
Development of an Atmospheric Pressure Chemical Ionization Interface for GC-MS



Abstract
:1. Introduction
2. Results and Discussion
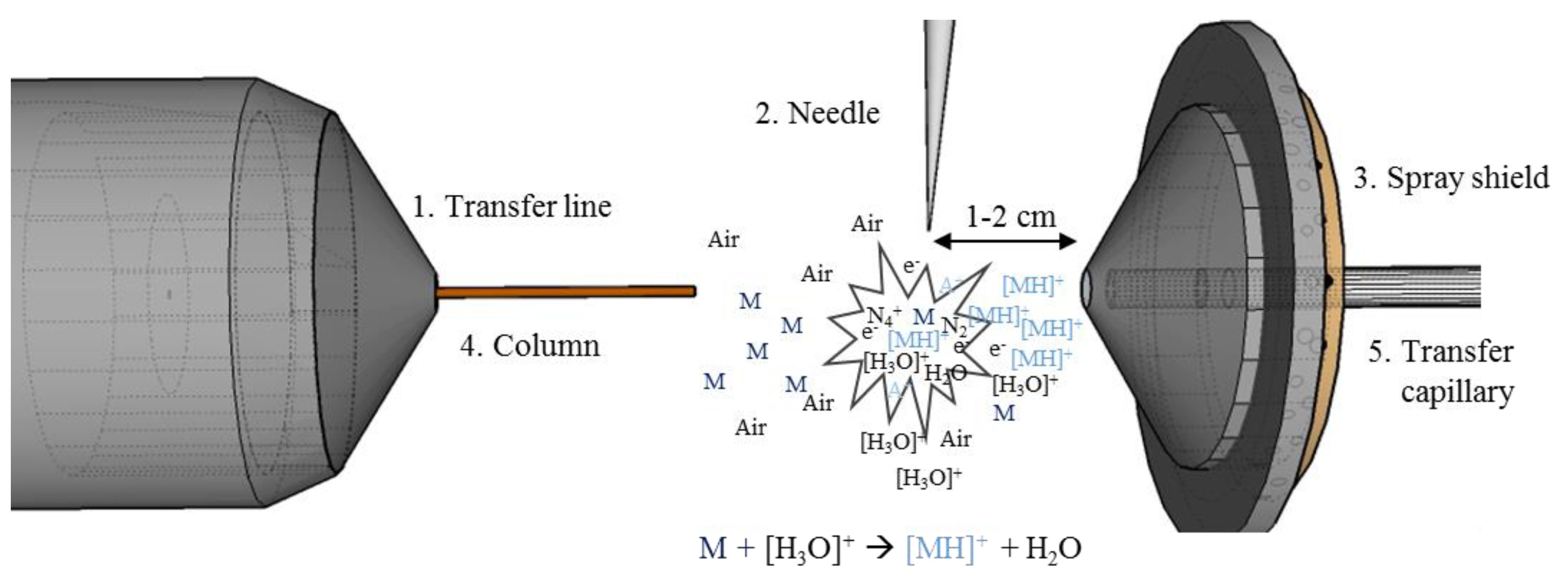
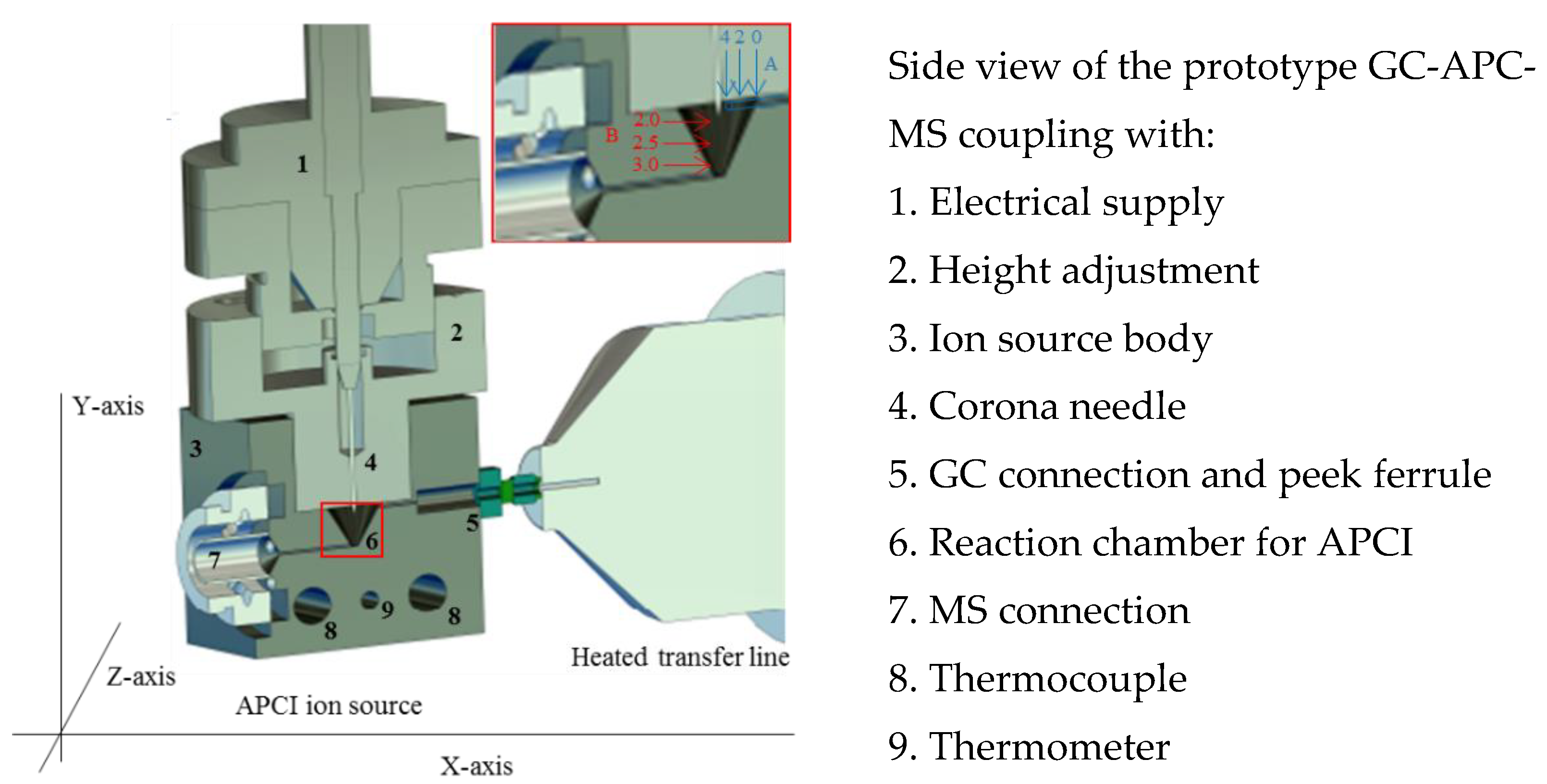
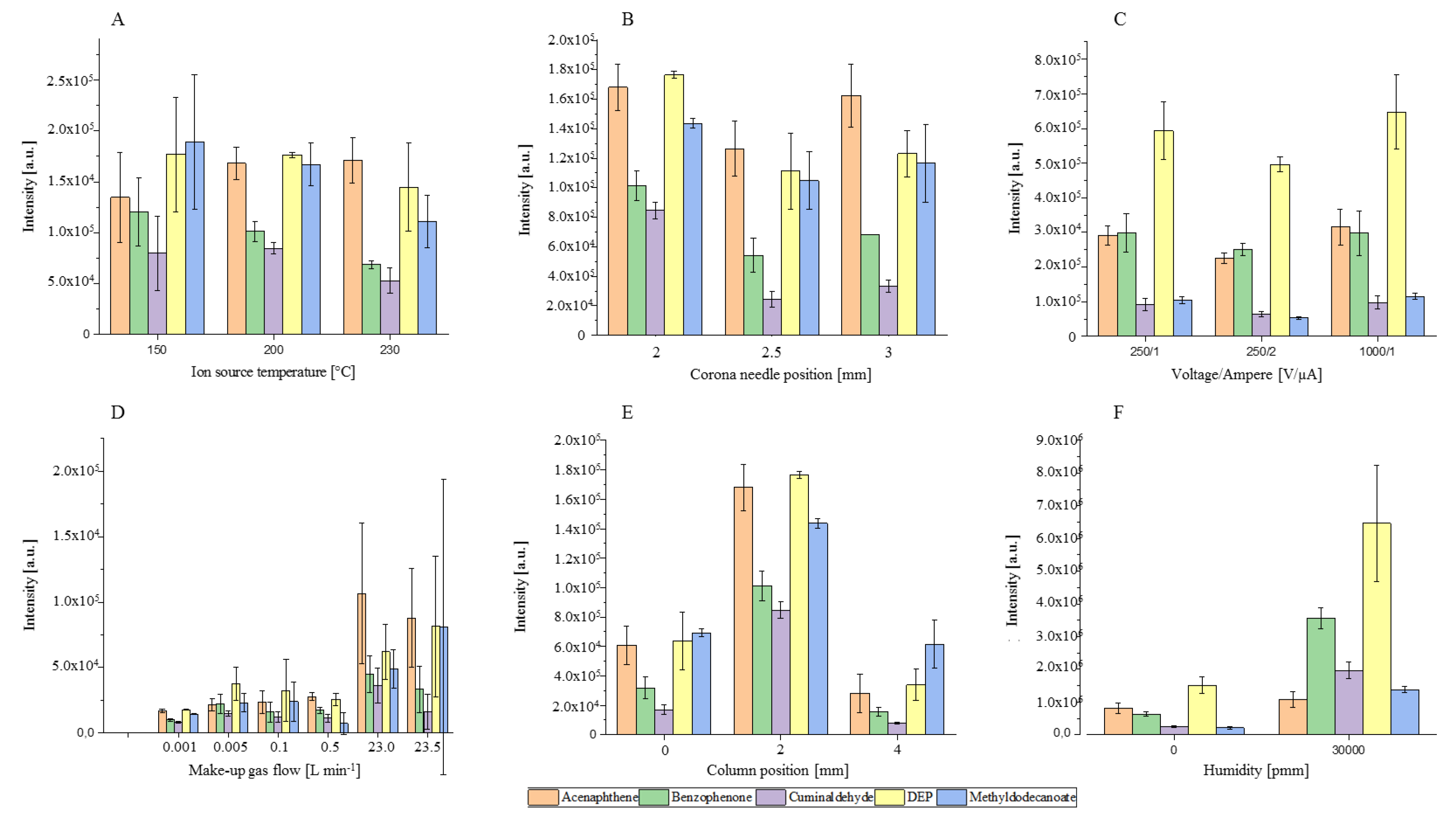
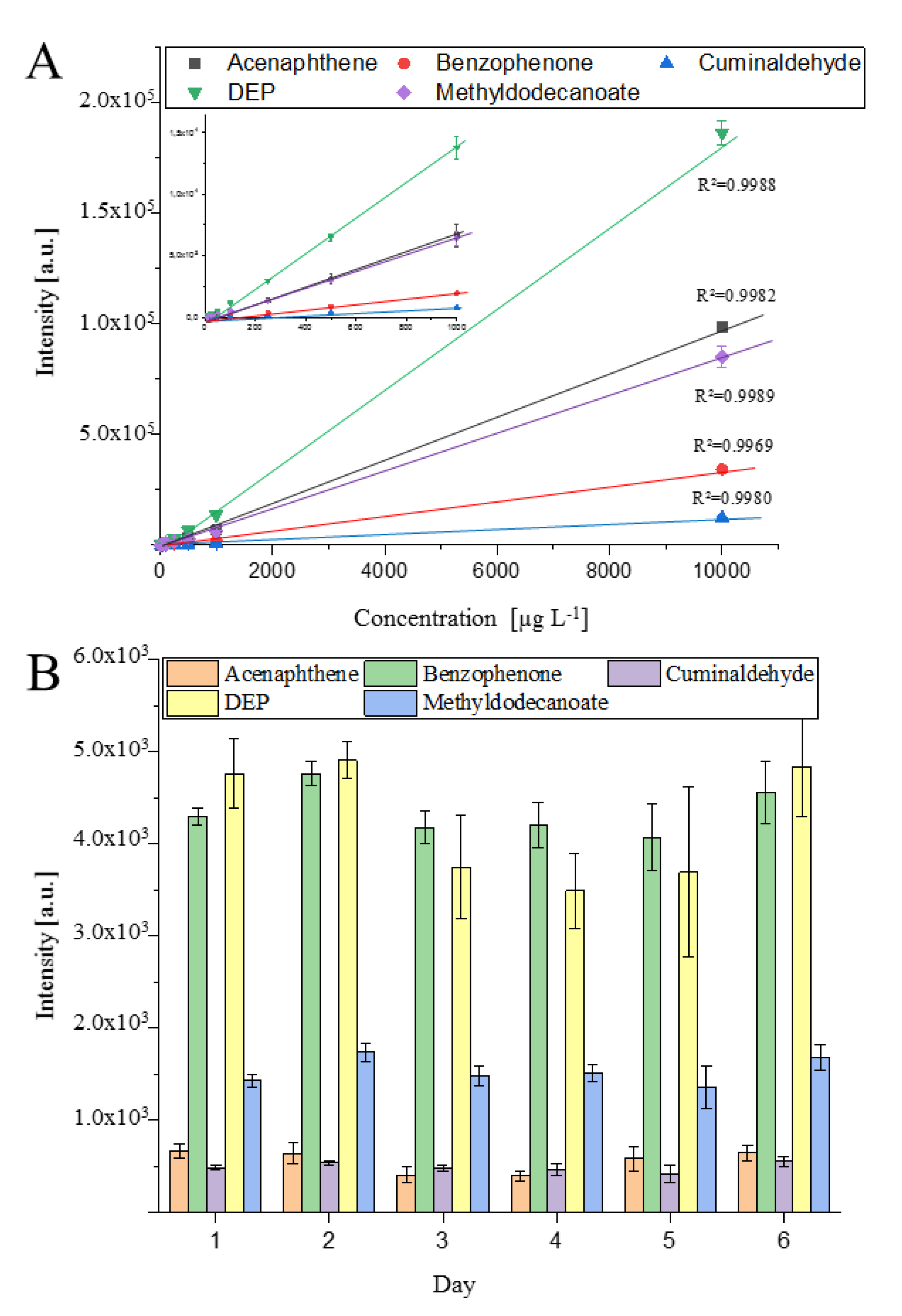
2.1. Development of the GC-APCI Ion Source
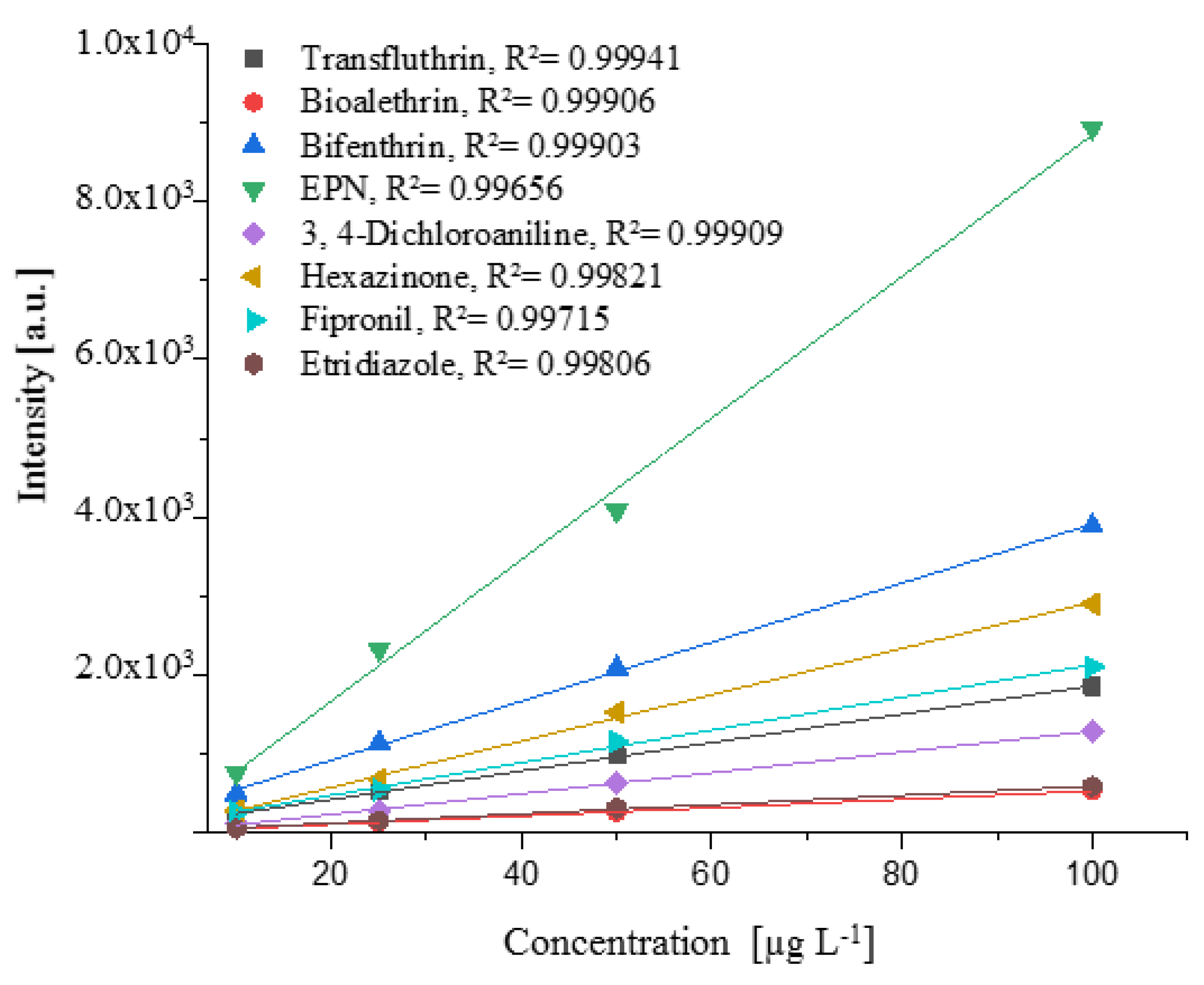
2.2. Target Analysis of Plant Protection Product Residues in Coffee Beans
3. Materials and Methods
3.1. Chemicals and Solutions
3.2. Instrumentation
3.3. Extraction Method and Evaluation of the Extraction Protocol
3.4. Analytical Methods
3.4.1. Influence of the Ion Source Parameters on the Ionization Behavior
3.4.2. Application of Plant Protection Products in Coffee Beans
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Horning, E.C.; Horning, M.G.; Carroll, D.I.; Dzidic, I.; Stillwell, R.N. New Picogram Detection System Based on A Mass Spectrometer with an External Ionization Source at Atmospheric Pressure. Anal. Chem. 1973, 45, 936–943. [Google Scholar] [CrossRef]
- Sparkman, O.D.; Penton, Z.; Kitson, F.G. Gas. Chromatography and Mass Spectrometry: A Practical Guide, 2nd ed.; Elsevier: Amsterdam, Boston, MA, USA, 2011. [Google Scholar]
- Portolés, T.; Mol, J.G.J.; Sancho, J.V.; Hernández, F. Advantages of Atmospheric Pressure Chemical Ionization in Gas Chromatography Tandem Mass Spectrometry: Pyrethroid Insecticides as A Case Study. Anal. Chem. 2012, 84, 9802–9810. [Google Scholar] [CrossRef] [PubMed]
- Vinaixa, M.; Schymanski, E.L.; Neumann, S.; Navarro, M.; Salek, R.M.; Yanes, O. Mass Spectral Databases For LC/Ms- And GC/Ms-Based Metabolomics: State of The Field and Future Prospects. Trac-Trends Anal. Chem. 2016, 78, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Adams, R.P.; Granat, M.; Hogge, L.R.; Rudloff, E.V. Identification of Lower Terpenoids From Gas-Chromatography-Mass Spectral Data by On-Line Computer Method. J. Chromatogr. Sci. 1979, 17, 75–81. [Google Scholar] [CrossRef]
- Stein, S.E. Estimating Probabilities of Correct Identification from Results of Mass Spectral Library Searches. J. Am. Soc. Mass Spectrom. 1994, 5, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Portolés, T.; Sancho, J.V.; Hernández, F.; Newton, A.; Hancock, P. Potential of Atmospheric Pressure Chemical Ionization Source in GC-QTOF Ms For Pesticide Residue Analysis. Org. Mass Spectrom. 2010, 45, 926–936. [Google Scholar] [CrossRef]
- Canellas, E.; Vera, P.; Domeño, C.; Alfaro, P.; Nerín, C. Atmospheric Pressure Gas Chromatography Coupled to Quadrupole-Time of Flight Mass Spectrometry as A Powerful Tool for Identification of Non Intentionally Added Substances In Acrylic Adhesives Used In Food Packaging Materials. J. Chromatogr. A 2012, 1235, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Portolés, T.; Cherta, L.; Beltran, J.; Hernández, F. Improved Gas Chromatography-Tandem Mass Spectrometry Determination of Pesticide Residues Making Use of Atmospheric Pressure Chemical Ionization. J. Chromatogr. A 2012, 1260, 183–192. [Google Scholar] [CrossRef]
- Mcewen, C.N.; Mckay, R.G. A Combination Atmospheric Pressure LC/MS-GC/Ms Ion Source: Advantages of Dual AP-LC/MS-GC/Ms Instrumentation. J. Am. Soc. Mass Spectrom. 2005, 16, 1730–1738. [Google Scholar] [CrossRef] [Green Version]
- Schiewek, R.; Lorenz, M.; Giese, R.; Brockmann, K.; Benter, T.; Gäb, S.; Schmitz, O.J. Development of A Multipurpose Ion Source For LC-Ms and GC-API Ms. Anal. Bioanal. Chem. 2008, 392, 87–96. [Google Scholar] [CrossRef]
- Klee, S.; Derpmann, V.; Wißdorf, W.; Klopotowski, S.; Kersten, H.; Brockmann, K.J.; Benter, T.; Albrecht, S.; Bruins, A.P.; Dousty, F.; et al. Are Clusters Important in Understanding the Mechanisms in Atmospheric Pressure Ionization? Part 1: Reagent Ion Generation and Chemical Control of Ion Populations. J. Am. Soc. Mass Spectrom. 2014, 25, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Bruins, A.P. Mass Spectrometry with Ion Sources Operating at Atmospheric Pressure. Mass Spectrom. Rev. 1991, 10, 53–77. [Google Scholar] [CrossRef]
- Rösch, A.; Beck, B.; Hollender, J.; Singer, H. Picogram Per Liter Quantification OF Pyrethroid And Organophosphate Insecticides in Surface Waters: A Result of Large Enrichment with Liquid-Liquid Extraction and Gas Chromatography Coupled to Mass Spectrometry Using Atmospheric Pressure Chemical Ionization. Anal. Bioanal. Chem. 2019, 411, 3151–3164. [Google Scholar] [PubMed]
- Halloum, W.; Cariou, R.; Dervilly-Pinel, G.; Jaber, F.; Le Bizec, B. Apci As an Innovative Ionization Mode Compared with EI And Ci For the Analysis of a Large Range of Organophosphate Esters Using GC-Ms/Ms. J. Mass Spectrom. JMS 2017, 52, 54–61. [Google Scholar] [CrossRef]
- Wachsmuth, C.J.; Dettmer, K.; Lang, S.A.; Mycielska, M.E.; Oefner, P.J. Continuous Water Infusion Enhances Atmospheric Pressure Chemical Ionization of Methyl Chloroformate Derivatives in Gas Chromatography Coupled to Time-Of-Flight Mass Spectrometry-Based Metabolomics. Anal. Chem. 2014, 86, 9186–9195. [Google Scholar] [CrossRef]
- Fang, J.; Zhao, H.; Zhang, Y.; Wong, M.; He, Y.; Sun, Q.; Xu, S.; Cai, Z. Evaluation of Gas Chromatography-Atmospheric Pressure Chemical Ionization Tandem Mass Spectrometry as An Alternative to Gas Chromatography Tandem Mass Spectrometry for The Determination of Polychlorinated Biphenyls and Polybrominated Diphenyl Ethers. Chemosphere 2019, 225, 288–294. [Google Scholar] [CrossRef]
- Kersten, H.; Kroll, K.; Haberer, K.; Brockmann, K.J.; Benter, T.; Peterson, A.; Makarov, A. Design Study of An Atmospheric Pressure Photoionization Interface For GC-Ms. J. Am. Soc. Mass Spectrom. 2016, 27, 607–614. [Google Scholar] [CrossRef]
- Húsková, R.; Matisová, E.; Hrouzková, S.; Svorc, L. Analysis of Pesticide Residues by Fast Gas Chromatography in Combination with Negative Chemical Ionization Mass Spectrometry. J. Chromatogr. A 2009, 1216, 6326–6334. [Google Scholar] [CrossRef]
- Dong, J.; Pan, Y.-X.; Lv, J.-X.; Sun, J.; Gong, X.-M.; Li, K. Multiresidue Method for The Determination of Pesticides in Fruits and Vegetables Using Gas Chromatography-Negative Chemical Ionization-Triple Quadrupole Tandem Mass Spectrometry. Chromatographia 2011, 74, 109–119. [Google Scholar] [CrossRef]
- Zacs, D.; Perkons, I.; Bartkevics, V. Evaluation of Analytical Performance of Gas Chromatography Coupled with Atmospheric Pressure Chemical Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry (GC-APCI-Ft-ICR-Ms) In the Target and Non-Targeted Analysis of Brominated and Chlorinated Flame Retardants in Food. Chemosphere 2019, 225, 368–377. [Google Scholar]
- Fang, J.; Zhao, H.; Zhang, Y.; Lu, M.; Cai, Z. Atmospheric Pressure Chemical Ionization in Gas Chromatography-Mass Spectrometry for the Analysis of Persistent Organic Pollutants. Trends Environ. Anal. Chem. 2020, 25, E00076. [Google Scholar] [CrossRef]
- Portolés, T.; Mol, J.G.J.; Sancho, J.V.; López, F.J.; Hernández, F. Validation of A Qualitative Screening Method for Pesticides in Fruits and Vegetables by Gas Chromatography Quadrupole-Time of Flight Mass Spectrometry with Atmospheric Pressure Chemical Ionization. Anal. Chim. Acta 2014, 838, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Cherta, L.; Portolés, T.; Beltran, J.; Pitarch, E.; Mol, J.G.J.; Hernández, F. Application of Gas Chromatography- (Triple Quadrupole) Mass Spectrometry with Atmospheric Pressure Chemical Ionization for The Determination of Multiclass Pesticides in Fruits and Vegetables. J. Chromatogr. A 2013, 1314, 224–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, F.; Cervera, M.I.; Portolés, T.; Beltrán, J.; Pitarch, E. The Role of GC-Ms/Ms With Triple Quadrupole in Pesticide Residue Analysis in Food and The Environment. Anal. Methods 2013, 5, 5875. [Google Scholar] [CrossRef] [Green Version]
- Portolés, T.; Mol, J.G.J.; Sancho, J.V.; Hernández, F. Use of Electron Ionization and Atmospheric Pressure Chemical Ionization in Gas Chromatography Coupled to Time-Of-Flight Mass Spectrometry for Screening and Identification of Organic Pollutants in Waters. J. Chromatogr. A 2014, 1339, 145–153. [Google Scholar] [CrossRef]
- Hernández, F.; Ibáñez, M.; Portolés, T.; Cervera, M.I.; Sancho, J.V.; López, F.J. Advancing Towards Universal Screening for Organic Pollutants in Waters. J. Hazard. Mater. 2015, 282, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portolés, T.; Rosales, L.E.; Sancho, J.V.; Santos, F.J.; Moyano, E. Gas Chromatography-Tandem Mass Spectrometry with Atmospheric Pressure Chemical Ionization for Fluorotelomer Alcohols and Perfluorinated Sulfonamides Determination. J. Chromatogr. A 2015, 1413, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Olmo-García, L.; Wendt, K.; Kessler, N.; Bajoub, A.; Fernández-Gutiérrez, A.; Baessmann, C.; Carrasco-Pancorbo, A. Exploring the Capability of LC-Ms and GC-Ms Multi-Class Methods to Discriminate Virgin Olive Oils from Different Geographical Indications and To Identify Potential Origin Markers. Eur. J. Lipid Sci. Technol. 2019, 354, 26. [Google Scholar] [CrossRef]
- Olmo-García, L.; Kessler, N.; Neuweger, H.; Wendt, K.; Olmo-Peinado, J.M.; Fernández-Gutiérrez, A.; Baessmann, C.; Carrasco-Pancorbo, A. Unravelling the Distribution OF Secondary Metabolites in Olea Europaea, L.: Exhaustive Characterization of Eight Olive-Tree Derived Matrices by Complementary Platforms (LC-ESI/APCI-Ms and GC-APCI-Ms). Molecules 2018, 23, 2419. [Google Scholar] [CrossRef] [Green Version]
- Bajoub, A.; Pacchiarotta, T.; Hurtado-Fernández, E.; Olmo-García, L.; García-Villalba, R.; Fernández-Gutiérrez, A.; Mayboroda, O.A.; Carrasco-Pancorbo, A. Comparing Two Metabolic Profiling Approaches (Liquid Chromatography AND Gas Chromatography Coupled to Mass Spectrometry) For Extra-Virgin Olive Oil Phenolic Compounds Analysis: A Botanical Classification Perspective. J. Chromatogr. A 2016, 1428, 267–279. [Google Scholar] [CrossRef]
- Bristow, T.; Harrison, M.; Sims, M. The Application of Gas Chromatography/Atmospheric Pressure Chemical Ionisation Time-Of-Flight Mass Spectrometry to Impurity Identification in Pharmaceutical Development. Rapid Commun. Mass Spectrom: RCM 2010, 24, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.; Bristow, T.; Ray, A.; Rome, K.; Sanderson, N.; Sims, M.; Cojocariu, C.; Silcock, P. Applicability of Gas Chromatography/Quadrupole-Orbitrap Mass Spectrometry in Support of Pharmaceutical Research and Development. Rapid Commun. Mass Spectrom: RCM 2016, 30, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Statement on The Available Outcomes of The Human Health Assessment in The Context of The Pesticides Peer Review of The Active Substance Chlorpyrifos. EFSA 2019, 17, 5809–5832.
- Schmidt, G.H. Pestizide Und Umweltschutz; Vieweg+Teubner Verlag: Wiesbaden, Germany, 1986. [Google Scholar]
- Organization, F.A.A. Fao Statistical Pocketbook; Food & Agriculture Org: Rome, Italy, 2019. [Google Scholar]
- Dettmer-Wilde, K.; Engewald, W. (Eds.) Practical Gas. Chromatography: A Comprehensive Reference; Springer: Berlin, Heidelberg, Germany, 2014. [Google Scholar]
- Lias, S.G.; Liebman, J.F.; Levin, R.D. Evaluated Gas Phase Basicities And Proton Affinities of Molecules; Heats of Formation of Protonated Molecules. J. Phys. Chem. Ref. Data 1984, 13, 695–808. [Google Scholar] [CrossRef]
- Upadhyay, S.K. Chemical Kinetics and Reaction Dynamics; Springer Netherlands: New Delhi, India, 2006. [Google Scholar]
- Nicol, G.; Sunner, J.; Kebarle, P. Kinetics and Thermodynamics of Protonation Reactions: H3O+ (H2O) H + B = Bh+ (H2O) B + (H − B + 1) H2O, Where B Is A Nitrogen, Oxygen or Carbon Base. Int. J. Mass Spectorm. Ion. Processes. 1988, 84, 135–155. [Google Scholar] [CrossRef]
- Sunner, J.; Ikonomou, M.G.; Kebarle, P. Sensitivity Enhancements Obtained at High Temperatures in Atmospheric Pressure Ionization Mass Spectrometry. Anal. Chem. 1988, 60, 1308–1313. [Google Scholar] [CrossRef]
- Sunner, J.; Nicol, G.; Kebarle, P. Factors Determining Relative Sensitivity of Analytes in Positive Mode Atmospheric Pressure Ionization Mass Spectrometry. Anal. Chem. 1988, 60, 1300–1307. [Google Scholar] [CrossRef]
- Bartosińska, E.; Borsuk-De Moor, A.; Siluk, D.; Markuszewski, M.J.; Wiczling, P. Ionization of Tocopherols and Tocotrienols In Atmospheric Pressure Chemical Ionization. Rapid Commun. Mass Spectrom: RCM 2018, 32, 919–927. [Google Scholar] [CrossRef]
- Tanaka, Y.; Otsuka, K.; Terabe, S. Evaluation of An Atmospheric Pressure Chemical Ionization Interface for Capillary Electrophoresis–Mass Spectrometry. J. Pharm. Biomed. Anal. 2003, 30, 1889–1895. [Google Scholar] [CrossRef]
- Harrison, A.G. Chemical Ionization Mass Spectrometry, Second Edition, 2nd ed.; Routledge: Boca Raton, FA, USA, 2018. [Google Scholar]
- Bohme, D.K.; Mackay, G.I.; Schiff, H.I. Determination of Proton Affinities from The Kinetics of Proton Transfer Reactions. Vii. The Proton Affinities of O2, H2, Kr, O, N2, Xe, CO2, Ch4, N2O, And Co. J. Chem. Phys. 2008, 73, 4976. [Google Scholar] [CrossRef]
- Lipok, C.; Hippler, J.; Schmitz, O.J. A Four Dimensional Separation Method Based On Continuous Heart-Cutting Gas Chromatography With Ion Mobility And High Resolution Mass Spectrometry. J. Chromatogr. A 2018, 1536, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Specker, H.; Kaiser, H. Bewertung Und Vergleich Von Analysenverfahren; Springer: Dortmund, Germany, 1956. [Google Scholar]
- Hurtado-Fernández, E.; Pacchiarotta, T.; Longueira-Suárez, E.; Mayboroda, O.A.; Fernández-Gutiérrez, A.; Carrasco-Pancorbo, A. Evaluation of Gas Chromatography-Atmospheric Pressure Chemical Ionization-Mass Spectrometry as An Alternative to Gas Chromatography-Electron Ionization-Mass Spectrometry: Avocado Fruit as Example. J. Chromatogr. A 2013, 1313, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Wachsmuth, C.J.; Almstetter, M.F.; Waldhier, M.C.; Gruber, M.A.; Nürnberger, N.; Oefner, P.J.; Dettmer, K. Performance Evaluation of Gas Chromatography-Atmospheric Pressure Chemical Ionization-Time-Of-Flight Mass Spectrometry for Metabolic Fingerprinting and Profiling. Anal. Chem. 2011, 83, 7514–7522. [Google Scholar] [CrossRef] [PubMed]
- Kaziur, W.; Salemi, A.; Jochmann, M.A.; Schmidt, T.C. Automated Determination of Picogram-Per-Liter Level of Water Taste and Odor Compounds Using Solid-Phase Microextraction Arrow Coupled with Gas Chromatography-Mass Spectrometry. Anal. Bioanal. Chem. 2019, 411, 2653–2662. [Google Scholar] [CrossRef]
- Saito-Shida, S.; Nagata, M.; Nemoto, S.; Akiyama, H. Quantitative Analysis OF Pesticide Residues in Tea by Gas Chromatography-Tandem Mass Spectrometry with Atmospheric Pressure Chemical Ionization. J. Chromatogr. B 2020, 1143, 122057. [Google Scholar] [CrossRef]
- Rahman, M.M.; Abd El-Aty, A.M.; Shim, J.-H. Matrix Enhancement Effect: A Blessing or A Curse for Gas Chromatography-A Review. Anal. Chim. Acta 2013, 801, 14–21. [Google Scholar] [CrossRef]
- Picó, Y. Food Contaminants and Residue Analysis, 1st ed.; Elsevier Professional: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Hoi, P.V.; Mol, A.P.J.; Oosterveer, P.; Van Den Brink, P.J.; Huong, P.T.M. Pesticide Use in Vietnamese Vegetable Production: A 10-Year Study. Int. J. Agric. Sustain. 2016, 14, 325–338. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | LOD Solution | Injection | Split | Mass on Column |
|---|---|---|---|---|
| (nM) | (µL) | (pg) | ||
| Acenaphthene | 30 | 1 | 10 | 0.5 |
| Benzophenone | 130 | 1 | 10 | 2.5 |
| Cuminaldehyde | 170 | 1 | 10 | 2.5 |
| Diethylphthalate | 40 | 1 | 10 | 1.0 |
| Methyldodecanoate | 50 | 1 | 10 | 1.0 |
| Class | Name | M1 (a.u.) | M2 (a.u.) | M3 (a.u.) | Mean (a.u.) | RSD (%) | Concentration (ng/g) |
|---|---|---|---|---|---|---|---|
| HME | Metalaxyl | 240 | 250 | 210 | 233 | 8.9 | 25 |
| ONP | Paclobutrazol | 310 | 280 | 290 | 293 | 5.2 | 239 |
| OPP | Edifenphos | 410 | 360 | 430 | 400 | 9.0 | 160 |
| OPP | Fonofos | 420 | 440 | 460 | 440 | 4.5 | 150 |
| OPP | Sulprofos | 190 | 165 | 180 | 178 | 7.0 | 376 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipok, C.; Uteschil, F.; Schmitz, O.J. Development of an Atmospheric Pressure Chemical Ionization Interface for GC-MS. Molecules 2020, 25, 3253. https://doi.org/10.3390/molecules25143253
Lipok C, Uteschil F, Schmitz OJ. Development of an Atmospheric Pressure Chemical Ionization Interface for GC-MS. Molecules. 2020; 25(14):3253. https://doi.org/10.3390/molecules25143253
Chicago/Turabian StyleLipok, Christian, Florian Uteschil, and Oliver J. Schmitz. 2020. "Development of an Atmospheric Pressure Chemical Ionization Interface for GC-MS" Molecules 25, no. 14: 3253. https://doi.org/10.3390/molecules25143253
APA StyleLipok, C., Uteschil, F., & Schmitz, O. J. (2020). Development of an Atmospheric Pressure Chemical Ionization Interface for GC-MS. Molecules, 25(14), 3253. https://doi.org/10.3390/molecules25143253