
The (+)-Brevipolide H Displays Anticancer Activity against Human Castration-Resistant Prostate Cancer: The Role of Oxidative Stress and Akt/mTOR/p70S6K-Dependent Pathways in G1 Checkpoint Arrest and Apoptosis

 and
and
Abstract
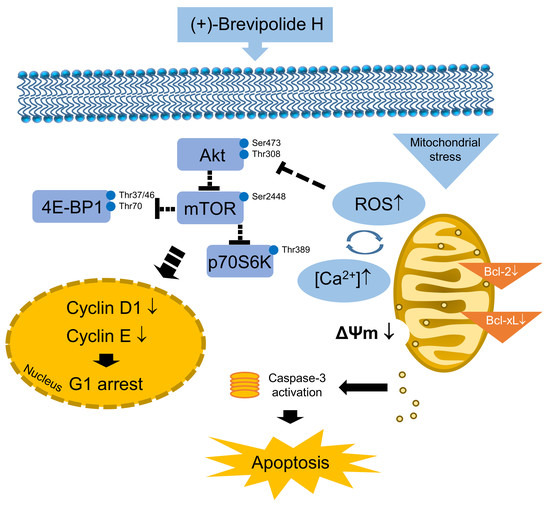
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The (+)-Brevipolide H Induces Anti-Proliferative Effect in PC-3 Cells with Long-Term Efficacy
2.2. The (+)-Brevipolide H Induces G1 Arrest of the Cell Cycle and Downregulates G1 Cyclins
2.3. The (+)-Brevipolide H Inhibits Akt/mTOR/p70S6K Signaling Pathways
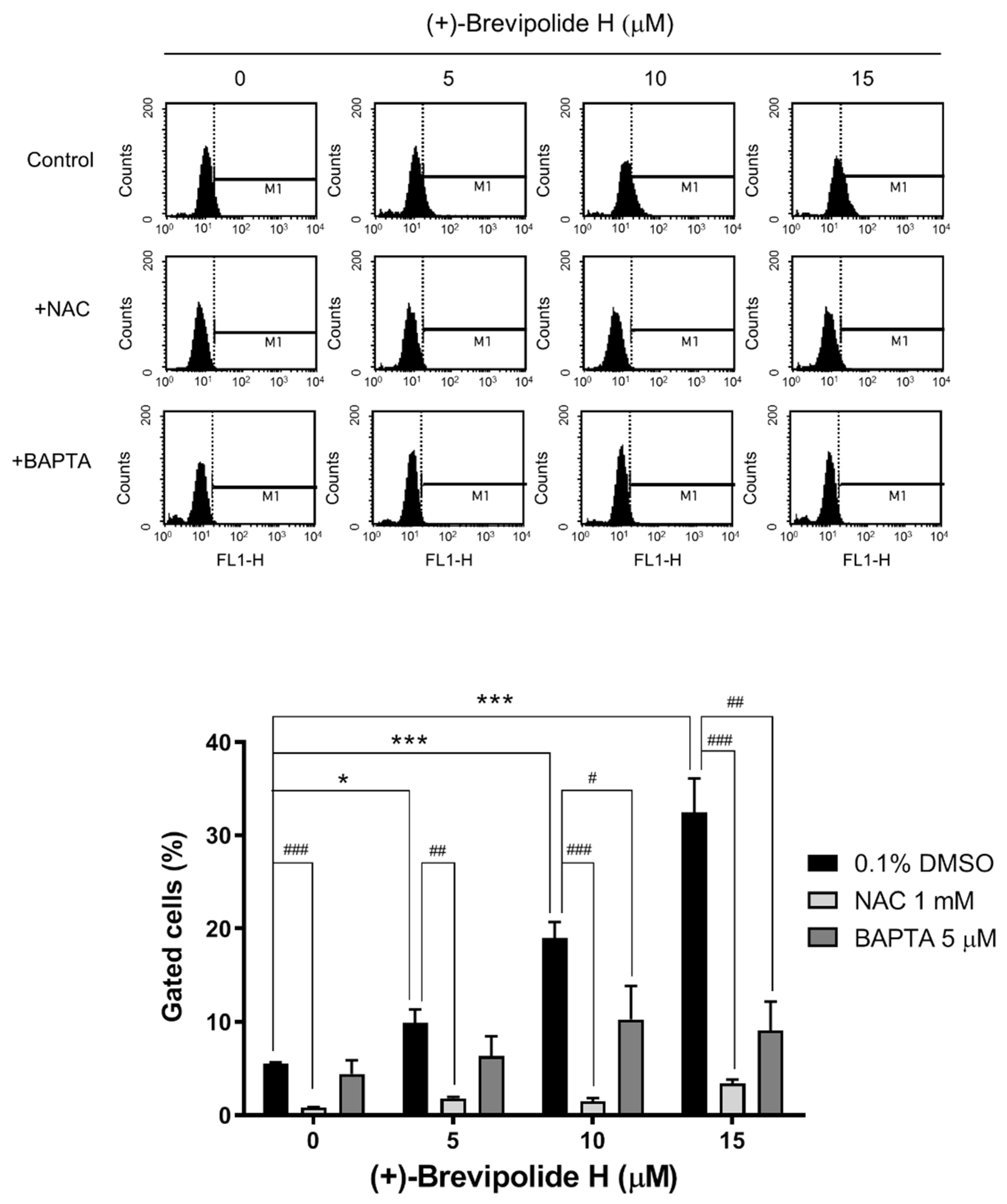
2.4. Crosstalk between ROS and Calcium Plays a Key Role in (+)-Brevipolide H-Mediated Signaling Pathways
2.5. The (+)-Brevipolide H Induced Downregulation of Anti-Apoptotic Bcl-2 Family Proteins and Loss of Mitochondrial Membrane Potential
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Sulforhodamine B (SRB) Assay
4.4. Colony Formation Assay
4.5. Cell Proliferation Assay with CFSE Staining
4.6. Cell Distribution Analysis with PI Staining
4.7. Measurement of Mitochondrial Membrane Potential Loss with JC-1 Staining
4.8. Western Blot Analysis
4.9. Confocal Immunofluorescence Microscopy
4.10. Transfection
4.11. Measurement of Reactive Oxygen Species (ROS) Production
4.12. Measurement of Intracellular Ca2+ Content
4.13. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CFSE | carboxyfluoresceinsuccinimidyl ester |
| CRPC | castration-resistant prostate cancer |
| DCF-DA | 2′,7′-dichlorodihydrofluorescein diacetate |
| DTT | DL-dithiothreitol |
| ER | endoplasmic reticulum |
| InsP3 | inositol trisphosphate |
| MAPK | mitogen-activated protein kinase |
| mTOR | mammalian target of rapamycin |
| Myr-Akt | myristoylated Akt |
| NAC | N-acetyl-L-cysteine |
| PI3K | phosphoinositide 3-kinase |
| PI | propidium iodide |
| PMSF | phenylmethanesulfonyl fluoride |
| PTEN | phosphatase and tensin homologue |
| ROS | reactive oxygen species |
| SERCA | sarco/endoplasmic reticulum Ca2+-ATPase |
| SRB | sulforhodamine B |
| TCA | trichloroacetic acid |
References
- Scher, H.I.; Heller, G. Clinical states in prostate cancer: Toward a dynamic model of disease progression. Urology 2000, 55, 323–327. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J.; Jang, H. A New View of Pathway-Driven Drug Resistance in Tumor Proliferation. Trends Pharmacol. Sci. 2017, 38, 427–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Befani, C.D.; Vlachostergios, P.J.; Hatzidaki, E.; Patrikidou, A.; Bonanou, S.; Simos, G.; Papandreou, C.N.; Liakos, P. Bortezomib represses HIF-1α protein expression and nuclear accumulation by inhibiting both PI3K/Akt/TOR and MAPK pathways in prostate cancer cells. J. Mol. Med. (Berl) 2012, 90, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.; Ghosh, P.; Kumavath, R. Strophanthidin Attenuates MAPK, PI3K/AKT/mTOR, and Wnt/β-Catenin Signaling Pathways in Human Cancers. Front. Oncol. 2020, 9, 1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, M.; Platonova, N.; Giannandrea, D.; Palano, M.T.; Basile, A.; Chiaramonte, R. Re-establishing Apoptosis Competence in Bone Associated Cancers via Communicative Reprogramming Induced Through Notch Signaling Inhibition. Front. Pharmacol. 2019, 10, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanankutty, A. PI3K/ Akt/ mTOR Pathway as a Therapeutic Target for Colorectal Cancer: A Review of Preclinical and Clinical Evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef]
- Marquard, F.E.; Jücker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2020, 172, 113729. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Bitting, R.L.; Armstrong, A.J. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr. Relat. Cancer 2013, 20, R83–R99. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Balunas, M.J.; Kim, J.A.; Lantvit, D.D.; Chin, Y.W.; Chai, H.; Sugiarso, S.; Kardono, L.B.; Fong, H.H.; Pezzuto, J.M.; et al. Bioactive 5,6-dihydro-alpha-pyrone derivatives from Hyptis brevipes. J. Nat. Prod. 2009, 72, 1165–1169. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Ortiz, G.A.; Cerda-García-Rojas, C.M.; Hernández-Rojas, A.; Pereda-Miranda, R. Absolute configuration and conformational analysis of (-)-brevipolides, bioactive 5,6-dihydro-α-pyrones from Hyptis brevipes. J. Nat. Prod. 2013, 76, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.N.; Hou, D.R. Enantioselective synthesis of (+)-brevipolide H. Org. Biomol. Chem. 2016, 14, 6762–6768. [Google Scholar] [CrossRef]
- Hegde, V.R.; Pu, H.; Patel, M.; Das, P.R.; Strizki, J.; Gullo, V.P.; Chou, C.C.; Buevich, A.V.; Chan, T.M. Three new compounds from the plant Lippia alva as inhibitors of chemokine receptor 5 (CCR5). Bioorg. Med. Chem. Lett. 2004, 14, 5339–5342. [Google Scholar] [CrossRef] [PubMed]
- Conley-LaComb, M.K.; Saliganan, A.; Kandagatla, P.; Chen, Y.Q.; Cher, M.L.; Chinni, S.R. PTEN loss mediated Akt activation promotes prostate tumor growth and metastasis via CXCL12/CXCR4 signaling. Mol. Cancer 2013, 12, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gul, A.; Leyland-Jones, B.; Dey, N.; De, P. A combination of the PI3K pathway inhibitor plus cell cycle pathway inhibitor to combat endocrine resistance in hormone receptor-positive breast cancer: A genomic algorithm-based treatment approach. Am. J. Cancer Res. 2018, 8, 2359–2376. [Google Scholar]
- Hinds, P.W.; Mittnacht, S.; Dulic, V.; Arnold, A.; Reed, S.I.; Weinberg, R.A. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 1992, 70, 993–1006. [Google Scholar] [CrossRef]
- Matsushime, H.; Ewen, M.E.; Strom, D.K.; Kato, J.Y.; Hanks, S.K.; Roussel, M.F.; Sherr, C.J. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell 1992, 71, 323–334. [Google Scholar] [CrossRef]
- Liao, D.J.; Thakur, A.; Wu, J.; Biliran, H.; Sarkar, F.H. Perspectives on c-Myc, Cyclin D1, and their interaction in cancer formation, progression, and response to chemotherapy. Crit. Rev. Oncog. 2007, 13, 93–158. [Google Scholar] [CrossRef]
- Pérez-Roger, I.; Solomon, D.L.; Sewing, A.; Land, H. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27(Kip1) binding to newly formed complexes. Oncogene 1997, 14, 2373–2381. [Google Scholar] [CrossRef] [Green Version]
- Baldin, V.; Lukas, J.; Marcote, M.J.; Pagano, M.; Draetta, G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993, 7, 812–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsubo, M.; Theodoras, A.M.; Schumacher, J.; Roberts, J.M.; Pagano, M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol. Cell Biol. 1995, 15, 2612–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.M.; Klinghoffer, R.; Prestwich, G.D.; Toker, A.; Kazlauskas, A. PDGF induces an early and a late wave of PI 3-kinase activity, and only the late wave is required for progression through G1. Curr. Biol. 1999, 9, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Showkat, M.; Beigh, M.A.; Andrabi, K.I. mTOR signaling in Protein Translation Regulation: Implications in Cancer Genesis and Therapeutic Interventions. Mol. Biol. Int. 2014, 2014, 686984. [Google Scholar] [CrossRef] [Green Version]
- Pause, A.; Belsham, G.J.; Gingras, A.C.; Donzé, O.; Lin, T.A.; Lawrence, J.C., Jr.; Sonenberg, N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef]
- Zhang, W.; Haines, B.B.; Efferson, C.; Zhu, J.; Ware, C.; Kunii, K.; Tammam, J.; Angagaw, M.; Hinton, M.C.; Keilhack, H.; et al. Evidence of mTOR Activation by an AKT-Independent Mechanism Provides Support for the Combined Treatment of PTEN-Deficient Prostate Tumors with mTOR and AKT Inhibitors. Transl. Oncol. 2012, 5, 422–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Chen, Y.T.; Chiu, W.Y.; Shen, M.R. Remodeling of calcium signaling in tumor progression. J. Biomed. Sci. 2013, 20, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fixemer, T.; Wissenbach, U.; Flockerzi, V.; Bonkhoff, H. Expression of the Ca2+-selective cation channel TRPV6 in human prostate cancer: A novel prognostic marker for tumor progression. Oncogene 2003, 22, 7858–7861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [Green Version]
- McGlacken, G.P.; Fairlamb, I.J. 2-Pyrone natural products and mimetics: Isolation, characterization and biological activity. Nat. Prod. Rep. 2005, 22, 369–385. [Google Scholar] [CrossRef]
- Garcia, I.J.P.; de Oliveira, G.C.; de Moura Valadares, J.M.; Banfi, F.F.; Andrade, S.N.; Freitas, T.R.; Dos Santos Monção Filho, E.; Lima Santos, H.; Júnior, G.M.V.; Chaves, M.H.; et al. New bufadienolides extracted from Rhinella marina inhibit Na,K-ATPase and induce apoptosis by activating caspases 3 and 9 in human breast and ovarian cancer cells. Steroids 2019, 152, 108490. [Google Scholar] [CrossRef]
- Li, Y.; Tang, H.; Tian, X.; Lin, H.; Wang, M.; Yao, M. Three new cytotoxic isomalabaricane triterpenes from the marine sponge Stelletta tenuis. Fitoterapia 2015, 106, 226–230. [Google Scholar] [CrossRef]
- Cimmino, A.; Mathieu, V.; Masi, M.; Baroncelli, R.; Boari, A.; Pescitelli, G.; Ferderin, M.; Lisy, R.; Evidente, M.; Tuzi, A.; et al. Evidente, Higginsianins A and B, Two Diterpenoid α-Pyrones Produced by Colletotrichum higginsianum, with in Vitro Cytostatic Activity. J. Nat. Prod. 2016, 79, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Coulup, S.K.; Huang, D.S.; Wong, H.L.; Georg, G.I. Identification of the Metabolic Profile of the α-Tubulin-Binding Natural Product (-)-Pironetin. J. Med. Chem. 2019, 62, 1684–1689. [Google Scholar] [CrossRef]
- Kondoh, M.; Usui, T.; Nishikiori, T.; Mayumi, T.; Osada, H. Apoptosis induction via microtubule disassembly by an antitumour compound, pironetin. Biochem. J. 1999, 340, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Potashnikova, D.M.; Saidova, A.A.; Tvorogova, A.V.; Sheval, E.V.; Vorobjev, I.A. Non-linear Dose Response of Lymphocyte Cell Lines to Microtubule Inhibitors. Front. Pharmacol. 2019, 10, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcelli, L.; Stolfa, D.; Stefanachi, A.; Di Fonte, R.; Garofoli, M.; Iacobazzi, R.M.; Silvestris, N.; Guarini, A.; Cellamare, S.; Azzariti, A. Synthesis and biological evaluation of N-biphenyl-nicotinic based moiety compounds: A new class of antimitotic agents for the treatment of Hodgkin Lymphoma. Cancer Lett. 2019, 445, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Antico Arciuch, V.G.; Elguero, M.E.; Poderoso, J.J.; Carreras, M.C. Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox. Signal. 2012, 16, 1150–1180. [Google Scholar] [CrossRef] [Green Version]
- Van den Bogert, C.; Muus, P.; Haanen, C.; Pennings, A.; Melis, T.E.; Kroon, A.M. Mitochondrial biogenesis and mitochondrial activity during the progression of the cell cycle of human leukemic cells. Exp. Cell Res. 1988, 178, 143–153. [Google Scholar] [CrossRef]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Richter, C. Reactive oxygen and nitrogen species regulate mitochondrial Ca2+ homeostasis and respiration. Biosci. Rep. 1997, 17, 53–66. [Google Scholar] [CrossRef] [Green Version]
- Grijalba, M.T.; Vercesi, A.E.; Schreier, S. Ca2+-induced increased lipid packing and domain formation in submitochondrial particles. A possible early step in the mechanism of Ca2+-stimulated generation of reactive oxygen species by the respiratory chain. Biochemistry 1999, 38, 13279–13287. [Google Scholar] [CrossRef]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Yoboue, E.D.; Sitia, R.; Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 2018, 9, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Marqués, M.; Carrera, A.C. Phosphoinositide 3-kinase activation in late G1 is required for c-Myc stabilization and S phase entry. Mol. Cell Biol. 2006, 26, 9116–9125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klippel, A.; Escobedo, M.A.; Wachowicz, M.S.; Apell, G.; Brown, T.W.; Giedlin, M.A.; Kavanaugh, W.M.; Williams, L.T. Activation of phosphatidylinositol 3-kinase is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformation. Mol. Cell Biol. 1998, 18, 5699–5711. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.T.; Wagner, L., 2nd; Yule, D.I.; Bhanumathy, C.; Joseph, S.K. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 2006, 281, 3731–3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [Green Version]
- Catz, S.D.; Johnson, J.L. BCL-2 in prostate cancer: A minireview. Apoptosis 2003, 8, 29–37. [Google Scholar] [CrossRef]
- Ren, W.; Joshi, R.; Mathew, P. Synthetic Lethality in PTEN-Mutant Prostate Cancer Is Induced by Combinatorial PI3K/Akt and BCL-XL nhibition. Mol. Cancer Res. 2016, 14, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Pinton, P.; Ferrari, D.; Magalhães, P.; Schulze-Osthoff, K.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J. Cell Biol. 2000, 148, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Vais, H.; Gu, W.; Foskett, J.K. Biphasic regulation of InsP3 receptor gating by dual Ca2+ release channel BH3-like domains mediates Bcl-xL control of cell viability. Proc. Natl. Acad. Sci. USA 2016, 113, E1953–E1962. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are available from the author Dr. Duen-Ren Hou. |








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheng, Y.-H.; Leu, W.-J.; Chen, C.-N.; Hsu, J.-L.; Liu, Y.-T.; Hsu, L.-C.; Hou, D.-R.; Guh, J.-H. The (+)-Brevipolide H Displays Anticancer Activity against Human Castration-Resistant Prostate Cancer: The Role of Oxidative Stress and Akt/mTOR/p70S6K-Dependent Pathways in G1 Checkpoint Arrest and Apoptosis. Molecules 2020, 25, 2929. https://doi.org/10.3390/molecules25122929
Sheng Y-H, Leu W-J, Chen C-N, Hsu J-L, Liu Y-T, Hsu L-C, Hou D-R, Guh J-H. The (+)-Brevipolide H Displays Anticancer Activity against Human Castration-Resistant Prostate Cancer: The Role of Oxidative Stress and Akt/mTOR/p70S6K-Dependent Pathways in G1 Checkpoint Arrest and Apoptosis. Molecules. 2020; 25(12):2929. https://doi.org/10.3390/molecules25122929
Chicago/Turabian StyleSheng, Yi-Hua, Wohn-Jenn Leu, Ching-Nung Chen, Jui-Ling Hsu, Ying-Tung Liu, Lih-Ching Hsu, Duen-Ren Hou, and Jih-Hwa Guh. 2020. "The (+)-Brevipolide H Displays Anticancer Activity against Human Castration-Resistant Prostate Cancer: The Role of Oxidative Stress and Akt/mTOR/p70S6K-Dependent Pathways in G1 Checkpoint Arrest and Apoptosis" Molecules 25, no. 12: 2929. https://doi.org/10.3390/molecules25122929
APA StyleSheng, Y. -H., Leu, W. -J., Chen, C. -N., Hsu, J. -L., Liu, Y. -T., Hsu, L. -C., Hou, D. -R., & Guh, J. -H. (2020). The (+)-Brevipolide H Displays Anticancer Activity against Human Castration-Resistant Prostate Cancer: The Role of Oxidative Stress and Akt/mTOR/p70S6K-Dependent Pathways in G1 Checkpoint Arrest and Apoptosis. Molecules, 25(12), 2929. https://doi.org/10.3390/molecules25122929