
Lipid and Polymer-Based Nanoparticle siRNA Delivery Systems for Cancer Therapy



Abstract
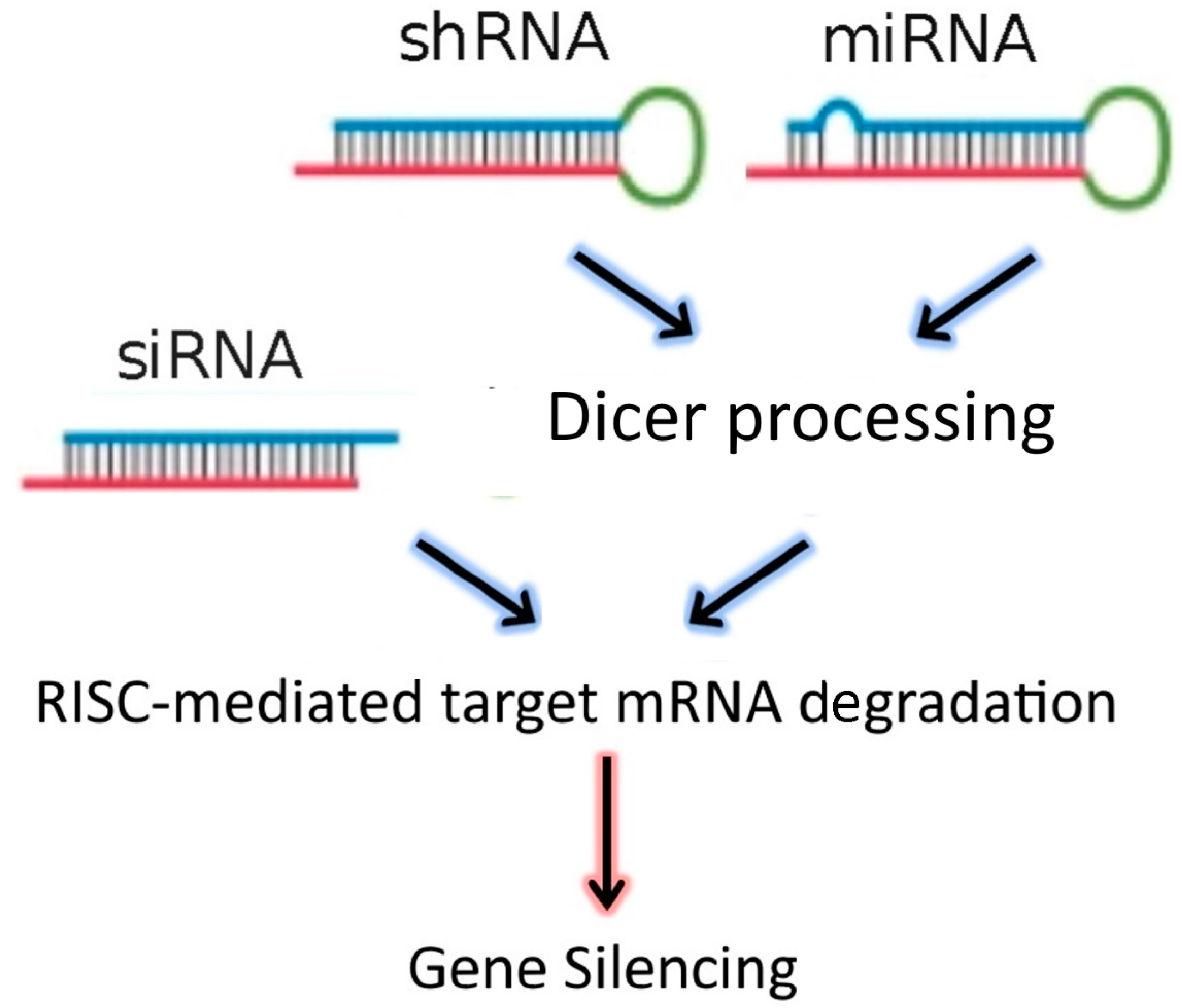
:1. Introduction
2. Challenges in siRNA Delivery: Physiological and Intracellular Barriers
2.1. Physiological Barriers That Limit NP Accumulation in Tumors
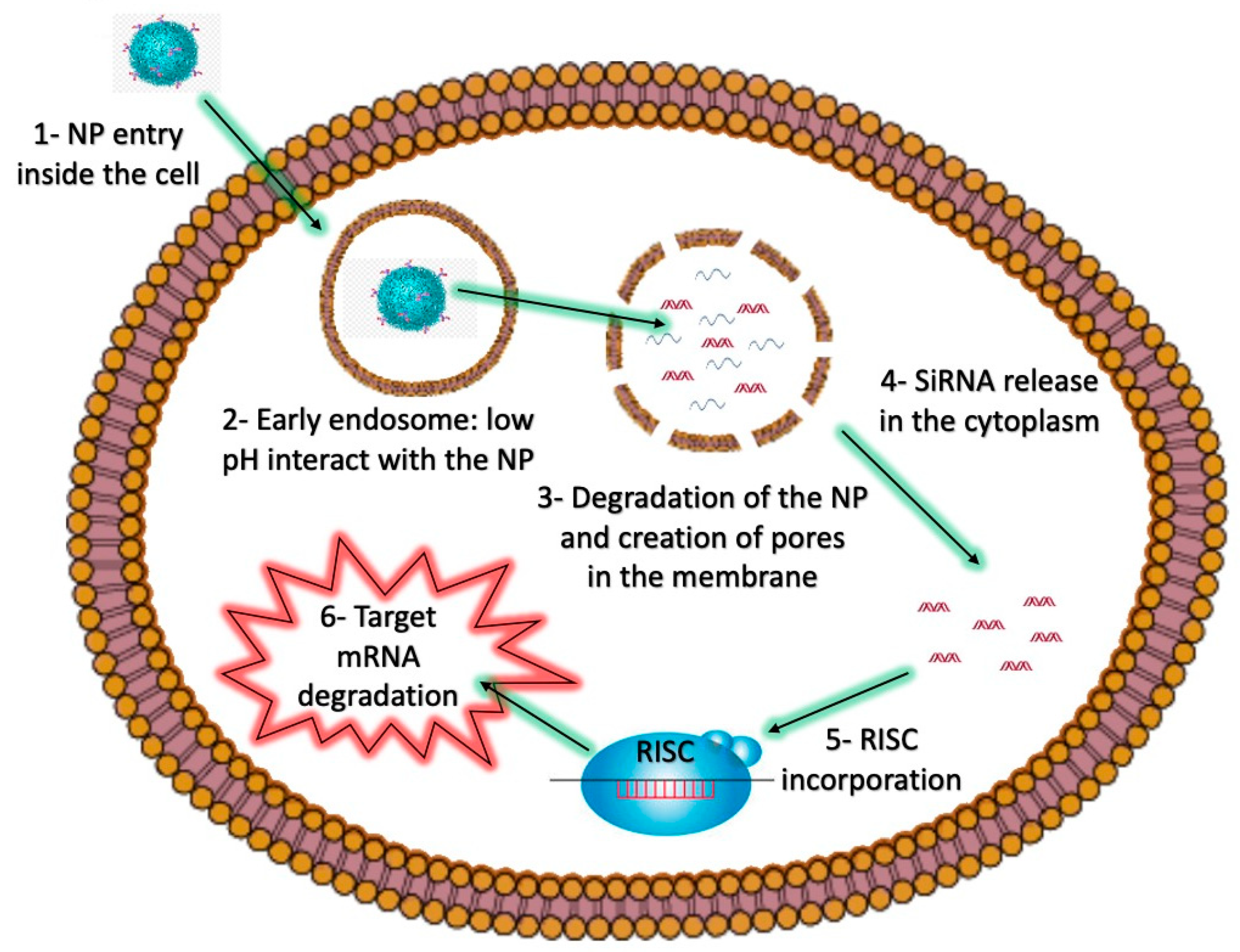
2.2. Intracellular Barriers and Endosomal Escape Strategies
3. Lipid and Polymer-Based siRNA Carriers for Cancer Therapy
3.1. Liposomes
3.2. PEI-Derived Nanosystems
3.3. PLL-Derived Nanosystems
3.4. Anti-Tumor Nanovaccines Enhanced by siRNAs
4. Ongoing Clinical Trials Using siRNA-Loaded NPs for Cancer Therapy
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Ameres, S.L.; Martinez, J.; Schroeder, R. Molecular Basis for Target RNA Recognition and Cleavage by Human RISC. Cell 2007, 130, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Robb, G.B.; Rana, T.M. RNA Helicase A Interacts with RISC in Human Cells and Functions in RISC Loading. Mol. Cell 2007, 26, 523–537. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Paddison, P.J.; Caudy, A.A.; Bernstein, E.; Hannon, G.J.; Conklin, D.S. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002, 16, 948–958. [Google Scholar] [CrossRef] [Green Version]
- Anfray, C.; Mainini, F.; Andón, F.T. Nanoparticles for immunotherapy. In Frontiers of Nanoscience; Elsevier Ltd.: Amsterdam, The Netherlands, 2020; Volume 16, pp. 265–306. [Google Scholar]
- Guan, J.; Sun, J.; Sun, F.; Lou, B.; Zhang, D.; Mashayekhi, V.; Sadeghi, N.; Storm, G.; Mastrobattista, E.; He, Z. Hypoxia-induced tumor cell resistance is overcome by synergistic GAPDH-siRNA and chemotherapy co-delivered by long-circulating and cationic-interior liposomes. Nanoscale 2017, 9, 9190–9201. [Google Scholar] [CrossRef]
- Yang, X.; Fan, B.; Gao, W.; Li, L.; Li, T.; Sun, J.; Peng, X.; Li, X.; Wang, Z.; Wang, B.; et al. Enhanced endosomal escape by photothermal activation for improved small interfering RNA delivery and antitumor effect. Int. J. Nanomed. 2018, 13, 4333–4344. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, D.W.; Davis, M.E. Effect of siRNA nuclease stability on the in vitro and in vivo kinetics of siRNA-mediated gene silencing. Biotechnol. Bioeng. 2007, 97, 909–921. [Google Scholar] [CrossRef]
- Siegler, E.L.; Kim, Y.J.; Wang, P. Nanomedicine targeting the tumor microenvironment: Therapeutic strategies to inhibit angiogenesis, remodel matrix, and modulate immune responses. J. Cell. Immunother. 2016, 2, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.Y.; Cheng, R.; Yang, Z.; Tian, Z.M. Nanotechnology for cancer therapy based on chemotherapy. Molecules 2018, 23, 826. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.G. Small-molecule delivery by nanoparticles for anticancer therapy. Trends Mol. Med. 2010, 16, 594–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naseri, N.; Ajorlou, E.; Asghari, F.; Pilehvar-Soltanahmadi, Y. An update on nanoparticle-based contrast agents in medical imaging. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Dacoba, T.G.; Olivera, A.; Torres, D.; Crecente-Campo, J.; Alonso, M.J. Modulating the immune system through nanotechnology. Semin. Immunol. 2017, 34, 78–102. [Google Scholar] [CrossRef] [PubMed]
- Acharya, R. The recent progresses in shRNA-nanoparticle conjugate as a therapeutic approach. Mater. Sci. Eng. C 2019, 104, 109928. [Google Scholar] [CrossRef]
- O’Neill, C.P.; Dwyer, R.M. Nanoparticle-Based Delivery of Tumor Suppressor microRNA for Cancer Therapy. Cells 2020, 9, 521. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Siegwart, D.J.; Anderson, D.G. Strategies, design, and chemistry in siRNA delivery systems. Adv. Drug Deliv. Rev. 2019, 144, 133–147. [Google Scholar] [CrossRef]
- Vicentini, F.T.M.D.C.; Borgheti-Cardoso, L.N.; Depieri, L.V.; De MacEdo Mano, D.; Abelha, T.F.; Petrilli, R.; Bentley, M.V.L.B. Delivery systems and local administration routes for therapeutic siRNA. Pharm. Res. 2013, 30, 915–931. [Google Scholar] [CrossRef]
- Du, X.; Wang, J.; Zhou, Q.; Zhang, L.; Wang, S.; Zhang, Z.; Yao, C. Advanced physical techniques for gene delivery based on membrane perforation. Drug Deliv. 2018, 25, 1516–1525. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Yu, M.; Zheng, J. Transport and interactions of nanoparticles in the kidneys. Nat. Rev. Mater. 2018, 3, 358–374. [Google Scholar] [CrossRef]
- Fadeel, B. Hide and seek: Nanomaterial interactions with the immune system. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Hussain, Z.; Khan, S.; Imran, M.; Sohail, M.; Shah, S.W.A.; de Matas, M. PEGylation: A promising strategy to overcome challenges to cancer-targeted nanomedicines: A review of challenges to clinical transition and promising resolution. Drug Deliv. Transl. Res. 2019, 9, 721–734. [Google Scholar] [CrossRef]
- Tengood, J.E.; Levy, R.J.; Stachelek, S.J. The use of CD47-modified biomaterials to mitigate the immune response. Exp. Biol. Med. 2016, 241, 1033–1041. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Wang, X.; Li, J.; Nie, Y.; Liao, G.; Yu, Y.; Li, C. Overcoming the Reticuloendothelial System Barrier to Drug Delivery with a “Don’t-Eat-Us” Strategy. ACS Nano 2019, 13, 13015–13026. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Greish, K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol. Biol. 2010, 624, 25–37. [Google Scholar] [PubMed]
- Stylianopoulos, T.; Poh, M.-Z.; Insin, N.; Bawendi, M.G.; Fukumura, D.; Munn, L.L.; Jain, R.K. Diffusion of particles in the extracellular matrix: The effect of repulsive electrostatic interactions. Biophys. J. 2010, 99, 1342–1349. [Google Scholar] [CrossRef] [Green Version]
- Lazarovits, J.; Chen, Y.Y.; Sykes, E.A.; Chan, W.C.W. Nanoparticle-blood interactions: The implications on solid tumour targeting. Chem. Commun. 2015, 51, 2756–2767. [Google Scholar] [CrossRef]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Walkey, C.D.; Olsen, J.B.; Guo, H.; Emili, A.; Chan, W.C.W. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J. Am. Chem. Soc. 2012, 134, 2139–2147. [Google Scholar] [CrossRef]
- Roncato, F.; Rruga, F.; Porcù, E.; Casarin, E.; Ronca, R.; Maccarinelli, F.; Realdon, N.; Basso, G.; Alon, R.; Viola, G.; et al. Improvement and extension of anti-EGFR targeting in breast cancer therapy by integration with the Avidin-Nucleic-Acid-Nano-Assemblies. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Parvani, J.G.; Gujrati, M.D.; Mack, M.A.; Schiemann, W.P.; Lu, Z.R. Silencing β3 integrin by targeted ECO/siRNA nanoparticles inhibits EMT and metastasis of triple-negative breast cancer. Cancer Res. 2015, 75, 2316–2325. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.-R.; Liu, L.-T.; Lu, Z.-X. Development of small interfering RNA delivery system using PEI-PEG-APRPG polymer for antiangiogenic vascular endothelial growth factor tumor-targeted therapy. Int. J. Nanomed. 2011, 6, 1661. [Google Scholar] [CrossRef] [Green Version]
- Zwicke, G.L.; Ali Mansoori, G.; Jeffery, C.J. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012, 3, 18496. [Google Scholar] [CrossRef]
- Yang, H.; Li, Y.; Li, T.; Xu, M.; Chen, Y.; Wu, C.; Dang, X.; Liu, Y. Multifunctional core/shell nanoparticles cross-linked polyetherimide-folic acid as efficient notch-1 siRNA carrier for targeted killing of breast cancer. Sci. Rep. 2014, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.Y.; Yoon, H.Y.; Kim, J.H.; Bae, S.M.; Park, R.W.; Kang, Y.M.; Kim, I.S.; Kwon, I.C.; Choi, K.; Jeong, S.Y.; et al. Smart nanocarrier based on PEGylated hyaluronic acid for cancer therapy. ACS Nano 2011, 5, 8591–8599. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Kanwar, J.R.; Athalya, P.K.; Janakiraman, N.; Khetan, V.; Kanwar, R.K.; Eluchuri, S.; Krishnakumar, S. EpCAM aptamer mediated cancer cell specific delivery of EpCAM siRNA using polymeric nanocomplex. J. Biomed. Sci. 2015, 22, 4. [Google Scholar] [CrossRef] [Green Version]
- Cupic, K.I.; Rennick, J.J.; Johnston, A.P.R.; Such, G.K. Controlling endosomal escape using nanoparticle composition: Current progress and future perspectives. Nanomedicine 2019, 14, 215–223. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Degors, I.M.S.; Wang, C.; Rehman, Z.U.; Zuhorn, I.S. Carriers break barriers in drug delivery: Endocytosis and endosomal escape of gene delivery vectors. Acc. Chem. Res. 2019, 52, 1750–1760. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Morris, V.B.; Labhasetwar, V. Effectiveness of small interfering RNA delivery via arginine-rich polyethylenimine-based polyplex in metastatic and doxorubicin-resistant breast cancer cells. J. Pharmacol. Exp. Ther. 2019, 370, 902–910. [Google Scholar] [CrossRef]
- Huang, H.; Yuan, S.; Ma, Z.; Ji, P.; Ma, X.; Wu, Z.; Qi, X. Genetic recombination of poly (l-lysine) functionalized apoferritin nanocages that resemble viral capsid nanometer-sized platforms for gene therapy. Biomater. Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rittner, K.; Benavente, A.; Bompard-Sorlet, A.; Heitz, F.; Divita, G.; Brasseur, R.; Jacobs, E. New basic membrane-destabilizing peptides for plasmid-based gene delivery in vitro and in vivo. Mol. Ther. 2002, 5, 104–114. [Google Scholar] [CrossRef]
- Koynova, R.; Wang, L.; MacDonald, R.C. An intracellular lamellar-nonlamellar phase transition rationalizes the superior performance of some cationic lipid transfection agents. Proc. Natl. Acad. Sci. USA 2006, 103, 14373–14378. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, H.; Akita, H.; Kogure, K.; Oishi, M.; Nagasaki, Y.; Kihira, Y.; Ueno, M.; Kobayashi, H.; Kikuchi, H.; Harashima, H. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007, 14, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Berg, K.; Weyergang, A.; Høgset, A.; Selbo, P.K. The Potential of Photochemical Internalization (PCI) for the Cytosolic Delivery of Nanomedicines. In Weissig/Organelle-Specific Nanotechnology; John Wiley and Sons: Hoboken, NJ, USA, 2010; pp. 311–322. [Google Scholar]
- Tynga, I.M.; Abrahamse, H. Nano-mediated photodynamic therapy for cancer: Enhancement of cancer specificity and therapeutic effects. Nanomaterials 2018, 8, 923. [Google Scholar]
- Judge, A.D.; Sood, V.; Shaw, J.R.; Fang, D.; Mcclintock, K.; Maclachlan, I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 2005, 23, 457–462. [Google Scholar] [CrossRef]
- Whitehead, K.A.; Dahlman, J.E.; Langer, R.S.; Anderson, D.G. Silencing or Stimulation? siRNA Delivery and the Immune System. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 77–96. [Google Scholar] [CrossRef] [Green Version]
- Hayes, M.E.; Drummond, D.C.; Hong, K.; Park, J.W.; Marks, J.D.; Kirpotin, D.B. Assembly of nucleic acid-lipid nanoparticles from aqueous-organic monophases. Biochim. Biophys. Acta-Biomembr. 2006, 1758, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Campbell, R.B.; Fukumura, D.; Brown, E.B.; Mazzola, L.M.; Izumi, Y.; Jain, R.K.; Torchilin, V.P.; Munn, L.L. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002, 62, 6831–6836. [Google Scholar]
- Lin, S.Y.; Zhao, W.Y.; Tsai, H.C.; Hsu, W.H.; Lo, C.L.; Hsiue, G.H. Sterically polymer-based liposomal complexes with dual-shell structure for enhancing the siRNA delivery. Biomacromolecules 2012, 13, 664–675. [Google Scholar] [CrossRef]
- Lee, J.; Cho, Y.J.; Lee, J.W.; Ahn, H.J. KSP siRNA/paclitaxel-loaded PEGylated cationic liposomes for overcoming resistance to KSP inhibitors: Synergistic antitumor effects in drug-resistant ovarian cancer. J. Control. Release 2020, 321, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Bi, X.; Yang, L.; Wu, S.; Yu, Y.; Jiang, B.; Zhang, A.; Lan, K.; Duan, S. Co-delivery of paclitaxel and PLK1-targeted siRNA using aptamer-functionalized cationic liposome for synergistic anti-breast cancer effects in vivo. J. Biomed. Nanotechnol. 2019, 15, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, F.; Jiang, X.; Zhao, X.; Wang, Y.; Kuai, Q.; Nie, G.; He, M.; Pan, Y.; Shi, W.; et al. Co-delivery of gemcitabine and Mcl-1 SiRNA via cationic liposome-based system enhances the efficacy of chemotherapy in pancreatic cancer. J. Biomed. Nanotechnol. 2019, 15, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhu, P.; Lee, S.K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef]
- Weinstein, S.; Toker, I.A.; Emmanuel, R.; Ramishetti, S.; Hazan-Halevy, I.; Rosenblum, D.; Goldsmith, M.; Abraham, A.; Benjamini, O.; Bairey, O.; et al. Harnessing RNAi-based nanomedicines for therapeutic gene silencing in B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E16–E22. [Google Scholar] [CrossRef] [Green Version]
- Kedmi, R.; Veiga, N.; Ramishetti, S.; Goldsmith, M.; Rosenblum, D.; Dammes, N.; Hazan-Halevy, I.; Nahary, L.; Leviatan-Ben-Arye, S.; Harlev, M.; et al. A modular platform for targeted RNAi therapeutics. Nat. Nanotechnol. 2018, 13, 214–219. [Google Scholar] [CrossRef]
- Pandey, A.P.; Sawant, K.K. Polyethylenimine: A versatile, multifunctional non-viral vector for nucleic acid delivery. Mater. Sci. Eng. C. Mater. Biol. Appl. 2016, 68, 904–918. [Google Scholar] [CrossRef]
- Tae, H.K.; Su, I.K.; Akaike, T.; Chong, S.C. Synergistic effect of poly (ethylenimine) on the transfection efficiency of galactosylated chitosan/DNA complexes. J. Control. Release 2005, 105, 354–366. [Google Scholar]
- Huh, M.S.; Lee, S.Y.; Park, S.; Lee, S.; Chung, H.; Lee, S.; Choi, Y.; Oh, Y.K.; Park, J.H.; Jeong, S.Y.; et al. Tumor-homing glycol chitosan/polyethylenimine nanoparticles for the systemic delivery of siRNA in tumor-bearing mice. J. Control. Release 2010, 144, 134–143. [Google Scholar] [CrossRef]
- Zhang, B.F.; Xing, L.; Cui, P.F.; Wang, F.Z.; Xie, R.L.; Zhang, J.L.; Zhang, M.; He, Y.J.; Lyu, J.Y.; Qiao, J.B.; et al. Mitochondria apoptosis pathway synergistically activated by hierarchical targeted nanoparticles co-delivering siRNA and lonidamine. Biomaterials 2015, 61, 178–189. [Google Scholar] [CrossRef]
- Roedig, H.; Damiescu, R.; Zeng-Brouwers, J.; Kutija, I.; Trebicka, J.; Wygrecka, M.; Schaefer, L. Danger matrix molecules orchestrate CD14/CD44 signaling in cancer development. Semin. Cancer Biol. 2020, 62, 31–47. [Google Scholar] [CrossRef]
- Ganesh, S.; Iyer, A.K.; Morrissey, D.V.; Amiji, M.M. Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials 2013, 34, 3489–3502. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, M.; Gooding, M.; Evans, J.C.; O’Driscoll, D.; Darcy, R.; O’Driscoll, C.M. Cyclodextrin-siRNA conjugates as versatile gene silencing agents. Eur. J. Pharm. Sci. 2018, 114, 30–37. [Google Scholar] [CrossRef]
- Wang, D.; Wang, T.; Xu, Z.; Yu, H.; Feng, B.; Zhang, J.; Guo, C.; Yin, Q.; Zhang, Z.; Li, Y. Cooperative Treatment of Metastatic Breast Cancer Using Host-Guest Nanoplatform Coloaded with Docetaxel and siRNA. Small 2016, 12, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, H.; Zhang, X. Design, preparation and application of nucleic acid delivery carriers. Biotechnol. Adv. 2014, 32, 804–817. [Google Scholar] [CrossRef]
- Buyens, K.; Meyer, M.; Wagner, E.; Demeester, J.; De Smedt, S.C.; Sanders, N.N. Monitoring the disassembly of siRNA polyplexes in serum is crucial for predicting their biological efficacy. J. Control. Release 2010, 141, 38–41. [Google Scholar] [CrossRef]
- Qi, R.; Liu, S.; Chen, J.; Xiao, H.; Yan, L.; Huang, Y.; Jing, X. Biodegradable copolymers with identical cationic segments and their performance in siRNA delivery. J. Control. Release 2012, 159, 251–260. [Google Scholar] [CrossRef]
- Xiao, H.; Qi, R.; Li, T.; Awuah, S.G.; Zheng, Y.; Wei, W.; Kang, X.; Song, H.; Wang, Y.; Yu, Y.; et al. Maximizing Synergistic Activity When Combining RNAi and Platinum-Based Anticancer Agents. J. Am. Chem. Soc. 2017, 139, 3033–3044. [Google Scholar] [CrossRef]
- Liu, Y.; Ai, K.; Liu, J.; Deng, M.; He, Y.; Lu, L. Dopamine-melanin colloidal nanospheres: An efficient near-infrared photothermal therapeutic agent for in vivo cancer therapy. Adv. Mater. 2013, 25, 1353–1359. [Google Scholar] [CrossRef]
- Sun, W.; Chen, X.; Xie, C.; Wang, Y.; Lin, L.; Zhu, K.; Shuai, X. Co-Delivery of Doxorubicin and Anti-BCL-2 siRNA by pH-Responsive Polymeric Vector to Overcome Drug Resistance in In Vitro and in Vivo HepG2 Hepatoma Model. Biomacromolecules 2018, 19, 2248–2256. [Google Scholar] [CrossRef]
- Wang, G.; Gao, X.; Gu, G.; Shao, Z.; Li, M.; Wang, P.; Yang, J.; Cai, X.; Li, Y. Polyethylene glycol–poly(ε-benzyloxycarbonyl-L-lysine)-conjugated VEGF siRNA for antiangiogenic gene therapy in hepatocellular carcinoma. Int. J. Nanomed. 2017, 12, 3591–3603. [Google Scholar] [CrossRef] [Green Version]
- van den Boorn, J.G.; Barchet, W.; Hartmann, G. Nucleic acid adjuvants: Toward an educated vaccine. Adv. Immunol. 2012, 114, 1–32. [Google Scholar]
- Liu, J.; Miao, L.; Sui, J.; Hao, Y.; Huang, G. Nanoparticle cancer vaccines: Design considerations and recent advances. Asian J. Pharm. Sci. 2019. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Y.; Ding, Z.Y.; Liu, J.Y. Safety and efficacy of therapeutic cancer vaccines alone or in combination with immune checkpoint inhibitors in cancer treatment. Front. Pharmacol. 2019, 10, 1184. [Google Scholar] [CrossRef]
- Huang, K.W.; Hsu, F.F.; Qiu, J.T.; Chern, G.J.; Lee, Y.A.; Chang, C.C.; Huang, Y.T.; Sung, Y.C.; Chiang, C.C.; Huang, R.L.; et al. Highly efficient and tumor-selective nanoparticles for dual-targeted immunogene therapy against cancer. Sci. Adv. 2020, 6, eaax5032. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, L.; Xu, Z.; Miao, L.; Huang, L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol. Ther. 2018, 26, 420–434. [Google Scholar] [CrossRef] [Green Version]
- Rubel, F.; Kern, J.S.; Technau-Hafsi, K.; Uhrich, S.; Thoma, K.; Häcker, G.; von Bubnoff, N.; Meiss, F.; von Bubnoff, D. Indoleamine 2,3-Dioxygenase Expression in Primary Cutaneous Melanoma Correlates with Breslow Thickness and Is of Significant Prognostic Value for Progression-Free Survival. J. Invest. Dermatol. 2018, 138, 679–687. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.Q.; Lu, S.; Zhang, L.X.; Ji, M.; Liu, S.Y.; Wang, S.W.; Liu, R.T. An indoleamine 2, 3-dioxygenase siRNA nanoparticle-coated and Trp2-displayed recombinant yeast vaccine inhibits melanoma tumor growth in mice. J. Control. Release 2018, 273, 1–12. [Google Scholar] [CrossRef]
- Mainini, F.; Larsen, D.S.; Webster, G.A.; Young, S.L.; Eccles, M.R. Bridging small molecules to modified bacterial microparticles using a disulphide linkage: MIS416 as a cargo delivery system. PLoS ONE 2015, 10, e0145403. [Google Scholar] [CrossRef] [Green Version]
- Mainini, F.; Larsen, D.S.; Webster, G.A.; Young, S.L.; Eccles, M.R. MIS416 as a siRNA Delivery System with the Ability to Target Antigen-Presenting Cells. Nucleic Acid Ther. 2018, 28, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Safety Study of CALAA-01 to Treat Solid Tumor Cancers-Full Text View-ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/study/NCT00689065 (accessed on 26 May 2020).
- Schultheis, B.; Strumberg, D.; Santel, A.; Vank, C.; Gebhardt, F.; Keil, O.; Lange, C.; Giese, K.; Kaufmann, J.; Khan, M.; et al. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 4141–4148. [Google Scholar] [CrossRef]
- Study with Atu027 in Patients With Advanced Solid Cancer-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT00938574 (accessed on 26 May 2020).
- Atu027 Plus Gemcitabine in Advanced or Metastatic Pancreatic Cancer (Atu027-I-02)-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01808638 (accessed on 9 April 2020).
- Dose Escalation Trial to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Intravenous ALN-VSP02 In Patients with Advanced Solid Tumors with Liver Involvement-Full Text View -ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00882180 (accessed on 26 May 2020).
- Multi-Center, Open Label, Extension Study of ALN-VSP02 in Cancer Patients Who Have Responded to ALN-VSP02 Treatment-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01158079 (accessed on 26 May 2020).
- Phase I, Multicenter, Dose Escalation Study of DCR-MYC in Patients with Solid Tumors, Multiple Myeloma, or Lymphoma-No Study Results Posted-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/results/NCT02110563 (accessed on 9 April 2020).
- EphA2 siRNA in Treating Patients with Advanced or Recurrent Solid Tumors-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01591356 (accessed on 26 May 2020).
- A Phase 2 Study of siG12D LODER in Combination with Chemotherapy in Patients with Locally Advanced Pancreatic Cancer-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01676259 (accessed on 9 April 2020).
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef]
- Cerqueira, N.; Pereira, S.; Fernandes, P.; Ramos, M. Overview of Ribonucleotide Reductase Inhibitors: An Appealing Target in Anti-Tumour Therapy. Curr. Med. Chem. 2005, 12, 1283–1294. [Google Scholar] [CrossRef]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Martinez, D.; Wood, D.L.; Fielman, B.; Sharma, M.; Janisch, L.A.; Brown, B.D.; et al. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 11006. [Google Scholar] [CrossRef]
- Wagner, M.J.; Mitra, R.; Mcarthur, M.J.; Baze, W.; Barnhart, K.; Wu, S.Y.; Rodriguez-Aguayo, C.; Zhang, X.; Coleman, R.L.; Lopez-Berestein, G.; et al. Preclinical mammalian safety studies of EPHARNA (DOPC Nanoliposomal EphA2-Targeted siRNA). Mol. Cancer Ther. 2017, 16, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Markosyan, N.; Li, J.; Sun, Y.H.; Richman, L.P.; Lin, J.H.; Yan, F.; Quinones, L.; Sela, Y.; Yamazoe, T.; Gordon, N.; et al. Tumor cell-intrinsic EPHA2 suppresses antitumor immunity by regulating PTGS2 (COX-2). J. Clin. Investig. 2019, 129, 3594–3609. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Chan, T.A.; Kroemer, G.; Wolchok, J.D.; López-Soto, A. The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med. 2018, 10, eaat7807. [Google Scholar] [CrossRef]
Sample Availability: Not available. |



{kind=link}
{kind=link}
| Barrier | Approach |
|---|---|
| Degradation by RNAses | Chemical modification of siRNAs, inclusion of siRNAs in NP-based delivery systems |
| Renal clearance | Inclusion of the siRNA in a nanocomplex with a HD >6 nm |
| Reticuloendothelial system | Addition of PEG to the nanocomplex to reduce protein corona formation and phagocytosis |
| Limited access into tumor tissue | Passive accumulation: limit NP size (<200 nm) to promote the EPR effect. Active targeting: Inclusion of a targeting ligand on the surface of the NPs |
| Name | Type | Target | Type of Cancer | Status | Reference |
|---|---|---|---|---|---|
| CALAA-01 | Cyclodextrin polymer-based NPs | RRM2 | Solid tumors | Completed | NCT00689065 [83] |
| Atu027 | Liposomes | Protein kinase N3 | Solid tumors, pancreatic carcinoma | Completed Completed Completed | NCT00938574 [84] NCT00938574 [85] NCT01808638 [86] |
| ALN-VSP | Lipid-based NPs | VEGF and KSP | Solid tumors | Completed Completed | NCT00882180 [87] NCT01158079 [88] |
| DCR-PHXC-101 | Lipid-based NPs | Myc | Solid tumors, multiple myeloma, non-Hodgkin’s lymphoma | Terminated | NCT02110563 [89] |
| SiRNA-EphA2 | Liposomes | EphA2 | Advanced cancers | Recruiting | NCT01591356 [90] |
| siG12D LODER | Biodegradable polymeric matrix | KRASG12D | Pancreatic ductal adenocarcinoma, pancreatic cancer | Recruiting | NCT01676259 [91] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mainini, F.; Eccles, M.R. Lipid and Polymer-Based Nanoparticle siRNA Delivery Systems for Cancer Therapy. Molecules 2020, 25, 2692. https://doi.org/10.3390/molecules25112692
Mainini F, Eccles MR. Lipid and Polymer-Based Nanoparticle siRNA Delivery Systems for Cancer Therapy. Molecules. 2020; 25(11):2692. https://doi.org/10.3390/molecules25112692
Chicago/Turabian StyleMainini, Francesco, and Michael R. Eccles. 2020. "Lipid and Polymer-Based Nanoparticle siRNA Delivery Systems for Cancer Therapy" Molecules 25, no. 11: 2692. https://doi.org/10.3390/molecules25112692
APA StyleMainini, F., & Eccles, M. R. (2020). Lipid and Polymer-Based Nanoparticle siRNA Delivery Systems for Cancer Therapy. Molecules, 25(11), 2692. https://doi.org/10.3390/molecules25112692