
New Microporous Lanthanide Organic Frameworks. Synthesis, Structure, Luminescence, Sorption, and Catalytic Acylation of 2-Naphthol

 ,
, 
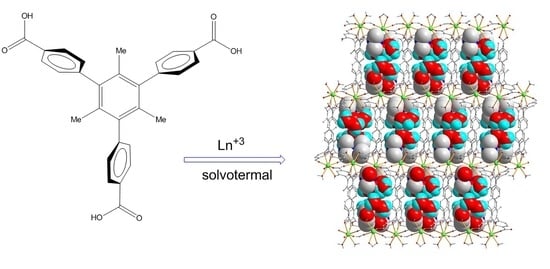
Abstract
:
1. Introduction
2. Results and Discussion
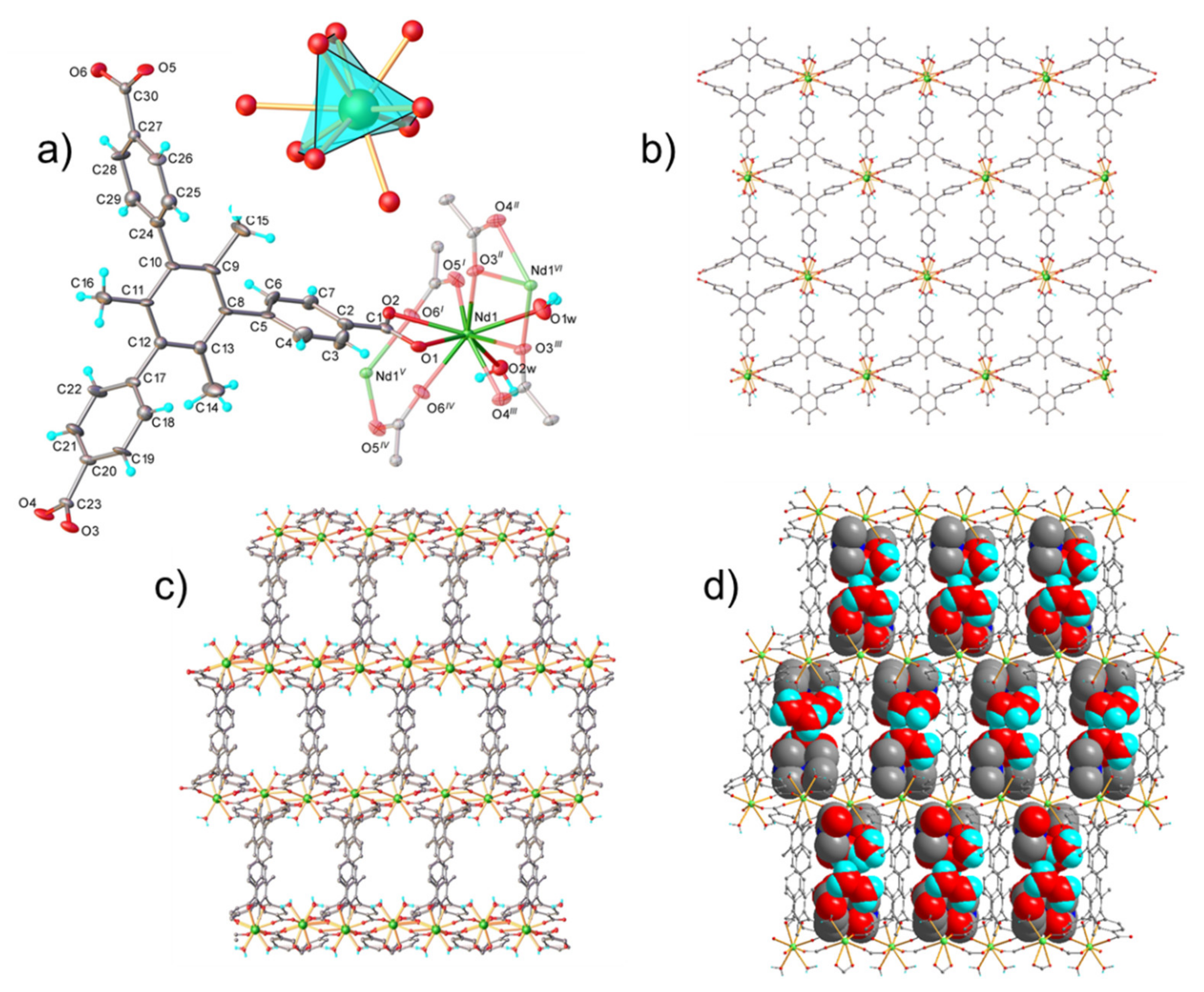
2.1. Structural Characterization
2.2. Infrared Spectroscopy Attenuated Total Reflectance (ATR)
2.3. Powder X-ray Diffraction
2.4. Thermogravimetric Analysis (TG/DTG)
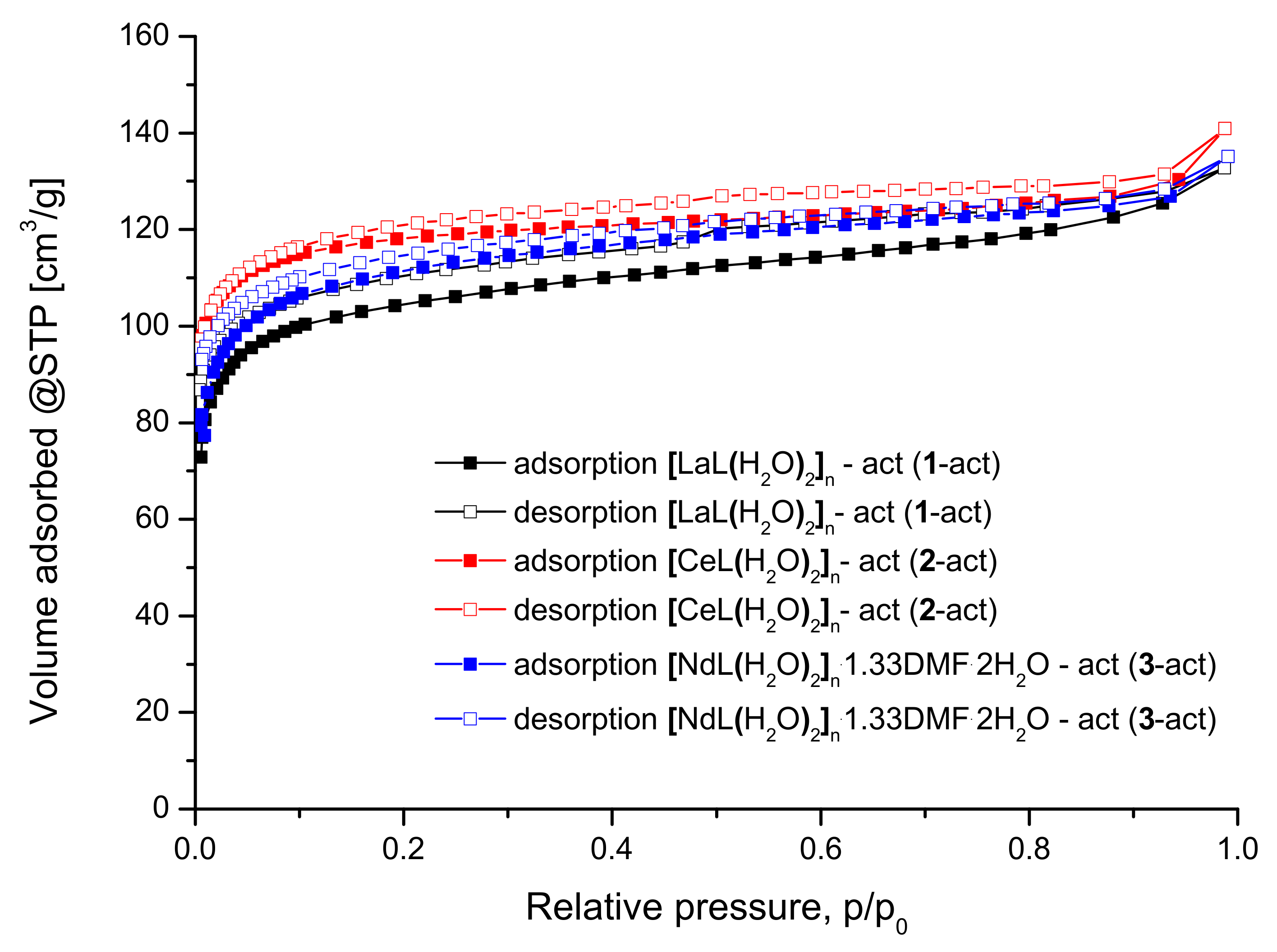
2.5. Sorption Properties
2.6. Luminescence
2.7. Catalytic Activity
3. Material and Methods
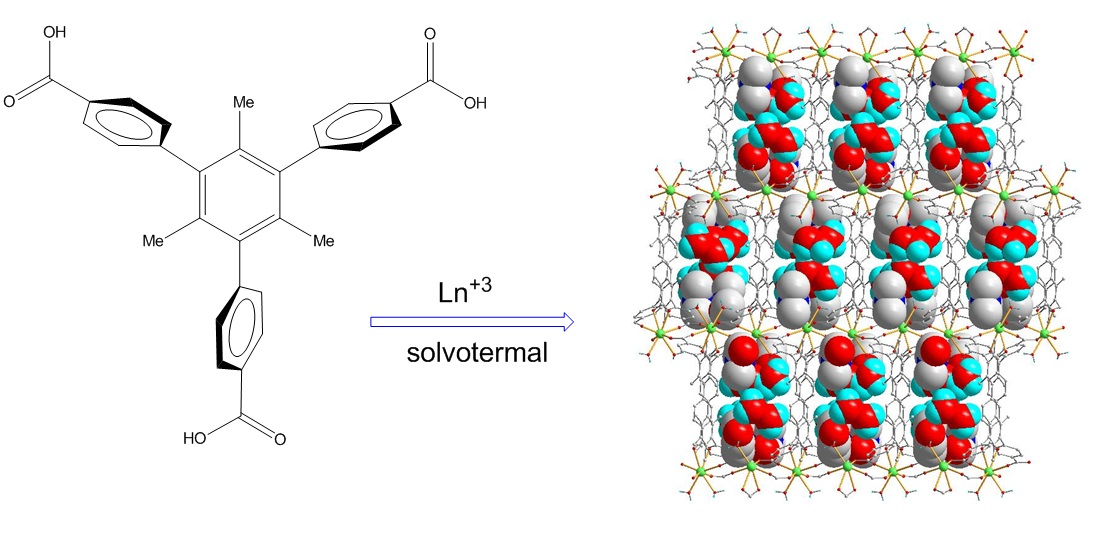
3.1. Synthesis of the Ligand 1,3,5-tris(4-carboxyphenyl)-2,4,6-trimethylbenzene (H3L)
3.2. Typical Synthesis of Ln-MOFs Where Ln = La (1), Ce (2), Nd (3), Eu (4), Gd (5), Dy (6), Ho (7)
3.3. Synthesis of 2-Naphthyl Acetate
3.4. Crystal Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chen, Y.; Ma, S. Microporous lanthanide metal-organic frameworks. Rev. Inorg. Chem. 2012, 32, 81–100. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, X.-Q.; Jiang, H.-L.; Sun, L.-B. Metal–Organic Frameworks for Heterogeneous Basic Catalysis. Chem. Rev. 2017, 117, 8129–8176. [Google Scholar] [CrossRef] [PubMed]
- Janiak, C.; Vieth, J.K. MOFs, MILs and more: Concepts, properties and applications for porous coordination networks (PCNs). New J. Chem. 2010, 34, 2366. [Google Scholar] [CrossRef]
- Zhou, H.-C.; Kitagawa, S. Metal–Organic Frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415–5418. [Google Scholar] [CrossRef] [Green Version]
- Slater, A.G.; Cooper, A.I. Function-led design of new porous materials. Science 2015, 348, aaa8075. [Google Scholar] [CrossRef] [PubMed]
- Cheong, V.F.; Moh, P.Y. Recent advancement in metal–organic framework: Synthesis, activation, functionalisation, and bulk production. Mater. Sci. Technol. 2018, 34, 1025–1045. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhang, J.-W.; Wang, X.; Wei, Y.-B. Synthesis, Crystal Structure and Catalytic Property of a New Cadmium Coordination Polymer. J. Clust. Sci. 2017, 38, 1294–1962. [Google Scholar] [CrossRef]
- Yoon, M.; Srirambalaji, R.; Kim, K. Homochiral Metal–Organic Frameworks for Asymmetric Heterogeneous Catalysis. Chem. Rev. 2011, 112, 1196–1231. [Google Scholar] [CrossRef]
- Petit, C. Present and future of MOF research in the field of adsorption and molecular separation. Curr. Opin. Chem. Eng. 2018, 20, 132–142. [Google Scholar] [CrossRef]
- Canepa, P.; Chabal, Y.J.; Thonhauser, T. When metal organic frameworks turn into linear magnets. Phys. Rev. B 2013, 87, 094407. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Xie, Y.-B.; Zhang, C.; Sun, J. Synthesis, structures, thermal and magnetic properties of a series of lanthanide [Ln = Sm, Gd, Er, Yb] complexes with 4-quinolineacarboxylate. J. Rare Earths 2009, 27, 12–17. [Google Scholar] [CrossRef]
- Lustig, W.P.; Mukherjee, S.; Rudd, N.D.; Desai, A.V.; Li, J.; Ghosh, S.K. Metal–organic frameworks: Functional luminescent and photonic materials for sensing applications. Chem. Soc. Rev. 2017, 46, 3242–3285. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Yu, J.; Li, Y.; Phua, S.F.Z.; Liu, G.; Lim, W.Q.; Yang, X.; Ganguly, R.; Dang, C.; Yang, C.; et al. Versatile bimetallic lanthanide metal-organic frameworks for tunable emission and efficient fluorescence sensing. Commun. Chem. 2018, 1, 12. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, J.; Chen, B.; Qian, G. Lanthanide Metal-Organic Frameworks for Luminescent Applications. In Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K.A., Jr., Eyring, L., Eds.; Elsevier BV: North-Holland, 2016; Volume 50, pp. 243–268. [Google Scholar]
- Shen, L.; Yang, L.; Fan, Y.; Wang, L.; Xu, J. Construction of a series of lanthanide metal–organic frameworks: Synthesis, structure, luminescence and white light emission. Cryst. Eng. Comm. 2015, 17, 9363–9369. [Google Scholar] [CrossRef]
- Huxford, R.C.; Della Rocca, J.; Lin, W. Metal–organic frameworks as potential drug carriers. Curr. Opin. Chem. Boil. 2010, 14, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Lu, K.; Liu, D.; Lin, W. Nanoscale Metal–Organic Frameworks for the Co-Delivery of Cisplatin and Pooled siRNAs to Enhance Therapeutic Efficacy in Drug-Resistant Ovarian Cancer Cells. J. Am. Chem. Soc. 2014, 136, 5181–5184. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Wang, J.; Chu, C.; Chen, W.; Wu, C.; Liu, G. Metal-Organic Framework-Based Stimuli-Responsive Systems for Drug Delivery. Adv. Sci. 2018, 6, 1801526. [Google Scholar] [CrossRef] [Green Version]
- Schoedel, A.; Yaghi, O.M. Porosity in metal–organic compounds. In Macrocyclic and Supramolecular Chemistry: How Izatt–Christensen Award Winners Shaped the Field, 1st ed.; Izatt, R.M., Ed.; John-Wiley & Sons: West Sussex, UK, 2016; pp. 200–220. [Google Scholar]
- McGuire, C.V.; Forgan, R.S. The surface chemistry of metal–organic frameworks. Chem. Commun. 2015, 51, 5199–5217. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, H.; Ko, N.; Go, Y.B.; Aratani, N.; Choi, S.B.; Choi, E.; Yazaydin, A.O.; Snurr, R.Q.; O’Keeffe, M.; Kim, J.; et al. Ultrahigh Porosity in Metal-Organic Frameworks. Science 2010, 329, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Chui, S.S. A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef] [PubMed]
- Ameloot, R.; Gobechiya, E.; Uji-I, H.; Martens, J.A.; Hofkens, J.; Alaerts, L.; Sels, B.F.; De Vos, D.E. Direct Patterning of Oriented Metal-Organic Framework Crystals via Control over Crystallization Kinetics in Clear Precursor Solutions. Adv. Mater. 2010, 22, 2685–2688. [Google Scholar] [CrossRef] [PubMed]
- Bano, S.; Tariq, S.R.; Ilyas, A.; Aslam, M.; Bilad, M.R.; Nizami, A.-S.; Khan, A.L. Synergistic solution of CO2 capture by novel lanthanide-based MOF-76 yttrium nanocrystals in mixed-matrix membranes. Energy Environ. 2019, 31, 692–712. [Google Scholar] [CrossRef]
- Cheng, P.-C.; Li, B.-H.; Tseng, F.-S.; Liang, P.-C.; Lin, C.-H.; Liu, W.-R. Synthesis, Structures and Electrochemical Properties of Lithium 1,3,5-Benzenetricarboxylate Complexes. Polymers 2019, 11, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Liu, F.; Zhang, L.-L.; Sun, D.; Wang, R.; Ju, Z.; Yuan, D.; Sun, D. Achieving a Rare Breathing Behavior in a Polycatenated 2 D to 3 D Net through a Pillar-Ligand Extension Strategy. Chem. Eur. J. 2013, 20, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Vergadou, V.; Pistolis, G.; Michaelides, A.; Varvounis, G.; Siskos, M.; Boukos, N.; Skoulika, S. Self-Organization of Four Symmetric Tri-phenyl-benzene Derivatives. Cryst. Growth Des. 2006, 6, 2486–2492. [Google Scholar] [CrossRef]
- Bajpai, A.; Venugopalan, P.; Moorthy, J.N. Self-Assembly of Rigid Three-Connecting Mesitylenetribenzoic Acid: Multifarious Supramolecular Synthons and Solvent-Induced Supramolecular Isomerism. Cryst. Growth Des. 2013, 13, 4721–4729. [Google Scholar] [CrossRef]
- Qiu, Y.-C.; Yuan, S.; Li, X.-X.; Du, D.-Y.; Wang, C.; Qin, J.-S.; Drake, H.F.; Lan, Y.-Q.; Jiang, L.; Zhou, H.-C. Face-Sharing Archimedean Solids Stacking for the Construction of Mixed-Ligand Metal–Organic Frameworks. J. Am. Chem. Soc. 2019, 141, 13841–13848. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Li, P.; Otake, K.-I.; Cui, Y.; Lyu, J.; Krzyaniak, M.; Zhang, Y.; Li, Z.; Liu, J.; et al. Vanadium Catalyst on Isostructural Transition Metal, Lanthanide, and Actinide Based Metal–Organic Frameworks for Alcohol Oxidation. J. Am. Chem. Soc. 2019, 141, 8306–8314. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, G.; Li, Z.; Sun, F.; Yang, Z.; Qiu, S. A lanthanide metal–organic framework with high thermal stability and available Lewis-acid metal sites. Chem. Commun. 2006, 3172–3174. [Google Scholar] [CrossRef]
- Choi, J.R.; Tachikawa, T.; Fujitsuka, M.; Majima, T. Europium-Based Metal—Organic Framework as a Photocatalyst for the One-Electron Oxidation of Organic Compounds. Langmuir 2010, 26, 10437–10443. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Farha, O.K.; Roberts, J.M.; Scheidt, K.A.; Nguyen, S.T.; Hupp, J.T. Metal–organic framework materials as catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Raizada, M.; Sama, F.; Ashafaq, M.; Shahid, M.; Khalid, M.; Ahmad, M.; Siddiqi, Z.A. Synthesis, structure and magnetic studies of lanthanide metal–organic frameworks (Ln-MOFs): Aqueous phase highly selective sensors for picric acid as well as the arsenic ion. Polyhedron 2018, 139, 131–141. [Google Scholar] [CrossRef]
- Zhao, X.; He, H.; Dai, F.; Sun, D.; Ke, Y. Supramolecular Isomerism in Honeycomb Metal−Organic Frameworks Driven by CH···π Interactions: Homochiral Crystallization from an Achiral Ligand through Chiral Inducement. Inorg. Chem. 2010, 49, 8650–8652. [Google Scholar] [CrossRef]
- Wang, B.; Lv, X.-L.; Feng, D.; Xie, L.-H.; Zhang, J.; Li, M.; Xie, Y.; Li, J.-R.J.; Zhou, H.-C. Highly Stable Zr(IV)-Based Metal—Organic Frameworks for the Detection and Removal of Antibiotics and Organic Explosives in Water. J. Am. Chem. Soc. 2016, 138, 6204–6216. [Google Scholar] [CrossRef]
- Zhao, X.; Dou, J.; Sun, D.; Cui, P.; Sun, D.; Wu, Q. A porous metal–organic framework (MOF) with unusual 2D→3D polycatenation based on honeycomb layers. Dalton Trans. 2012, 41, 1928–1930. [Google Scholar] [CrossRef]
- Bumstead, A.M.; Cordes, D.B.; Dawson, D.M.; Chakarova, K.; Mihaylov, M.; Hobday, C.L.; Düren, T.; Hadjiivanov, K.I.; Slawin, A.M.Z.; Ashbrook, S.E.; et al. Modulator-Controlled Synthesis of Microporous STA-26, an Interpenetrated 8,3-Connected Zirconium MOF with the the-i Topology, and its Reversible Lattice Shift. Chem. Eur. J. 2018, 24, 6115–6126. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.-F.; Vermeulen, N.A.; Howarth, A.J.; Li, P.; Sarjeant, A.A.; Hupp, J.T.; Farha, O.K. Adding to the Arsenal of Zirconium-Based Metal-Organic Frameworks: The Topology as a Platform for Solvent-Assisted Metal Incorporation. Eur. J. Inorg. Chem. 2016, 2016, 4349–4352. [Google Scholar] [CrossRef]
- Gustafsson, M.; Li, Z.; Zhu, G.; Qiu, S.; Grins, J.; Zou, X. A porous chiral lanthanide Metal-Organic Framework with high thermal stability. In Zeolites and Related materials: Trends, Targets and Chalenges; Gedeon, A., Massiani, P., Babonneau, F., Eds.; Elsevier BV: Amsternam, The Netherlands, 2008; Volume 174, pp. 451–454. [Google Scholar]
- Thommes, M. Physical Adsorption Characterization of Nanoporous Materials. Chem. Ing. Tech. 2010, 82, 1059–1073. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.S.; Dey, S.; Attallah, A.G.; Krause-Rehberg, R.; Janiak, C.; Holdt, H. Insights into the pores of microwave-assisted metal–imidazolate frameworks showing enhanced gas sorption. Dalton Trans. 2017, 46, 4824–4833. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.S.; Bhunia, A.; Kelling, A.; Schilde, U.; Janiak, C.; Holdt, H. A supramolecular Co(ii)14-metal-organic cube in a hydrogen-bonded network and a Co(ii)-organic framework with a flexible methoxy substituent. Chem. Commun. 2014, 50, 5441–5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, S.S.; Dey, S.; Baburin, I.A.; Kelling, A.; Schilde, U.; Seifert, G.; Janiak, C.; Holdt, H.-J. Syntheses of two imidazolate-4-amide-5-imidate linker-based hexagonal metal–organic frameworks with flexible ethoxy substituent. Cryst. Eng. Comm. 2013, 15, 9394. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.S.; Bhunia, A.; Baburin, I.A.; Jäger, C.; Kelling, A.; Schilde, U.; Seifert, G.; Janiak, C.; Holdt, H.-J. Gate effects in a hexagonal zinc-imidazolate-4-amide-5-imidate framework with flexible methoxy substituents and CO2 selectivity. Chem. Commun. 2013, 49, 7599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alrefai, A.; Mondal, S.S.; Wruck, A.; Kelling, A.; Schilde, U.; Brandt, P.; Janiak, C.; Schönfeld, S.; Weber, B.; Rybakowski, L.; et al. Hydrogen-bonded supramolecular metal-imidazolate frameworks: Gas sorption, magnetic and UV/Vis spectroscopic properties. J. Incl. Phenom. Macrocycl. Chem. 2019, 94, 155–165. [Google Scholar] [CrossRef]
- Mondal, S.S.; Dey, S.; Attallah, A.G.; Bhunia, A.; Kelling, A.; Schilde, U.; Krause-Rehberg, R.; Janiak, C.; Holdt, H. Missing Building Blocks Defects in a Porous Hydrogen-bonded Amide-Imidazolate Network Proven by Positron Annihilation Lifetime Spectroscopy. ChemistrySelect 2016, 1, 4320–4325. [Google Scholar] [CrossRef]
- Mondal, S.S.; Bhunia, A.; Attallah, A.G.; Matthes, P.R.; Kelling, A.; Schilde, U.; Müller-Buschbaum, K.; Krause-Rehberg, R.; Janiak, C.; Holdt, H. Study of the Discrepancies between Crystallographic Porosity and Guest Access into Cadmium-Imidazolate Frameworks and Tunable Luminescence Properties by Incorporation of Lanthanides. Chem. Eur. J. 2016, 22, 6905–6913. [Google Scholar] [CrossRef]
- Binnemans, K. Lanthanide-Based Luminescent Hybrid Materials. Chem. Rev. 2009, 109, 4283–4374. [Google Scholar] [CrossRef] [Green Version]
- You, L.-X.; Li, Z.-G.; Ding, F.; Wang, S.-J.; Ren, B.-Y.; Sun, Y.-G. Synthesis, structure and luminescence properties of lanthanide coordination polymers using in situ decarboxylation of a H3cppdc ligand. Inorg. Chem. Commun. 2014, 46, 340–343. [Google Scholar] [CrossRef]
- Dechnik, J.; Mühlbach, F.; Dietrich, D.; Wehner, T.; Gutmann, M.; Lühmann, T.; Meinel, L.; Janiak, C.; Müller-Buschbaum, K. Luminescent Metal-Organic Framework Mixed-Matrix Membranes from Lanthanide Metal-Organic Frameworks in Polysulfone and Matrimid. Eur. J. Inorg. Chem. 2016, 2016, 4408–4415. [Google Scholar] [CrossRef]
- Kumar, M.; Sheikh, H.N.; Franconetti, A.; Zaręba, J.K.; Sahoo, S.C.; Frontera, A.; Franconetti, A. 2,5-Furandicarboxylic acid as a linker for lanthanide coordination polymers: The role of heteroaromatic π–π stacking and hydrogen bonding. New J. Chem. 2019, 43, 2179–2195. [Google Scholar] [CrossRef]
- Pagis, C.; Ferbinteanu, M.; Rothenberg, G.; Tanase, S.; Grecea, S.T. Lanthanide-Based Metal Organic Frameworks: Synthetic Strategies and Catalytic Applications. ACS Catal. 2016, 6, 6063–6072. [Google Scholar] [CrossRef]
- Rahman, A.; Uahengo, V.; Likius, D.S.; Mupa, M. Selective Acetylation of 2-Naphthol to 2-Naphthyl Acetate with Ni Homogeneous Catalysts: An Environmentally Friendly Protocol. Sci. J. Chem. 2017, 5, 47. [Google Scholar] [CrossRef] [Green Version]
- Herbst, A.; Khutia, A.; Janiak, C. Brønsted Instead of Lewis Acidity in Functionalized MIL-101Cr MOFs for Efficient Heterogeneous (nano-MOF) Catalysis in the Condensation Reaction of Aldehydes with Alcohols. Inorg. Chem. 2014, 53, 7319–7333. [Google Scholar] [CrossRef]
- Chughtai, A.H.; Ahmad, N.; Younus, H.A.; Laypkov, A.; Verpoort, F. Metal–organic frameworks: Versatile heterogeneous catalysts for efficient catalytic organic transformations. Chem. Soc. Rev. 2015, 44, 6804–6849. [Google Scholar] [CrossRef] [Green Version]
- Remya, V.R.; Kurian, M. Synthesis and catalytic applications of metal–organic frameworks: A review on recent literature. Int. Nano Lett. 2018, 9, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Liu, L.; Chen, Z.; Li, T.; Jin, T. Rapid and Efficient Method for Acetylation of Alcohols and Phenols with Acetic Anhydride Catalyzed by Silica Sulfate. Synth. Commun. 2006, 36, 1221–1227. [Google Scholar] [CrossRef]
- CrysAlisPro Software system, version 1.171.38.46; Rigaku Corporation: Oxford, UK, 2015.
- Dolomanov, O.; Bourhis, L.J.; Gildea, R.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ln-MOFs—Activated Compounds | BET Surface Area [m2/g] | Total Pore Volume [cm3/g] |
|---|---|---|
| [LaL(H2O)2]n—act (1-act) | 405/395 * | 0.19 |
| [CeL(H2O)2]n—act (2-act) | 467/460 * | 0.20 |
| [NdL(H2O)2]n—act (3-act) | 426/404 * | 0.20 |
| [EuL(H2O)2]n—act (4-act) | 114 | 0.08 |
| [GdL(H2O)2]n—act (5-act) | 348 | 0.18 |
| [DyL(H2O)2]n—act (6-act) [DyL(H2O)2]n—act (6-act) | 202 (Figure S35a) 298 (Figure S35b) | 0.16 0.16 |
| [HoL(H2O)2]n—act (7-act) | 286 | 0.15 |
| Entry | Substrate | Conversion * | Time | Catalyst | Run Number |
|---|---|---|---|---|---|
| 1 | 2-naphthol | 88 | 24 | 1 | 1 |
| 2 | 2-naphthol | 98 | 24 | 3 | 1 |
| 3 | 2-naphthol | 96 | 24 | 3 | 2 |
| 4 | 2-naphthol | 92 | 24 | 3 | 3 |
| 5 | 2-naphthol | 65 65 | 5 24 | 3 - | 1 2 |
| 6 ** | 2-naphthol | 80 72 | 24 24 | 3 3 | 1 2 |
| Compound | 3984 (1) | 3971 (2) | 3972 (3) |
|---|---|---|---|
| empirical formula | C30H25LaO8 | C30H25CeO8 | C34H38.33N1.33NdO11.33 |
| Fw | 652.41 | 653.62 | 791.21 |
| space group | Pnna | Pnna | P21/n |
| a [Å] | 9.3345(5) | 9.5029(6) | 9.5200(13) |
| b [Å] | 16.6700(9) | 16.6504(10) | 16.473(3) |
| c [Å] | 27.2304(19) | 27.1399(15) | 27.389(6) |
| a [°] | 90 | 90 | 90 |
| B [°] | 90 | 90 | 93.098(18) |
| g [°] | 90 | 90 | 90 |
| V [Å3] | 4237.2(4) | 4294.3(4) | 4288.9(13) |
| Z | 4 | 4 | 4 |
| rcalcd [g cm−3] | 1.023 | 1.011 | 1.225 |
| Crystal size [mm] | 0.20 × 0.20 × 0.10 | 0.40 × 0.20 × 0.20 | 0.70 × 0.60 × 0.18 |
| T [K] | 180 | 293 | 180 |
| μ [mm−1] | 1.040 | 1.092 | 1.260 |
| 2È range | 5.11 to 50.054 | 4.542 to 50.052 | 2.886 to 50.054 |
| Reflections collected | 9929 | 9514 | 11840 |
| Independent reflections | 3744 [Rint = 0.0515] | 3780 [Rint = 0.0952] | 11840 [Rint = 0.1178] |
| Data/restraints/parameters | 3744/54/181 | 3780/12/180 | 11840/48/439 |
| R1[a] | 0.0852 | 0.0538 | 0.0651 |
| wR2[b] | 0.2280 | 0.0983 | 0.1431 |
| GOF [c] | 1.048 | 1.002 | 1.040 |
| Largest diff. peak/hole/e Å−3 | 1.16/−1.45 | 1.23/−1.58 | 1.40/−1.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bejan, D.; Bahrin, L.G.; Shova, S.; Marangoci, N.L.; Kökҫam-Demir, Ü.; Lozan, V.; Janiak, C. New Microporous Lanthanide Organic Frameworks. Synthesis, Structure, Luminescence, Sorption, and Catalytic Acylation of 2-Naphthol. Molecules 2020, 25, 3055. https://doi.org/10.3390/molecules25133055
Bejan D, Bahrin LG, Shova S, Marangoci NL, Kökҫam-Demir Ü, Lozan V, Janiak C. New Microporous Lanthanide Organic Frameworks. Synthesis, Structure, Luminescence, Sorption, and Catalytic Acylation of 2-Naphthol. Molecules. 2020; 25(13):3055. https://doi.org/10.3390/molecules25133055
Chicago/Turabian StyleBejan, Dana, Lucian Gabriel Bahrin, Sergiu Shova, Narcisa Laura Marangoci, Ülkü Kökҫam-Demir, Vasile Lozan, and Christoph Janiak. 2020. "New Microporous Lanthanide Organic Frameworks. Synthesis, Structure, Luminescence, Sorption, and Catalytic Acylation of 2-Naphthol" Molecules 25, no. 13: 3055. https://doi.org/10.3390/molecules25133055
APA StyleBejan, D., Bahrin, L. G., Shova, S., Marangoci, N. L., Kökҫam-Demir, Ü., Lozan, V., & Janiak, C. (2020). New Microporous Lanthanide Organic Frameworks. Synthesis, Structure, Luminescence, Sorption, and Catalytic Acylation of 2-Naphthol. Molecules, 25(13), 3055. https://doi.org/10.3390/molecules25133055