,2R,3S,4S,5R)-5-(O-benzyloxycarbonyl)-l-tyrosylamino-2,4-dibenzyloxy-1-(amido-l-prolyl-l-phenylalanylcarbonyl) Cyclopentane (5b)
3.1.9. (1S,2R,3S,4S,5R)-5-(l-tyrosylamino)-1-(amido-Prolyl-l-phenylalanyloxycarbonyl) Cyclopentane (5a)
3.2. Conformational Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TBAF | Tetrabuthyl Ammonium Fluoride |
| THF | Tetrahydrofuran |
| TFA | Trifluoroacetic Acid |
| TBTU | 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate |
| ESI-HRMS | Electrospray Ionization High Resolution Mass Spectrometry |
| DIEA | N,N-Diisopropylethylamine |
| DMF | for N,N-Dimethylformamide |
| NOE | Nucelar Overhouse Effect |
| DOWEX | DOWEX* ion exchange resins |
| COSY | COrrelation SpectroscopY |
| TOCSY | TOtal Correlation SpectroscopY |
| ROESY | Rotating-frame Overhauser Effect SpectroscopY |
| HSQC | Heteronuclear Simple Quantum Coherence |
| HMBC | Heteronuclear Multiple Bond Correlation |
| RMSD | Root-Mean-Square Deviation |
References
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Chang, K.J.; Killian, A.; Hazum, E.; Cuatrecasas, P.; Chang, J.K. Morphiceptin (H-Tyr-Pro-Phe-Pro-NH2): A potent and specific agonist for Morphine (μ) receptors. Science 1981, 212, 75–77. [Google Scholar] [CrossRef]
- Brantl, V.; Teschemacher, H.; Blasig, J.; Henschen, A.; Lottspeich, F. Novel opioid peptides derived from casein (beta-casomorphins). I. Isolation from bovine casein peptone. Physiol. Chem. 1979, 360, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Janecka, A.; Fichna, J.; Mirowski, M.; Janecki, T. Structure-activity relationship, conformation and pharmacology studies of morphiceptin analogues-selective μ-opioid receptor ligands. Mini Rev. Med. Chem. 2002, 2, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Antošová, Z.; Macková, M.; Král, V.; Macek, T. Therapeutic application of peptides and proteins: Parenteral forever? Trends Biotechnol. 2009, 27, 628–635. [Google Scholar] [CrossRef]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, M.T.; Francis, J.N.; Redman, J.S.; Kay, M.S. Protease-Resistant Peptide Design—Empowering Nature’s Fragile Warriors Against HIV. Biopolymers 2012, 98, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Horne, W.S.; Gellman, S.H. Foldamers with Heterogeneous Backbones. Acc. Chem. Res. 2008, 41, 1399–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, N.; Harrison, J.E.; Blackburn, L.A.; Martin, J.N.; Deeks, S.G.; Doms, R.W. Clinical resistance to Enfuvirtide does not affect susceptibility of human immunodeficiency virus type 1 to other classes of entry inhibitors. J. Virol. 2007, 81, 3240–3250. [Google Scholar] [CrossRef] [Green Version]
- Cheng, R.P.; Gellman, S.H.; DeGrado, W.F. β-Peptides: From Structure to Function. Chem. Rev. 2001, 101, 3219–3232. [Google Scholar] [CrossRef]
- Cabrele, C.; Martinek, T.A.; Reiser, O.; Berlicki, Ł. Peptides Containing β-Amino Acid Patterns: Challenges and Successes in Medicinal Chemistry. J. Med. Chem. 2014, 57, 9718–9739. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.-I.; Purcell, A.W.; Devi, R.; Lew, R.; Rossjohn, J.; Smith, A.I.; Perlmutter, P. β-Amino acid-containing hybrid peptides—new opportunities in peptidomimetics. Org. Biomol. Chem. 2007, 5, 2884–2890. [Google Scholar] [CrossRef]
- Kuhl, A.; Hahn, M.G.; Dumić, M.; Mittendorf, J. Alicyclic β-amino acids in Medicinal Chemistry. Aminol. Acid. 2005, 29, 89–100. [Google Scholar] [CrossRef]
- Fülöp, F.; Martinek, T.A.; Tóth, G.K. Application of alicyclic β-amino acids in peptide chemistry. Chem. Soc. Rev. 2006, 35, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Bailey, C.W.; Claridge, T.D.W.; Davies, S.G.; Ling, K.B.; Odell, B.; Rees, T.L.; Roberts, P.M.; Russell, A.J.; Smith, A.D.; et al. A systematic study of the solid state and solution phase conformational preferences of β-peptides derived from transpentacin. Tetrahedron Asymm. 2010, 21, 1797–1815. [Google Scholar] [CrossRef]
- Konishi, M.; Nishio, M.; Saitoh, K.; Miyaki, T.; Oki, T.; Kawaguchi, H. Cispentacin, a new antifungal antibiotic. I. Production, isolation, physico-chemical properties and structure. J. Antibiot. 1989, 42, 1749–1755. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Iwamoto, T.; Tsujii, E.; Ezaki, M.; Fujie, A.; Okuhara, M.; Kohsaka, M.; Imanaka, H.; Kawabata, K. FR109615, a new antifungal antibiotic from Streptomyces setonii. Taxonomy, fermentation, isolation, physico-chemical properties and biological activity. J. Antibiot. 1990, 43, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zadina, J.E.; Hackler, L.; Ge, L.J.; Kastin, A.J. A potent and selective endogenous agonist for the μ-opiate receptor. Nature 1997, 386, 499–502. [Google Scholar] [CrossRef]
- Janecka, A.; Fichna, J.; Wiercioch, R.; Mirowski, M. Synthesis of novel morphiceptin analogues modified in position 3 and their μ-opioid receptors in experimental mammary adenocarcinoma. Bioorg. Med. Chem. 2003, 11, 3855–3860. [Google Scholar] [CrossRef]
- Fichna, J.; Chung, N.; Costentin, J.; Schiller, P.W.; Janecka, A. [Dmt1, D-1-Nal3]morphiceptin, a novel opioid peptide analog with high analgesic activity. Peptides 2008, 29, 633–638. [Google Scholar] [CrossRef]
- Giordano, C.; Sansone, A.; Masi, A.; Lucente, G.; Punzi, P.; Mollica, A.; Pinnen, F.; Feliciani, F.; Cacciatore, I.; Davis, P.; et al. Synthesis and activity of endomorphin-2 and morphiceptin analogues with proline surrogates in position 2. Eur. J. Med. Chem. 2010, 45, 4594–4600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, T.; Pröbsti, A.; Schiller, P.W.; Goodman, M. Biological and conformational studies of [Val4]morphiceptin and [D-Val4]morphiceptin analogs incorporating cis-2-aminocyclopentane carboxylic acid as a peptidomimetic for proline. Int. J. Pept. Protein Res. 1991, 37, 364–381. [Google Scholar] [CrossRef] [PubMed]
- Mierke, D.F.; Nossner, G.; Schiller, P.W.; Goodman, M. Morphiceptin analogs containing 2 aminocyclopentane carboxylic acid as a peptidomimetic for proline. Int. J. Pept. Protein Res. 1990, 35, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Soengas, R.G.; Estévez, J.C.; Estévez, R. Stereocontrolled Transformation of Nitrohexofuranoses into Cyclopentylamines via 2-Oxabicyclo[2.2.1]heptanes: Incorporation of Polyhydroxylated Carbocyclic β-Amino Acids into Peptides. J. Org. Lett. 2003, 5, 1423–1425. [Google Scholar] [CrossRef] [PubMed]
- Soengas, R.G.; Pampín, M.B.; Estévez, J.C.; Estévez, R.J. Stereocontrolled transformation of nitrohexofuranoses into cyclopentylamines via 2-oxabicyclo[2.2.1]heptanes. Part 2: Synthesis of (1S,2R,3S,4S,5R)-3,4,5-trihydroxy-2-aminocyclopentanecarboxylic acid. Tetrahedron Asymm. 2005, 16, 205–211. [Google Scholar] [CrossRef]
- Estévez, A.M.; Soengas, R.G.; Otero, J.M.; Estévez, J.C.; Nash, R.J.; Estévez, R.J.; Sussman, F. Stereocontrolled transformation of nitrohexofuranoses into cyclopentylamines via 2-oxabicyclo[2.2.1]heptanes. III: Synthesis of enantiopure methyl (1S,2S,3R,4S,5R)-2-amino-3,4,5-trihydroxycyclopentanecarboxylate. Tetrahedron Asymm. 2010, 21, 21–26. [Google Scholar] [CrossRef]
- Fernández, F.; PAmpín, B.; González, M.A.; Estévez, J.C.; Estévez, R.J.; Sussman, F. Stereocontrolled transformation of nitrohexofuranoses into cyclopentylamines via 2-oxabicyclo[2.2.1]heptanes. Part VI: Synthesis and incorporation of the novel polyhydroxylated 5-aminocyclopent-1-enecarboxylic acids into peptides. Tetrahedron Asymm. 2010, 21, 2021–2026. [Google Scholar] [CrossRef]
- Estévez, A.M.; Soengas, R.G.; Tato, R.; Thomas, P.; Estévez, J.C.; Estévez, R.J.; Sussman, F. Studies on the stereocontrolled transformation of nitrohexofuranoses into 2-oxabicyclo[2.2.1]heptanes. V: Synthesis of enantiopure methyl (1R,2R,4S)-2-amino-4-hydroxycyclopentanecarboxylate. Tetrahedron Asymm. 2010, 21, 116–122. [Google Scholar] [CrossRef]
- Estévez, A.M.; Soengas, R.G.; Thomas, P.; Alegre, M.; Balo, R.; Estévez, J.C.; Estévez, R.J. Stereocontrolled transformation of nitrohexofuranoses into cyclopentylamines via 2-oxabicyclo[2.2.1]heptanes. Part 6: Synthesis and incorporation into peptides of the first reported 2,3-dihydroxycyclopentanecarboxylic acid. Tetrahedron Asymm. 2014, 25, 583–590. [Google Scholar] [CrossRef]
- Benedek, G.; Palkó, M.; Wéber, E.; Martinek, T.A.; Forró, E.; Fülöp, F. Efficient synthesis of hydroxy-substituted cispentacin derivatives. Eur. J. Org. Chem. 2008, 3724–3730. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Kuszewski, J.J.; Clore, G.M. Using Xplor-NIH for NMR molecular structure determination. Progr. NMR Spectrosc. 2006, 48, 47–62. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLano, W.L. The PyMOL molecular graphics system. 2002. Available online: http://www.pymol.org (accessed on 1 June 2020).
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the μ-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity-a rapid access to atomic charges. Tetrahedron 1980, 35, 3219–3228. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Maestro, Version 11.8; Schrödinger, LLC: New York, NY, USA, 2018.
Sample Availability: Samples of the compounds are not available from the authors. |







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soengas, R.; Lorca, M.; Pampín, B.; Sánchez-Pedregal, V.M.; Estévez, R.J.; Estévez, J.C. New Morphiceptin Peptidomimetic Incorporating (1S,2R,3S,4S,5R)-2-Amino-3,4,5-trihydroxycyclopen-tane-1-carboxylic acid: Synthesis and Structural Study. Molecules 2020, 25, 2574. https://doi.org/10.3390/molecules25112574
Soengas R, Lorca M, Pampín B, Sánchez-Pedregal VM, Estévez RJ, Estévez JC. New Morphiceptin Peptidomimetic Incorporating (1S,2R,3S,4S,5R)-2-Amino-3,4,5-trihydroxycyclopen-tane-1-carboxylic acid: Synthesis and Structural Study. Molecules. 2020; 25(11):2574. https://doi.org/10.3390/molecules25112574
Chicago/Turabian StyleSoengas, Raquel, Marcos Lorca, Begoña Pampín, Víctor M. Sánchez-Pedregal, Ramón J. Estévez, and Juan C. Estévez. 2020. "New Morphiceptin Peptidomimetic Incorporating (1S,2R,3S,4S,5R)-2-Amino-3,4,5-trihydroxycyclopen-tane-1-carboxylic acid: Synthesis and Structural Study" Molecules 25, no. 11: 2574. https://doi.org/10.3390/molecules25112574
APA StyleSoengas, R., Lorca, M., Pampín, B., Sánchez-Pedregal, V. M., Estévez, R. J., & Estévez, J. C. (2020). New Morphiceptin Peptidomimetic Incorporating (1S,2R,3S,4S,5R)-2-Amino-3,4,5-trihydroxycyclopen-tane-1-carboxylic acid: Synthesis and Structural Study. Molecules, 25(11), 2574. https://doi.org/10.3390/molecules25112574