
An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study of the Halogen Bond with Explicit Analysis of Electron Correlation




Abstract
:1. Introduction
2. Results and Discussion
2.1. Minima Configurations
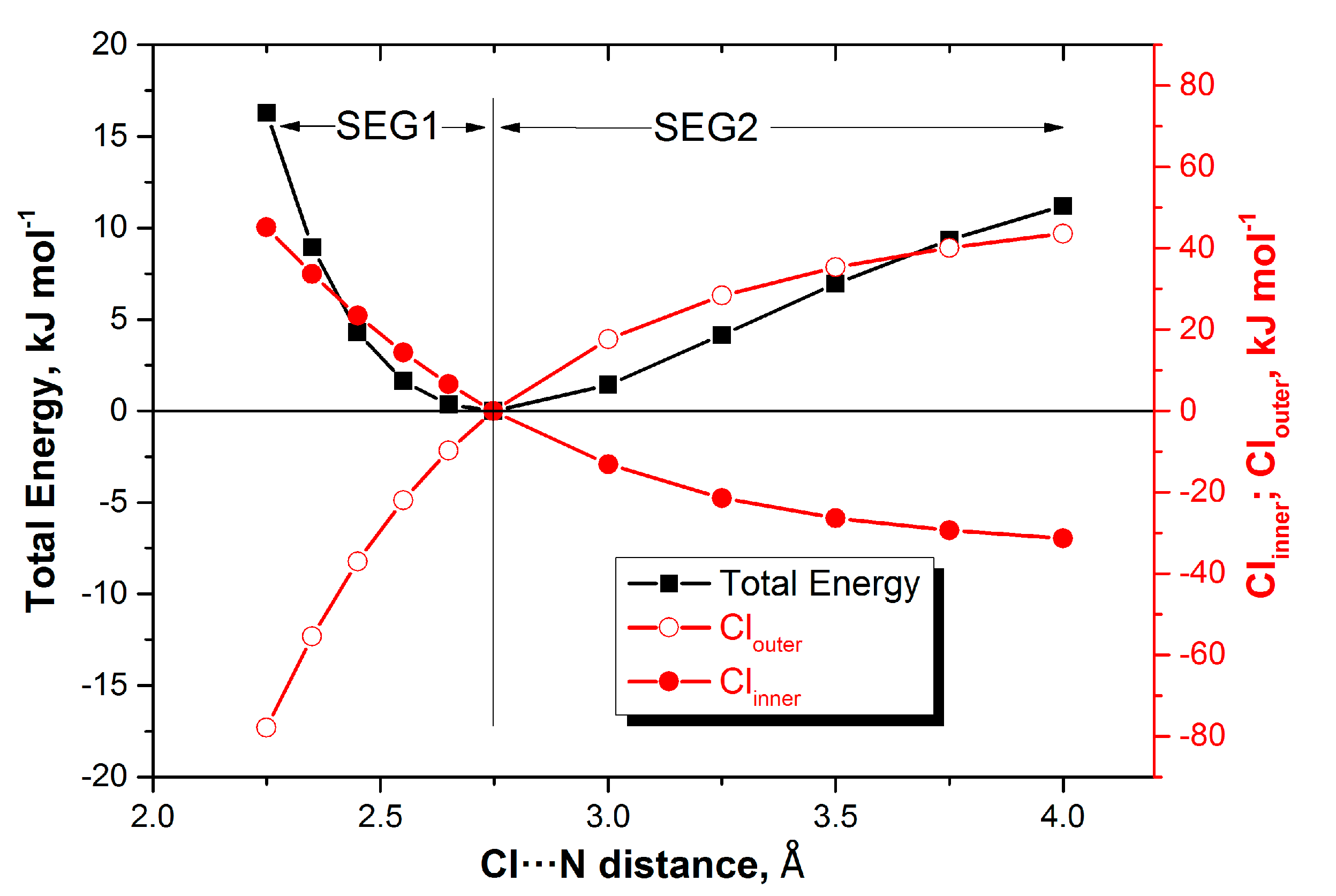
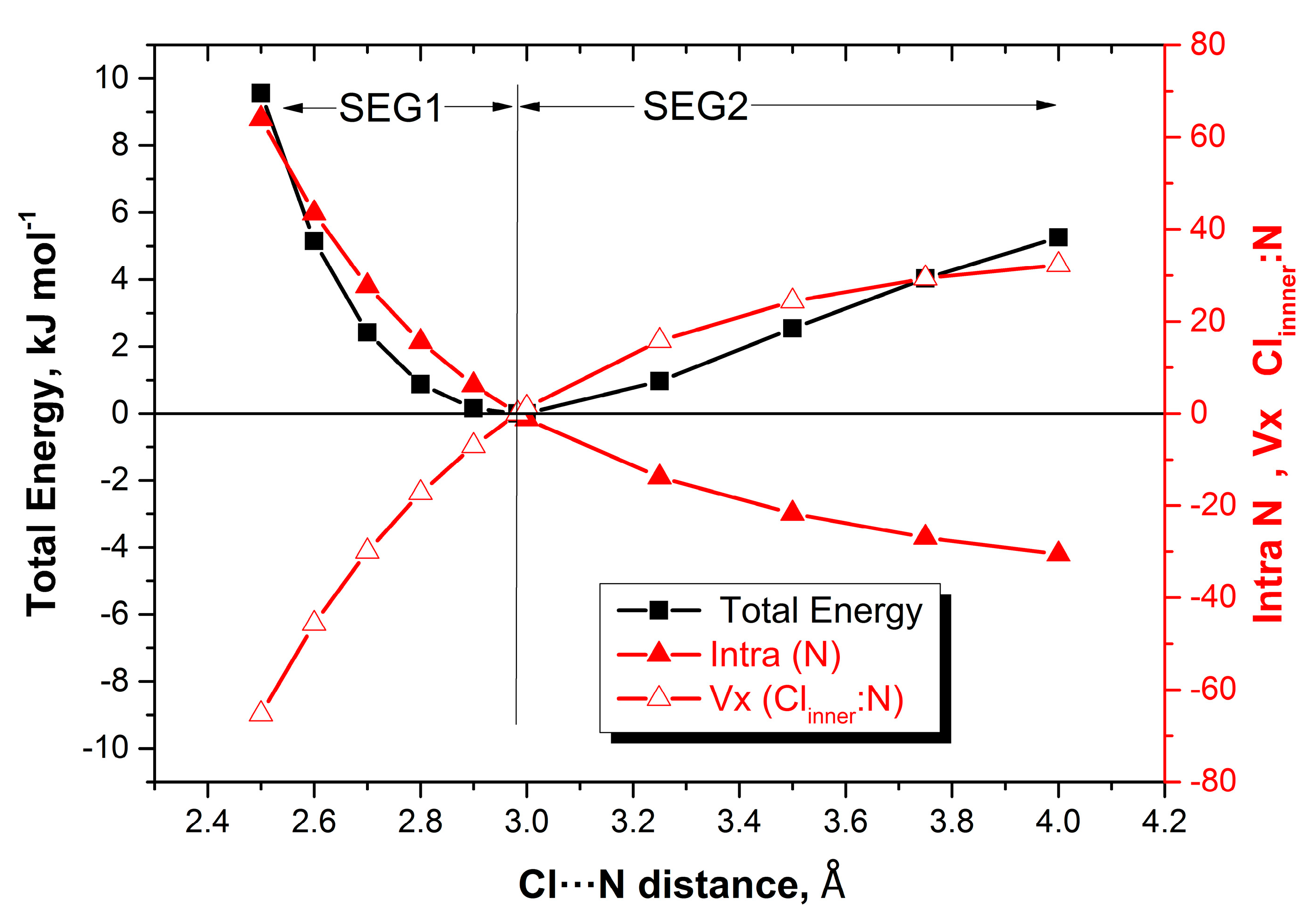
2.2. Energy Profiles
3. Computational Methods
3.1. Energy Profiles
3.2. Interacting Quantum Atoms (IQA)
3.3. Partitioning of Electron Correlation
3.4. The Relative Energy Gradient (REG) Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Costa, P.J. The halogen bond: Nature and applications. Phys. Sci. Rev. 2017, 2, 1–16. [Google Scholar] [CrossRef]
- Tognetti, V.; Joubert, L. Following halogen bonds formation with bader’s atoms-in-molecules theory. In Challenges and Advances in Computational Chemistry and Physics dedicated to “Applications of Topological Methods in Molecular Chemistry”; Chauvin, R., Lepetit, C., Alikhani, E., Silvi, B., Eds.; Springer: Basel, Switzerland, 2016; pp. 435–459. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bond: A long overlooked interaction. In Topics in Curr Chem: Halogen Bonding; Metrangolo, P., Resnati, G., Eds.; Springer: Basel, Switzerland, 2015; Volume 358, pp. 1–18. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiraju Gautam, R.; Ho, P.S.; Kloo, L.; Legon Anthony, C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure. Appl. Chem. 2013, 85, 1711. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Frontera, A. Not only hydrogen bonds: Other noncovalent interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Mulliken, R.S. Structures of complexes formed by halogen molecules with aromatic and with oxygenated solvents1. J. Am. Chem. Soc. 1950, 72, 600–608. [Google Scholar] [CrossRef]
- Wang, C.; Danovich, D.; Mo, Y.; Shaik, S. On the nature of the halogen bond. J. Chem. Theory Comput. 2014, 10, 3726–3737. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Danovich, D.; Shaik, S.; Mo, Y. Halogen bonds in novel polyhalogen monoanions. Chem. A Eur. J. 2017, 23, 8719–8728. [Google Scholar] [CrossRef] [PubMed]
- Thirman, J.; Engelage, E.; Huber, S.M.; Head-Gordon, M. Characterizing the interplay of Pauli repulsion, electrostatics, dispersion and charge transfer in halogen bonding with energy decomposition analysis. Phys. Chem. Chem. Phys. 2018, 20, 905–915. [Google Scholar] [CrossRef]
- Huber, S.M.; Jimenez-Izal, E.; Ugalde, J.M.; Infante, I. Unexpected trends in halogen-bond based noncovalent adducts. Chem. Commun. 2012, 48, 7708–7710. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen and halogen bonds are ruled by the same mechanisms. Phys. Chem. Chem. Phys. 2013, 15, 7249–7259. [Google Scholar] [CrossRef] [PubMed]
- Řezáč, J.; de la Lande, A. On the role of charge transfer in halogen bonding. Phys. Chem. Chem. Phys. 2017, 19, 791–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting quantum atoms: A correlated energy decomposition scheme based on the quantum theory of atoms in molecules. J. Chem. Theor. Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.; Martín Pendás, A.; Popelier, P.L.A. Extension of the interacting quantum atoms (IQA) approach to B3LYP level density functional theory. Phys. Chem. Chem. Phys. 2016, 18, 20986–21000. [Google Scholar] [CrossRef] [Green Version]
- Syzgantseva, O.A.; Tognetti, V.; Joubert, L. On the physical nature of halogen bonds: A QTAIM study. J. Phys. Chem. A 2013, 117, 8969–8980. [Google Scholar] [CrossRef]
- Riley, K.E.; Hobza, P. Investigations into the nature of halogen bonding including symmetry adapted perturbation theory analyses. J. Chem. Theory Comput. 2008, 4, 232–242. [Google Scholar] [CrossRef]
- Riley, K.E.; Murray, J.S.; Fanfrlík, J.; Řezáč, J.; Solá, R.J.; Concha, M.C.; Ramos, F.M.; Politzer, P. Halogen bond tunability II: The varying roles of electrostatic and dispersion contributions to attraction in halogen bonds. J. Mol. Model. 2013, 19, 4651–4659. [Google Scholar] [CrossRef]
- Riley, K.E.; Murray, J.S.; Politzer, P.; Concha, M.C.; Hobza, P. Br···O complexes as probes of factors affecting halogen bonding: Interactions of bromobenzenes and bromopyrimidines with acetone. J. Chem. Theory Comput. 2009, 5, 155–163. [Google Scholar] [CrossRef]
- Stone, A.J. Are halogen bonded structures electrostatically driven? J. Am. Chem. Soc. 2013, 135, 7005–7009. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. The intrinsic strength of the halogen bond: Electrostatic and covalent contributions described by coupled cluster theory. Phys. Chem. Chem. Phys. 2016, 18, 33031–33046. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-hole bond vs π-hole bond: A comparison based on halogen bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.P.; Swart, M. Intramolecular halogen–halogen bonds? Phys. Chem. Chem. Phys. 2013, 15, 11543–11553. [Google Scholar] [CrossRef] [Green Version]
- McDonagh, J.L.; Vincent, M.A.; Popelier, P.L.A. Partitioning dynamic electron correlation energy: Viewing Møller-Plesset correlation energies through Interacting Quantum Atom (IQA) energy partitioning. Chem. Phys. Lett. 2016, 662, 228–234. [Google Scholar] [CrossRef]
- McDonagh, J.L.; Silva, A.F.; Vincent, M.A.; Popelier, P.L.A. Quantifying electron correlation of the chemical bond. J. Phys. Chem. Lett. 2017, 8, 1937–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.F.; Vincent, M.A.; McDonagh, J.L.; Popelier, P.L.A. The transferability of topologically partitioned electron correlation energies in water clusters. ChemPhysChem 2017, 18, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, J.L.; Silva, A.F.; Vincent, M.A.; Popelier, P.L.A. Machine learning of dynamic electron correlation energies from topological atoms. J. Chem. Theor. Comput. 2018, 14, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.A.; Silva, A.F.; McDonagh, J.L.; Popelier, P.L.A. The effects of higher orders of perturbation theory on the correlation energy of atoms and bonds in molecules. Int. J. Quant. Chem. 2018, 118, e25519. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.F.; Popelier, P.L.A. MP2-IQA: Upscaling the analysis of topologically partitioned electron correlation. J. Mol. Mod. 2018, 24, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Tognetti, V.; Silva, A.F.; Vincent, M.A.; Joubert, L.; Popelier, P.L.A. Decomposition of Møller–Plesset energies within the quantum theory of atoms-in-molecules. J. Phys. Chem. A 2018, 122, 7748–7756. [Google Scholar] [CrossRef]
- Vincent, M.A.; Silva, A.F.; Popelier, P.L.A. Atomic partitioning of the MPn (n = 2, 3, 4) dynamic electron correlation energy by the interacting quantum atoms method: A fast and accurate electrostatic potential integral approach. J. Comp. Chem. 2019, 40, 2793–2800. [Google Scholar] [CrossRef] [Green Version]
- Chávez-Calvillo, R.; García-Revilla, M.; Francisco, E.; Martín Pendás, A.; Rocha-Rinza, T. Dynamical correlation within the interacting quantum atoms method through coupled cluster theory. Comput. Theor. Chem. 2015, 1053, 90–95. [Google Scholar] [CrossRef]
- Holguin-Gallego, F.J.; Chavez-Calvillo, R.; Garcia-Revilla, M.; Francisco, E.; Martin Pendas, A.; Rocha-Rinza, T. Electron correlation in the interacting quantum atoms partition via coupled-cluster lagrangian densities. J. Comput. Chem. 2016, 37, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Casalz-Sainz, J.L.; Guevara-Vela, J.M.; Francisco, E.; Rocha-Rinza, T.; Martin Pendas, A. Where does electron correlation lie? Some answers from a real space partition. ChemPhysChem 2017, 18, 3553–3561. [Google Scholar] [CrossRef] [PubMed]
- Thacker, J.C.R.; Popelier, P.L.A. The ANANKE relative energy gradient (REG) method to automate IQA analysis over configurational change. Theor. Chem. Acc. 2017, 136, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popelier, P.L.A.; Maxwell, P.I.; Thacker, J.C.R.; Alkorta, I. A relative energy gradient (REG) study of the planar and perpendicular torsional energy barriers in biphenyl. Theor. Chem. Acc. 2019, 138, 12. [Google Scholar] [CrossRef] [Green Version]
- Alkorta, I.; Thacker, J.C.R.; Popelier, P.L.A. An interacting quantum atom (IQA) study of model SN2 reactions (X ···CH3X, X. = F. Cl, Br and I). J. Comp. Chem. 2018, 39, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orangi, N.; Eskandari, K.; Thacker, J.C.R.; Popelier, P.L.A. Directionality of halogen bonds: An interacting quantum atoms (IQA) and relative energy gradient (REG) study. ChemPhysChem 2019, 20, 1922–1930. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Rozas, I.; Elguero, J. Charge-transfer complexes between dihalogen compounds and electron donors. J. Phys. Chem. A 1998, 102, 9278–9285. [Google Scholar] [CrossRef]
- Cooke, S.A.; Cotti, G.; Evans, C.M.; Holloway, J.H.; Legon, A.C. Rotational spectrum and properties of a gas-phase complex of molecular fluorine and hydrogen cyanide. Chem. Phys. Lett. 1996, 262, 308–314. [Google Scholar] [CrossRef]
- Bloemink, H.I.; Hinds, K.; Holloway, J.H.; Legon, A.C. Characterisation of a pre-reactive intermediate in gas-phase mixtures of fluorine and ammonia: The rotational spectrum of the H3N…F2 complex. Chem. Phys. Lett. 1995, 245, 598–604. [Google Scholar] [CrossRef]
- Legon, A.C.; Thorn, J.C. Identification and characterisation of the gas-phase complex HCN⋯Cl2 by rotational spectroscopy. J. Chem. Soc. Faraday Trans. 1993, 89, 4157–4162. [Google Scholar] [CrossRef]
- Legon, A.C.; Lister, D.G.; Thorn, J.C. Non-reactive interaction of ammonia and molecular chlorine: Rotational spectrum of the ‘charge-transfer’ complex H3N⋯Cl2. J. Chem. Soc. Faraday Trans. 1994, 90, 3205–3212. [Google Scholar] [CrossRef]
- Cooke, S.A.; Cotti, G.; Hinds, K.; Holloway, J.H.; Legon, A.C.; Lister, D.G. Rotational spectrum and molecular properties of the dinitrogen–chlorine monofluoride complex. J. Chem. Soc. Faraday Trans. 1996, 92, 2671–2676. [Google Scholar] [CrossRef]
- Hinds, K.; Legon, A.C.; Holloway, J.H. Rotational spectrum and properties of a complex of hydrogen cyanide and chlorine monofluoride. Mol. Phys. 1996, 88, 673–682. [Google Scholar] [CrossRef]
- Bloemink, H.I.; Evans, C.M.; Holloway, J.H.; Legon, A.C. Is the gas-phase complex of ammonia and chlorine monofluoride H3N…ClF or [H3NCl]+…F−? Evidence from rotational spectroscopy. Chem. Phys. Lett. 1996, 248, 260–268. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical bonds without bonding electron density—Does the difference electron-density analysis suffice for a description of the chemical bond? Angew. Chem. Int. Ed. Engl. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving H⋯F systems. Part I: The transit region between pure closed-shell and shared-shell interactions. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Buckingham, A.D.; Fowler, P.W. A model for the geometries of van der Waals complexes. Can. J. Chem. 1985, 63, 2018–2025. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. Halogen bonding versus hydrogen bonding: A molecular orbital perspective. Chem. Open 2012, 1, 96–105. [Google Scholar] [CrossRef]
- Li, Q.; Xu, X.; Liu, T.; Jing, B.; Li, W.; Cheng, J.; Gong, B.; Sun, J. Competition between hydrogen bond and halogen bond in complexes of formaldehyde with hypohalous acids. Phys. Chem. Chem. Phys. 2010, 12, 6837–6843. [Google Scholar] [CrossRef]
- Riley, K.E.; Hobza, P. The relative roles of electrostatics and dispersion in the stabilization of halogen bonds. Phys. Chem. Chem. Phys. 2013, 15, 17742–17751. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, P.C.; Pople, J.A. Influence of polarization functions on molecular-orbital hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- GAUSSIAN, Version: 16, Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Popelier, P.L.A. Quantum chemical topology. In The Chemical Bond–100 Years Old and Getting Stronger; Mingos, M., Ed.; Springer: Basel, Switzerland, 2016; pp. 71–117. [Google Scholar]
- Wilson, A.L.; Popelier, P.L.A. Exponential relationships capturing atomistic short-range repulsion from the interacting quantum atoms (IQA) method. J. Phys. Chem. A 2016, 120, 9647–9659. [Google Scholar] [CrossRef]
- Symons, B.C.B.; Williamson, D.J.; Brooks, C.M.; Wilson, A.L.; Popelier, P.L.A. Does the intra-atomic deformation energy of interacting quantum atoms represent steric energy? Chem. Open 2019, 8, 560–570. [Google Scholar]
- Keith, T.A. AIMAll; 17.11.14; TK Gristmill Software: Overland Park, KS, USA, 2017; Available online: http://aim.tkgristmill.com/ (accessed on 8 June 2020).
- Popelier, P.L.A. A method to integrate an atom in a molecule without explicit representation of the interatomic surface. Comput. Phys. Commun. 1998, 108, 180–190. [Google Scholar] [CrossRef]
- Thacker, J.C.R.; Vincent, M.A.; Popelier, P.L.A. Using the relative energy gradient method with interacting quantum atoms to determine the reaction mechanism and catalytic effects in the peptide hydrolysis in HIV–1 Protease. Chem. Eur. J. 2018, 14, 11200–11210. [Google Scholar] [CrossRef] [Green Version]
- Thacker, J.C.R.; Popelier, P.L.A. Fluorine gauche effect explained by electrostatic polarization instead of hyperconjugation: An interacting quantum atoms (IQA) and relative energy gradient (REG) study. J. Phys. Chem. A 2018, 122, 1439–1450. [Google Scholar] [CrossRef] [Green Version]
- Backhouse, O.J.; Thacker, J.C.R.; Popelier, P.L.A. A re-evaluation of factors controlling the nature of complementary hydrogen-bonded networks. ChemPhysChem 2019, 20, 555–564. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | N⋯X Dist. Exp. | N⋯X Dist. Calc. | Binding Energy |
|---|---|---|---|
| HCN:F2 | 2.803 [42] | 2.794 | −5.5 |
| H3N:F2 | 2.708 [43] | 2.684 | −8.5 |
| HCN:Cl2 | 2.915 [44] | 2.982 | −9.6 |
| H3N:Cl2 | 2.724 [45] | 2.748 | −17.7 |
| N2:ClF | 2.920 [46] | 2.969 | −6.9 |
| HCN:ClF | 2.639 [47] | 2.684 | −18.8 |
| H3N:ClF | 2.376 [48] | 2.359 | −39.6 |
| N··X Distance (Å) | ρBCP | ∇2ρBCP | HBCP | |
|---|---|---|---|---|
| HCN:F2 | 2.794 | 0.008 | 0.039 | 0.002 |
| H3N:F2 | 2.684 | 0.013 | 0.053 | 0.002 |
| HCN:Cl2 | 2.983 | 0.010 | 0.044 | 0.002 |
| H3N:Cl2 | 2.748 | 0.021 | 0.070 | 0.002 |
| N2:ClF | 2.969 | 0.009 | 0.042 | 0.002 |
| HCN:ClF | 2.684 | 0.019 | 0.079 | 0.002 |
| H3N:ClF | 2.359 | 0.047 | 0.137 | −0.003 |
| SEG1 | ||||||
| HCN:F2 | H3N:F2 | HCN:Cl2 | H3N:Cl2 | N2:ClF | HCN:ClF | H3N:ClF |
| Finner | Finner | Clinner | Clinner | N4 | C4 | H * |
| (2.94) | (3.54) | (2.95) | (2.56) | (1.55) | (2.33) | (1.35) |
| Fouter | Fouter | Clouter | Clouter | Fouter | Fouter | Clinner |
| (−3.08) | (−3.72) | (−3.40) | (−4.49) | (−1.64) | (−1.27) | (−1.70) |
| SEG2 | ||||||
| HCN:F2 | H3N:F2 | HCN:Cl2 | H3N:Cl2 | N2:ClF | HCN:ClF | H3N:ClF |
| Fouter | Fouter | Clouter | Clouter | N3 | N3 | N3 |
| (3.60) | (4.00) | (3.55) | (3.49) | (1.50) | (2.29) | (1.38) |
| Finner | Finner | Clinner | Clinner | N4 | C4 | H * |
| (−3.34) | (−3.74) | (−2.78) | (−2.51) | (−2.42) | (−2.38) | (−0.84) |
| SEG1 | ||||||
| HCN:F2 | H3N:F2 | HCN:Cl2 | H3N:Cl2 | N2:ClF | HCN:ClF | H3N:ClF |
| Intra_n3 | Intra_n3 | Intra_cl2 | Intra_n3 | Intra_n3 | Intra_n3 | Intra_n3 |
| (6.36) | (6.65) | (6.50) | (7.78) | (5.56) | (7.81) | (7.15) |
| Intra_f2 | Intra_f2 | Intra_n3 | Intra_cl2 | Intra_cl2 | Inter_cl2_c4 | Inter_f1_cl2 |
| (6.27) | (6.14) | (6.35) | (6.30) | (4.59) | (4.01) | (4.09) |
| Intra_f1 | Intra_f1 | Inter_n3_c4 | Intra_cl1 | Inter_n3_c4 | Intra_f1 | |
| (−3.39) | (−5.04) | (−2.58) | (−4.97) | (−3.25) | (−2.17) | |
| Inter_f2_n3 | Inter_f2_n3 | Inter_cl2_n3 | Inter_cl2_n3 | Inter_cl2_n3 | Inter_cl2_n3 | Inter_cl2_n3 |
| (−9.22) | (−10.34) | (−11.35) | (−13.74) | (−8.71) | (−12.71) | (−12.11) |
| SEG2 | ||||||
| HCN:F2 | H3N:F2 | HCN:Cl2 | H3N:Cl2 | N2:ClF | HCN:ClF | H3N:ClF |
| Inter_f2_n3 | Inter_f2_n3 | Inter_cl2_n3 | Inter_cl2_n3 | Inter_cl2_n3 | Inter_cl2_n3 | Inter_cl2_n3 |
| (6.47) | (7.46) | (11.87) | (10.54) | (9.93) | (13.03) | (9.80) |
| Intra_f1 | Intra_f1 | Inter_n3_c4 | Intra_cl1 | Inter_n3_c4 | ||
| (3.48) | (4.17) | (4.34) | (3.58) | (4.35) | ||
| Intra_n3 | Intra_n3 | Intra_n3 | Intra_n3 | Intra_n3 | Intra_n3 | Inter_f1_n3 |
| (−2.62) | (−3.25) | (−5.72) | (−4.91) | (−2.82) | (−4.89) | (−2.04) |
| Intra_f2 | Intra_f2 | Intra_cl2 | Intra_cl2 | Intra_cl2 | Inter_cl2_c4 | Intra_n3 |
| (−5.93) | (−6.55) | (−6.60) | (−5.95) | (−4.86) | (−5.81) | (−4.41) |
| SEG1 | ||||||
| HCN:F2 | H3N:F2 | HCN:Cl2 | H3N:Cl2 | N2:ClF | HCN:ClF | H3N:ClF |
| Intra_n3 | Vx_f1_f2 | Intra_cl2 | Intra_n3 | Intra_n3 | Intra_n3 | Intra_n3 |
| (6.36) | (6.80) | (6.50) | (7.78) | (5.56) | (7.81) | (7.15) |
| Intra_f2 | Intra_n3 | Intra_n3 | Intra_cl2 | Intra_cl2 | Vcl_cl2_c4 | Vx_f1_cl2 |
| (6.27) | (6.65) | (6.35) | (6.30) | (4.59) | (4.22) | (4.43) |
| Intra_f1 | Intra_f1 | Vcl_cl2_n3 | Vcl_cl2_n3 | Vcl_f1_cl2 | Vcl_cl2_n3 | Vcl_cl2_n3 |
| (−3.39) | (−5.04) | (−4.91) | (−5.52) | (−2.44) | (−5.07) | (−3.61) |
| Vx_f2_n3 | Vx_f2_n3 | Vx_cl2_n3 | Vx_cl2_n3 | Vx_cl2_n3 | Vx_cl2_n3 | Vx_cl2_n3 |
| (−7.20) | (−8.06) | (−6.43) | (−8.22) | (−7.61) | (−7.64) | (−8.50) |
| SEG2 | ||||||
| HCN:F2 | H3N:F2 | HCN:Cl2 | H3N:Cl2 | N2:ClF | HCN:ClF | H3N:ClF |
| Vx_f2_n3 | Vx_f2_n3 | Vx_cl2_n3 | Vx_cl2_n3 | Vx_cl2_n3 | Vcl_cl2_n3 | Vx_cl2_n3 |
| (4.81) | (5.65) | (6.07) | (5.80) | (6.98) | (7.93) | (5.33) |
| Intra_f1 | Intra_f1 | Vcl_cl2_n3 | Vcl_cl2_n3 | Vcl_cl2_n3 | Vcl_n3_c4 a | Vcl_cl2_n3 |
| (3.48) | (4.17) | (5.80) | (4.74) | (2.92) | (5.50) | (4.47) |
| Intra_n3 | Intra_n3 | Intra_n3 | Intra_n3 | Intra_n3 | Intra_n3 | Vx_f1_cl2 |
| (−2.62) | (−3.25) | (−5.72) | (−4.91) | (−2.82) | (−4.89) | (−2.49) |
| Intra_f2 | Intra_f2 | Intra_cl2 | Intra_cl2 | Intra_cl2 | Vcl_cl2_c4 | Intra_n3 |
| (−5.93) | (−6.55) | (−6.60) | (−5.95) | (−4.86) | (−5.93) | (−4.41) |
| SEG1 | ||||||
| HCN:F2 | H3N:F2 | HCN:Cl2 | H3N:Cl2 | N2:ClF | HCN:ClF | H3N:ClF |
| Vc_f1_n3 | Vc_f1_n3 | Vc_cl1_n3 | Vc_cl1_n3 | Vc_f1_n3 | Vc_f1_n3 | Vc_cl2_n3 |
| (0.18) | (0.21) | (0.11) | (0.13) | (0.12) | (0.09) | (0.07) |
| Vc_f1_n3 | ||||||
| (0.07) | ||||||
| Vc_f2_n3 | Vc_cl2_n3 | Vc_cl2_h4 | ||||
| (−0.43) | (−0.25) | (−0.04) | ||||
| Vc_f2_n3 | Vc_f1_f2 | Vc_cl2_n3 | Vc_cl1_cl2 | Vc_cl2_n3 | Vc_cl2_n3 | Vc_f1_cl2 |
| (−0.75) | (−0.43) | (−0.41) | (−0.33) | (−0.69) | (−0.40) | (−0.33) |
| SEG2 | ||||||
| HCN:F2 | H3N:F2 | HCN:Cl2 | H3N:Cl2 | N2:ClF | HCN:ClF | H3N:ClF |
| Vc_f2_n3 | Vc_f2_n3 | Vc_cl2_n3 | Vc_cl2_n3 | Vc_cl2_n3 | Vc_cl2_n3 | Vc_cl2_n3 |
| (0.89) | (0.60) | (0.88) | (0.45) | (1.05) | (0.57) | (0.24) |
| Vc_f1_n3 | ||||||
| (−0.22) | ||||||
| Vc_f1_n3 | Vc_f1_n3 | Vc_cl1_n3 | Vc_cl1_n3 | Vc_n3_n4 | Vc_f1_n3 | Vc_f1_n3 |
| (−0.27) | (−0.18) | (−0.17) | (−0.10) | (−0.29) | (−0.10) | (−0.07) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkorta, I.; Silva, A.F.; Popelier, P.L.A. An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study of the Halogen Bond with Explicit Analysis of Electron Correlation. Molecules 2020, 25, 2674. https://doi.org/10.3390/molecules25112674
Alkorta I, Silva AF, Popelier PLA. An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study of the Halogen Bond with Explicit Analysis of Electron Correlation. Molecules. 2020; 25(11):2674. https://doi.org/10.3390/molecules25112674
Chicago/Turabian StyleAlkorta, Ibon, Arnaldo F. Silva, and Paul L. A. Popelier. 2020. "An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study of the Halogen Bond with Explicit Analysis of Electron Correlation" Molecules 25, no. 11: 2674. https://doi.org/10.3390/molecules25112674
APA StyleAlkorta, I., Silva, A. F., & Popelier, P. L. A. (2020). An Interacting Quantum Atoms (IQA) and Relative Energy Gradient (REG) Study of the Halogen Bond with Explicit Analysis of Electron Correlation. Molecules, 25(11), 2674. https://doi.org/10.3390/molecules25112674