
Superelectrophiles: Recent Advances

Abstract
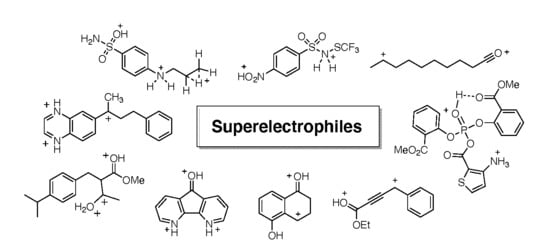
:
1. Introduction
2. New Types of Superelectrophiles
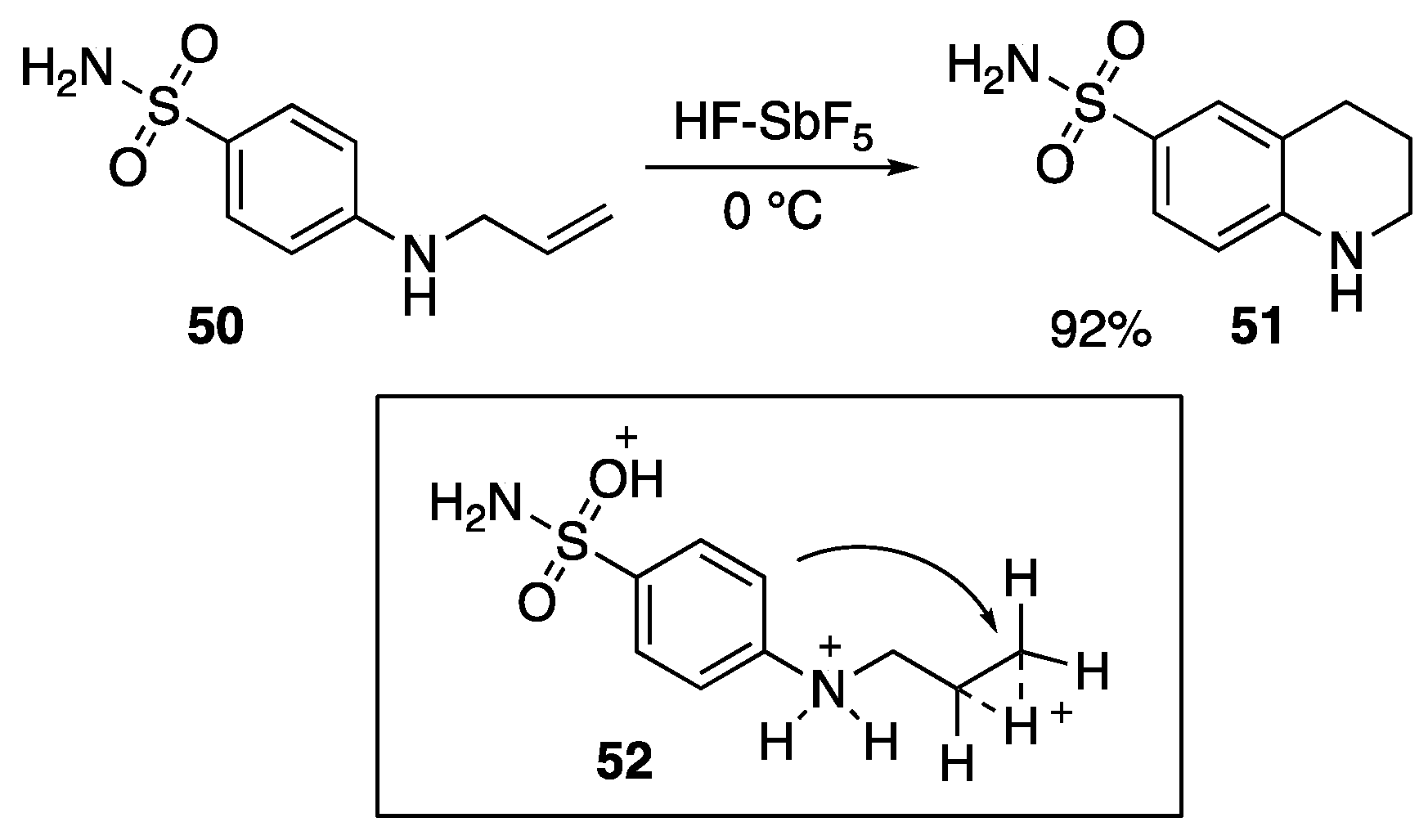
3. Anti-Markovnikov-Type Addition Chemistry
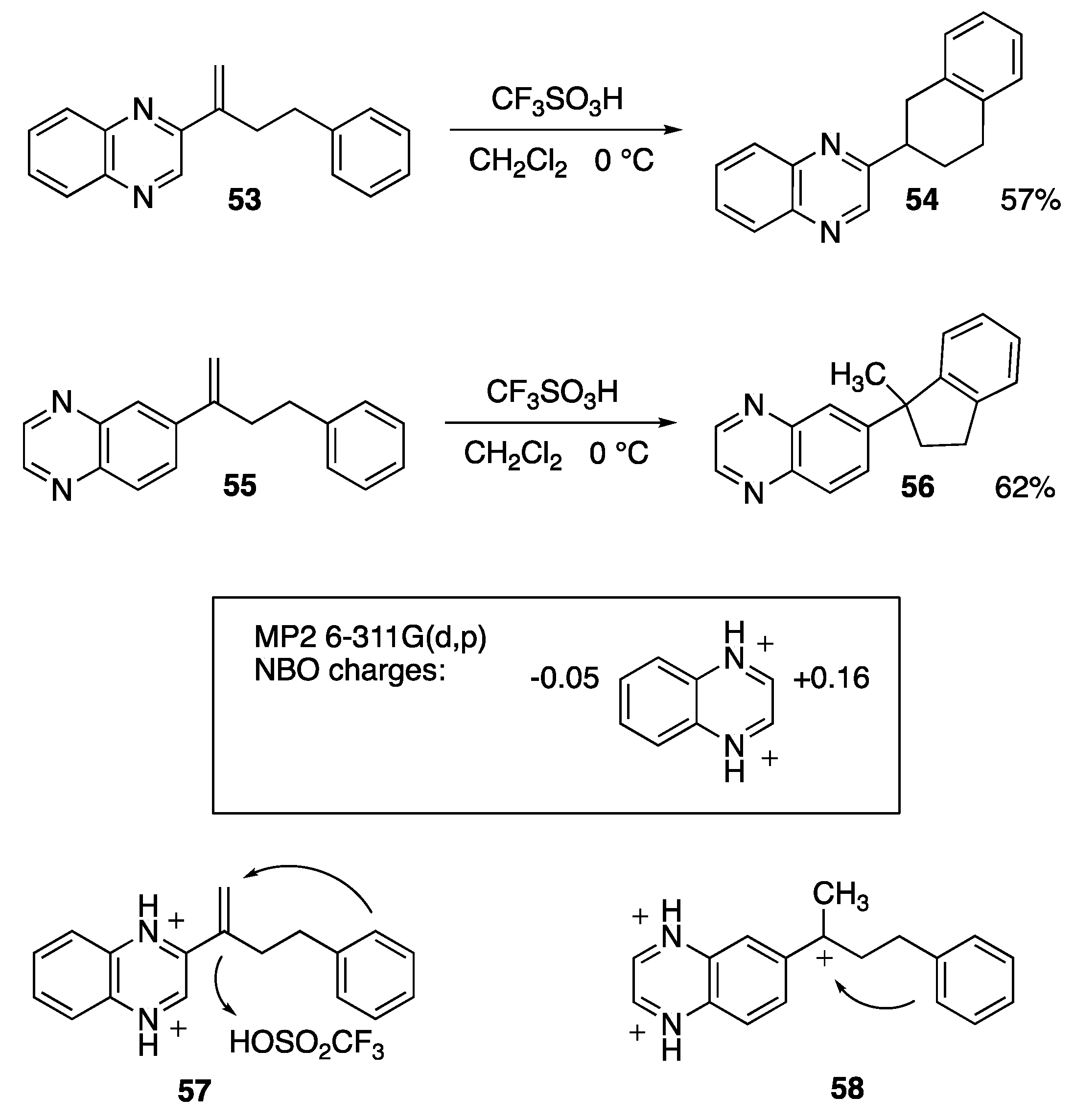
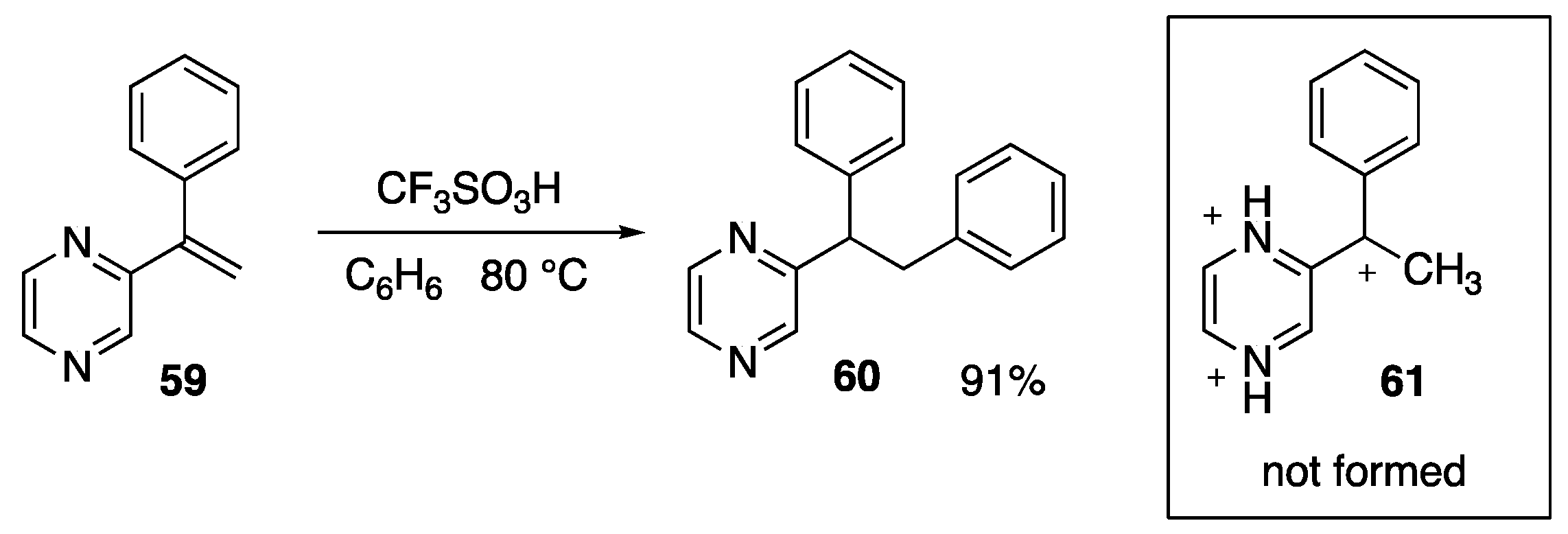
4. Superelectrophilic Pericyclic Reactions
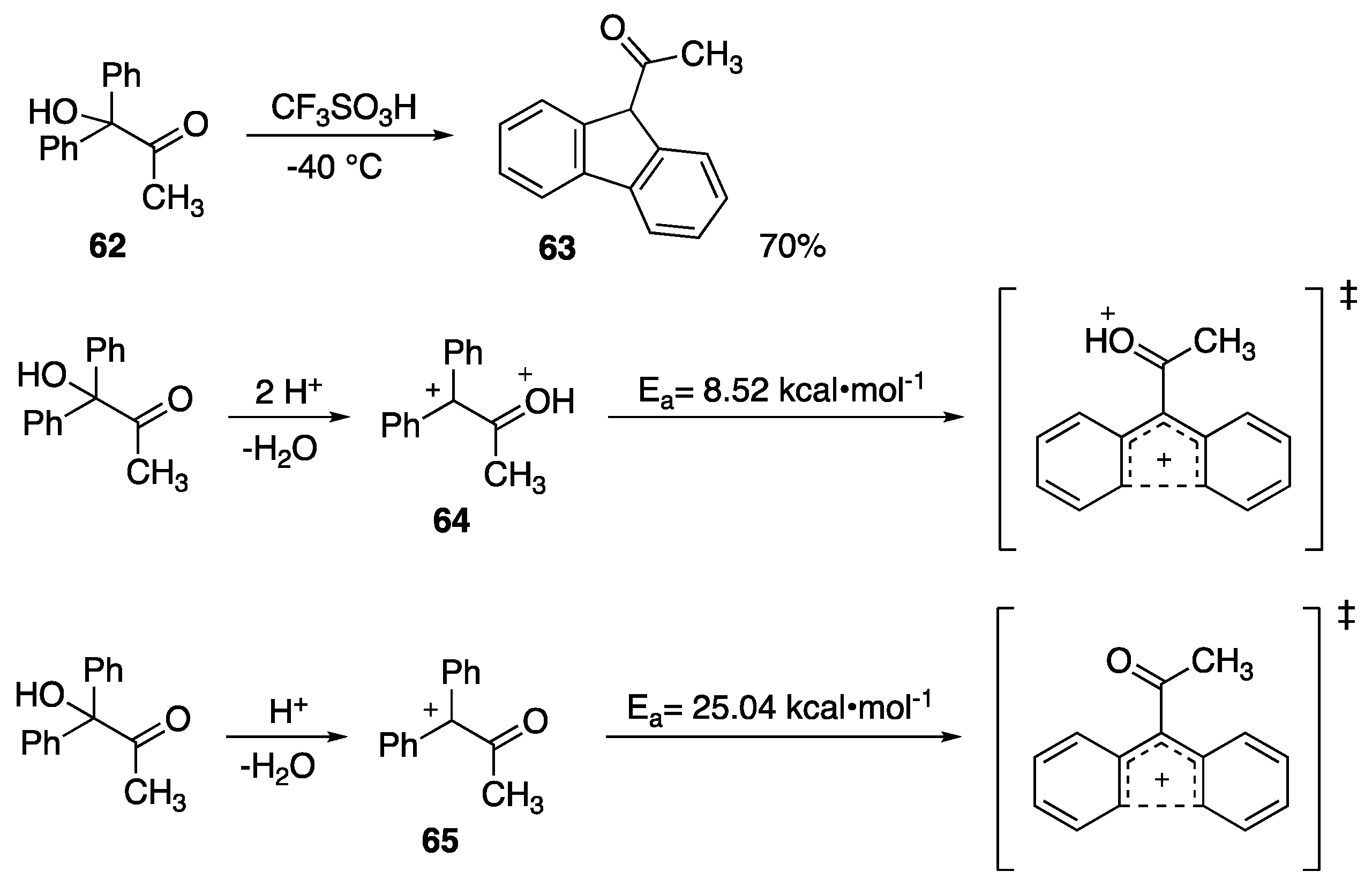
5. Carbonyl-Based Superelectrophiles
6. Activation of sp3 C-H Bonds
7. Superelectrophiles Involving Nitro Groups
8. Summary and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Staskun, B. The Conversion of Benzoylacetanilides into 2- and 4-Hydroxyquinolines. J. Org. Chem. 1964, 29, 1153–1157. [Google Scholar] [CrossRef]
- Olah, G.A.; Germain, A.; Lin, H.C.; Forsyth, D. Electrophilic reactions at single bonds. XVIII. Indication of rrotosolvated de facto substituting agents in the reactions of alkanes with acetylium and nitronium ions in superacidic media. J. Am. Chem. Soc. 1975, 97, 2928–2929. [Google Scholar] [CrossRef]
- Olah, G.A.; Klumpp, D.A. Superelectrophiles and Their Chemistry; Wiley: New York, NY, USA, 2007. [Google Scholar]
- Olah, G.A. Superelectrophiles. Angew. Chem. Int. Ed. Engl. 1993, 32, 767–788. [Google Scholar] [CrossRef]
- Klumpp, D.A.; Kennedy, S. Superelectrophiles in ring-forming reactions. ARKIVOC 2018, 215–232. [Google Scholar] [CrossRef]
- Michelet, B.; Carreyre, H.; Lecornue, F.; Mingot, A.; Thibaudeau, S. Superelectrophilic activation in superacid HF/SbF5: Expanding molecular diversity in nitrogen-containing compounds series by fluorination. J. Fluor. Chem. 2018, 214, 68–79. [Google Scholar] [CrossRef]
- Irena, S.A. Recent achievements in intermolecular sp3 C-H Bond Functionalization of Organic Compounds by Superelectrophilic Trihalomethyl Metal Complex. J. Organomet. Chem. 2015, 793, 54–77. [Google Scholar]
- Klumpp, D.A. molecular rearrangements of superelectrophiles. Beilstein J. Org. Chem. 2011, 7, 346–363. [Google Scholar] [CrossRef]
- Roithova, J. Superelectrophilic chemistry in the gas phase. Pure Applied Chem. 2011, 83, 1499–1506. [Google Scholar] [CrossRef]
- Klumpp, D.A. Superelectrophiles in hetrocyclic ring-forming reactions. ARKIVOC 2009, 63–80. Available online: https://quod.lib.umich.edu/cgi/p/pod/dod-idx/superelectrophiles-in-heterocyclic-ring-forming-reactions.pdf?c=ark;idno=5550190.0010.102;format=pdf (accessed on 17 July 2020).
- Klumpp, D.A. Superelectrophiles: Charge-charge repulsive effects. Chem. Eur. J. 2008, 14, 2004–2015. [Google Scholar] [CrossRef]
- Rusanov, A.L.; Chebotarev, V.P.; Lovkov, S.S. Superelectrophiles in the synthesis of condensation monomers and polymers. Russ. Chem. Rev. 2008, 77, 547–553. [Google Scholar] [CrossRef]
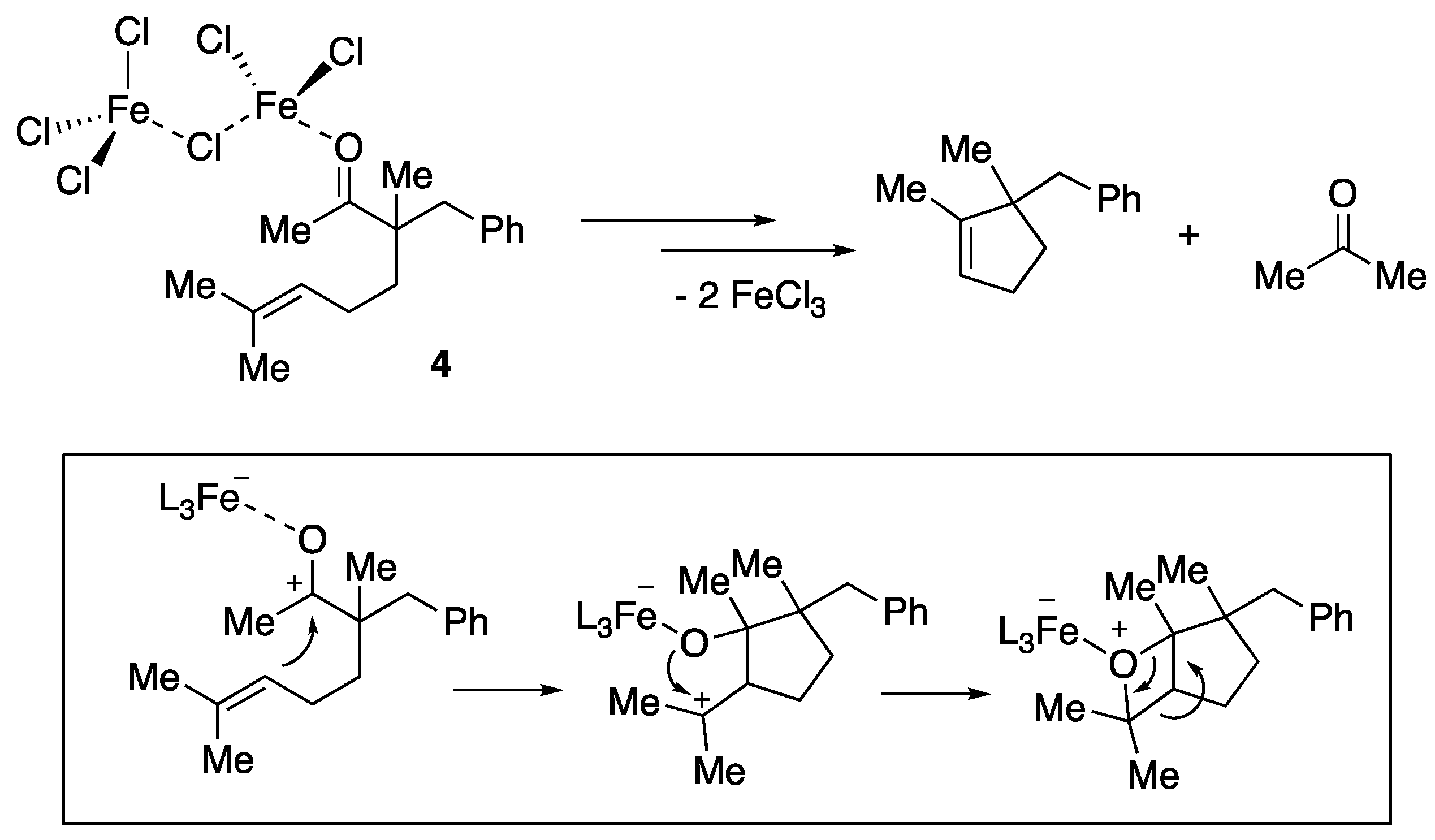
- Albright, H.; Riehl, P.S.; McAtee, C.C.; Reid, J.P.; Ludwig, J.R.; Karp, L.A.; Zimmerman, P.M.; Sigman, M.S.; Schindler, C.S. Catalytic carbonyl-olefin metathesis of aliphatic ketones: Iron(III) homo-dimers as lewis acidic superelectrophiles. J. Am. Chem. Soc. 2019, 141, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Albright, H.; Vonesh, H.L.; Schindler, C.S. Superelectrophilic Fe(III)-ion pairs as stronger lewis acid catalysts for (E)-selective intermolecular carbonyl-olefin metathesis. Org. Lett. 2020, 22, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Negishi, E. Bimetallic catalytic systems containing Ti, Zr, Ni, and Pd. Their application to selective organic synthesis. Pure Appl. Chem. 1981, 53, 2333–2356. [Google Scholar] [CrossRef]
- DeHaan, F.P.; Brown, H.C. Catalytic halides. XXXI. Directive effect in aromatic substitutions. 59. Kinetic of gallium chloride catalyzed methylation of benzene in excess of methyl chloride. J. Am. Chem. Soc. 1969, 91, 4844–4850. [Google Scholar] [CrossRef]
- Evans, D.A.; Chapman, K.T.; Bisaha, J. Asymmetric Diels-A;der Cycloaddition Reaction with Chiral.Alpha beta.-unsaturated N-acyloxazolines. J. Am. Chem. Soc. 1988, 110, 1238–1256. [Google Scholar] [CrossRef]
- Yang, S.; Bour, C.; Gandon, V. Superelectrophilic gallium(III) homodimers in gallium chloride-mediated methylation of benzene: A theoretical study. Acs Catal. 2020, 10, 3027–3033. [Google Scholar] [CrossRef]
- Djurovic, A.; Vayer, M.; Li, Z.; Guillot, R.; Baltaze, J.-P.; Gandon, V.; Bour, C. Synthesis of medium-sized carbocyles by gallium-catalyzed tandem carbonyl-olefin methatesis/transfer hydrogenation. Org. Lett. 2019, 21, 8132–8137. [Google Scholar] [CrossRef]
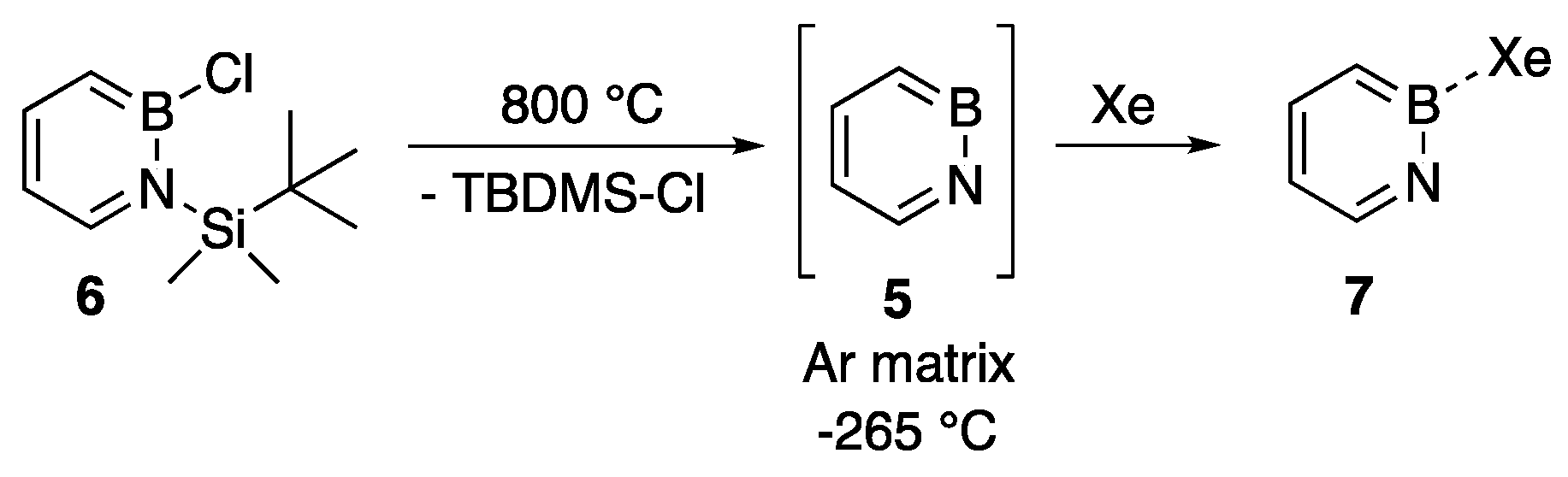
- Edel, K.; Ishibashi, J.S.A.; Liu, S.-Y.; Bettinger, H.F. Superelectrophilicity of 1,2-azaborine: Formation of xenon and carbon monooxide adducts. Angew. Chem. Int. Ed. 2019, 58, 4061–4064. [Google Scholar] [CrossRef]
- Mayer, M.; van Lessen, V.; Rohdenburg, M.; Hou, G.-L.; Exner, R.M.; Apra, E.; Azov, V.A.; Grabowsky, S.; Xantheas, S.S.; Asmis, K.R.; et al. Rational design of an argon-binding superelectrophile. Proc. Natl. Acad. Sci. USA 2019, 116, 8167–8172. [Google Scholar] [CrossRef] [Green Version]
- Rohdenburg, M.; Mayer, M.; Grellmann, M.; Jenne, C.; Borrmann, T.; Kleemiss, F.; Azov, V.A.; Asmis, K.R.; Grabowsky, S.; Warneke, J. Superelectrophilic behavior of an anion demonstrated by the spontaneous binding of noble gases to [B12 Cl11 ]−. Angew. Chem. Int. Edit. 2017, 56, 7980–7985. [Google Scholar] [CrossRef] [PubMed]
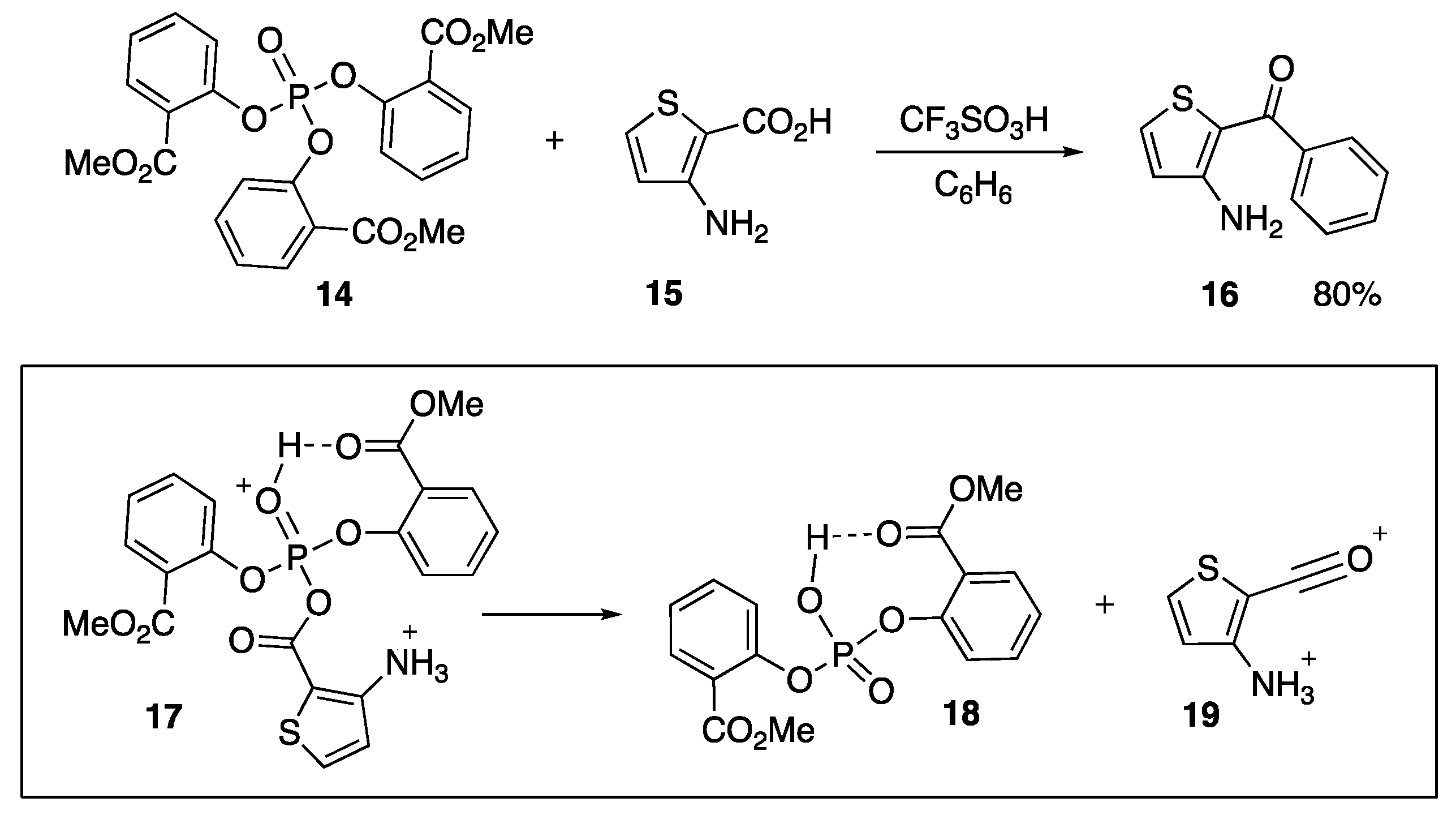
- Sumita, A.; Otani, Y.; Ohwada, T. Electrophilic activation of aminocarboxylic acids by phosphate ester promotes Friedel-Crafts acylation by overcoming charge-charge repulsion. Org. Biomol. Chem. 2017, 15, 9398–9407. [Google Scholar] [CrossRef] [PubMed]
- Sumita, A.; Otani, Y.; Ohwada, T. Chemoselective Generation of Acyl Phosphates, Acylium Ion Equivalents from Carboxylic Acids and an Organophosphate Ester in the Presence of a Brønsted Acid. Chem. Commun. 2017, 53, 1482–1485. [Google Scholar] [CrossRef]
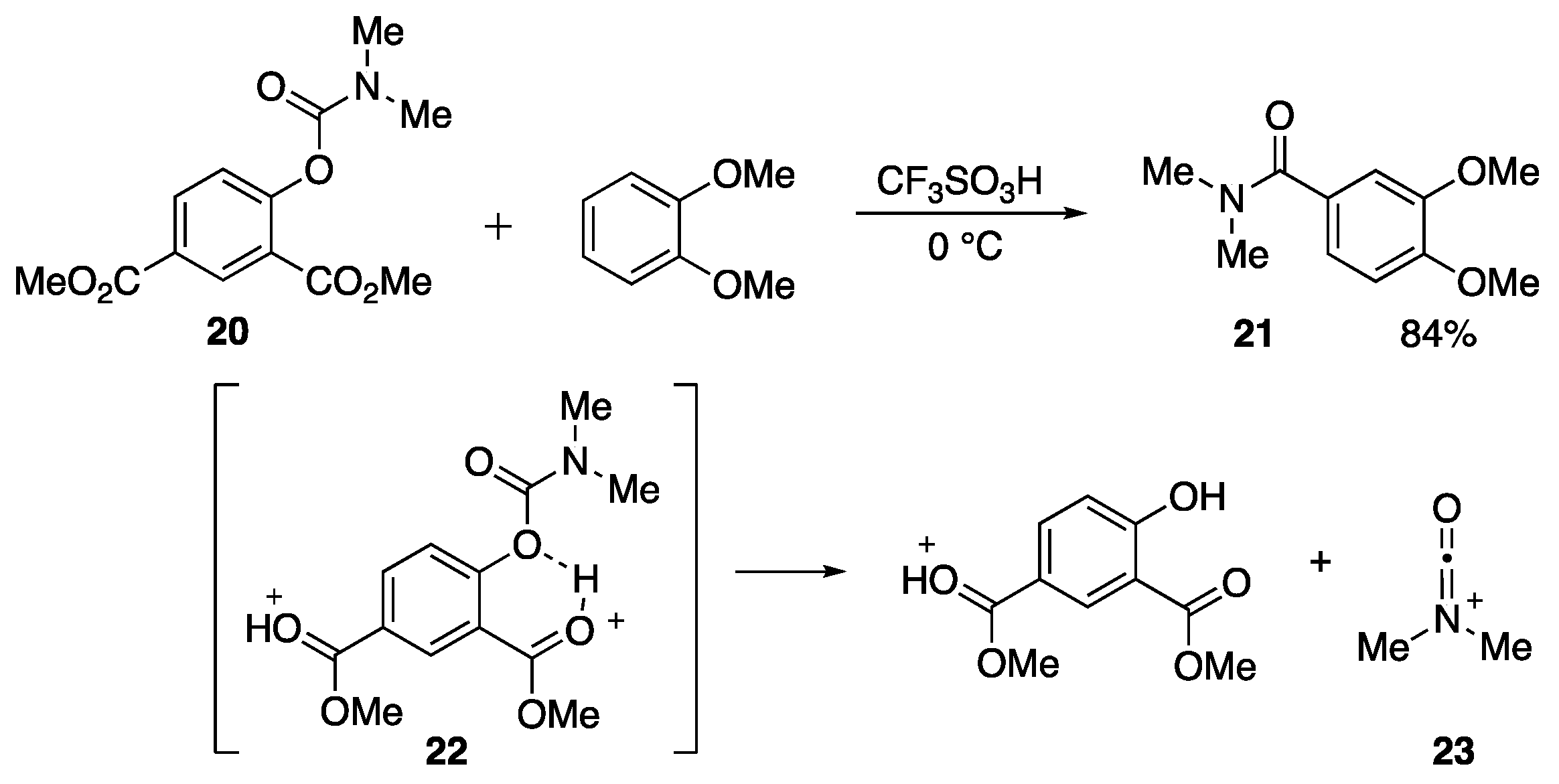
- Sumita, A.; Otani, Y.; Ohwada, T. Tandem buildup of complexity of aromatic molecules through multiple successive electrophile generation in one pot, controlled by varying the reaction temperature. Org. Biomol. Chem. 2016, 14, 1680–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
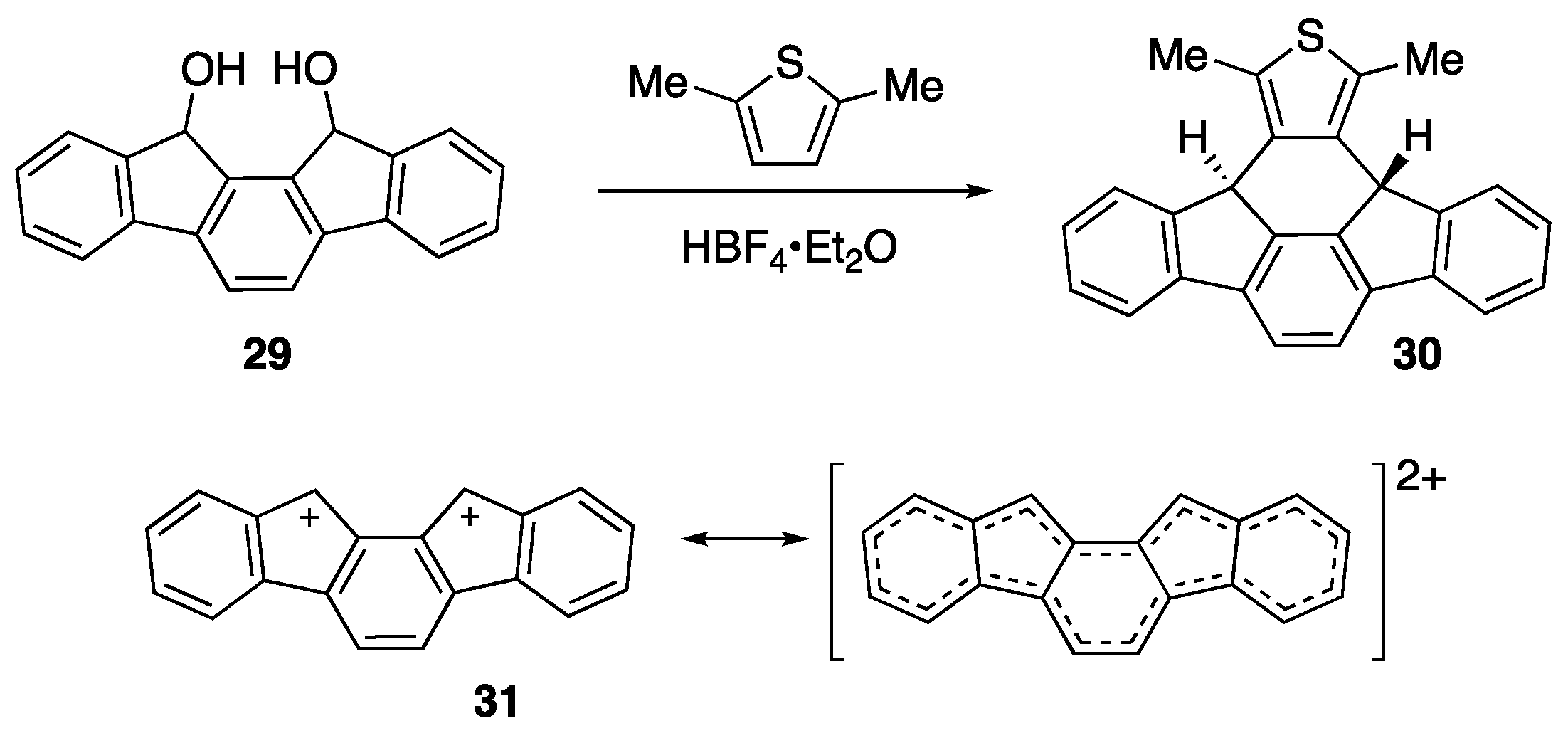
- Mills, N.S.; McClintock, S.P. Relationship of charge to antiaromaticity in bis-fluorenyl dications and fluorophenyl monocations: experimental support for theoretical calculations. Chem. Commun. 2012, 48, 8099–8101. [Google Scholar] [CrossRef]
- Mills, N.S.; Tirla, C.; Benish, M.A.; Rakowitz, A.J.; Bebell, L.M.; Hurd, C.M.M.; Bria, A.L.M. Dications of fluorenylidenes. The relationship between redox potentials and antiaromaticity for meta – and para-substituted diphenylmethylidenefluorenes. J. Org. Chem. 2005, 70, 10709–10716. [Google Scholar] [CrossRef]
- McClintock, S.P.; Mils, N.S. Magic acid free generation of antiaromatic dications at room temperature. J. Org. Chem. 2011, 76, 10254–10257. [Google Scholar] [CrossRef]
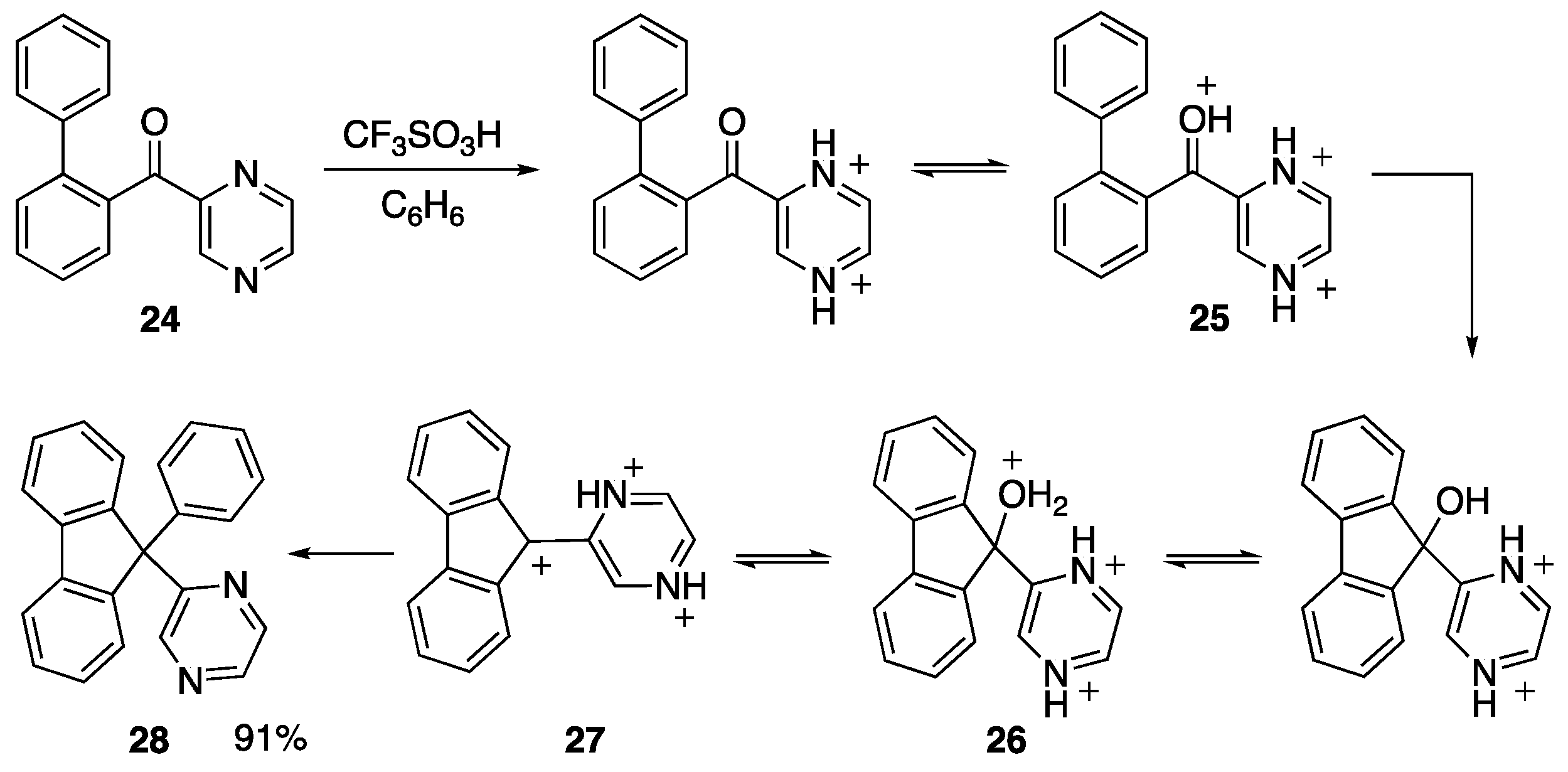
- Stentzel, M.R.; Klumpp, D.A. Preparation of functionalized fluorenes using dicationic electrophiles. Tetrahedron Lett. 2019, 60, 1675–1677. [Google Scholar] [CrossRef]
- Gasonoo, M.; Sumita, A.; Giuffre, K.; Boblak, K.; Ohwada, T.; Klumpp, D.A. Synthesis of heterocycle-containing 9,9-diarylfluorenes using superelectrophiles. J. Org. Chem. 2017, 82, 6044–6053. [Google Scholar] [CrossRef] [PubMed]
- Sumita, A.; Ohwada, T.; Boblak, K.; Gasonoo, M.; Klumpp, D.A. Use of charge-charge repulsion to enhance π-electron delocalization into anti-aromatic and aromatic systems. Chem. Eur. J. 2017, 23, 2566–2570. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zheng, X.; Lin, D.; Yuan, H.; Yin, Z.; Yang, L.; Yu, Y.; Wang, S.; Xie, L.-H.; Huang, W. Superelectrophilic-initiated C-H functionalization at the β-position of thiophenes: A one-pot synthesis of trans-stereospecific saddle-shaped cyclic compounds. J. Org. Chem. 2019, 84, 10701–10709. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Wei, Y.; Ou, C.; Huang, H.; Xie, L.; Tang, L.; Huang, W. carbon cationic relay via superelectrophiles: synthesis of spiro-diazafluorenes. Org. Lett. 2016, 18, 6220–6223. [Google Scholar] [CrossRef] [PubMed]
- Koltunov, K.Y. Condensation of naphthalenediols with benzene in the presence of aluminum bromide: an efficient synthesis of 5-, 6-, and 7-Hydroxy-4-phenyl-1- and 2-tetralones. Tetrahedron Lett. 2008, 49, 3891–3894. [Google Scholar] [CrossRef]
- Zhu, Z.; Koltunov, K.Y. A convenient synthesis of 8-Hydroxy-1-tetralones from naphthalene-1,8-diol. Mendeleev Commun. 2016, 26, 79–80. [Google Scholar] [CrossRef]
- Koltunov, K.Y.; Prakash, G.K.S.; Rasul, G.; Olah, G.A. Superacid catalyzed reactions of 5-amino-1-napthol with benzene and cyclohexane. Tetrahedron 2002, 58, 5423–5426. [Google Scholar] [CrossRef]
- Koltunov, K.Y.; Prakash, G.K.S.; Rasul, G.; Olah, G.A. Reactions of 2-, 3-, and 4-quinolinols with cyclohexane and benzene in superacids. Heterocycles 2004, 62, 757–772. [Google Scholar]
- Koltunov, K.Y.; Chernov, A.N.; Prakash, G.K.S.; Olah, G.A. Condensation of 2-naphthol and naphthalenediols with o-dichlorobenzene in the presence of aluminum halides. Chem. Pharm. Bull. 2012, 60, 722–727. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Salnikov, G.E.; Koltunov, K.Y. Cascade reaction of 2,3-naphalenediol with benzene in the presence of aluminum halides. Tetrtahedron Letters 2019, 60, 857–859. [Google Scholar] [CrossRef]
- Zhu, Z.; Genaev, A.M.; Salnikov, G.E.; Koltunov, K.Y. Mechanistic investigation of superelectrophilic activation of 1,1′-bi-2-naphtols in the presence of aluminum halides. Org. Biomol. Chem. 2019, 17, 3971–3977. [Google Scholar] [CrossRef]
- Genaev, A.M.; Salnikov, G.E.; Shernyukov, A.V.; Zhu, Z.; Koltunov, K.Y. protonation behavior of 1,1′-bi-2-naphthol and insights into its acid-catalyzed atropisomerization. Org. Lett. 2017, 19, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Genaev, A.M.; Shchegoleva, L.N.; Salnikov, G.E.; Shernyukov, A.V.; Shundrin, L.A.; Shundrin, I.K.; Zhu, Z.; Koltunov, K.Y. Acid-catalyzed versus thermally induced C1–C1′ Bond cleavage in 1,1′-bi-2-naphthol: An experimental and theoretical study. J. Org. Chem. 2019, 84, 7238–7243. [Google Scholar] [CrossRef]
- Devleshova, N.A.; Lozovskiy, S.V.; Vasilyev, A.V. Reactions of alkyl 4-hydroxybut-2-ynoates with arenes under superelectrophilic activation with triflic acid or HUSY zeolite: Alternative propargylation or allenylation of arenes, and synthesis of furan-2-ones. Tetrahedron 2019, 75, 130517–130529. [Google Scholar] [CrossRef]
- Ryabukhin, D.S.; Lisakova, A.D.; Zalivatskaya, A.S.; Boyarskaya, I.A.; Starova, G.L.; Trifonov, R.E.; Ostrovskii, V.A.; Vasilyev, A.V. Transformations of 5-(2-Arylethynyl)-2-methyl-2H-tetrazoles under superelectrophilic activation. Tetrahedron 2018, 74, 1838–1849. [Google Scholar] [CrossRef]
- Saulnier, S.; Golovanov, A.A.; Vasilyev, A.V. A controlled tandem transformation of conjugated enynones with arenes under superelectrophilic activation leading to aryl-substituted dienones and indenes. Rsc Adv. 2016, 6, 103546–103555. [Google Scholar] [CrossRef]
- Ryabukhin, D.S.; Fukin, G.K.; Vasilyev, A.V. Multi-channel transformation of 1,3-Diarylpropynones under superelectrophilic activation conditions: Concurrence of intra- and itermolecular reactions. Experimental and theoretical study. Tetrahedron 2014, 70, 7865–7873. [Google Scholar] [CrossRef]
- Naredla, R.R.; Zheng, C.; Nilsson, L.S.O.; Klumpp, D.A. Charge delocalization and enhanced acidity in tricationic superelectrophiles. J. Am. Chem. Soc. 2011, 133, 13169–13175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasonoo, M.; Naredla, R.R.; Nilsson, L.S.O.; Klumpp, D.A. Tetra- and pentacationic electrophiles and their chemistry. J. Org. Chem. 2016, 81, 11758–11765. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.H.; Gasonoo, M.; Klumpp, D.A. Superelectrophilic carbocations: Preparation and reactions of a substrate with six ionizable groups. Beil. J. Org. Chem. 2019, 15, 1515–1520. [Google Scholar] [CrossRef]
- Compain, G.; Martin-Mingot, A.; Frapper, G.; Bachmann, C.; Jouannetaud, M.-P.; Thibaudeau, S. Anti-Markovnikov additions to n-allylic derivatives involving ammonium–carbenium superelectrophiles. Chem. Commun. 2012, 48, 5877–5879. [Google Scholar] [CrossRef]
- Compain, G.; Bonneau, C.; Martin-Mingot, A.; Thibaudeau, S. Selective anti-Markovnikov cyclization and hydrofluorination reaction in superacid HF/SbF5: A tool in the design of nitrogen-containing (fluorinated) polycyclic systems. J. Org. Chem. 2013, 78, 4463–4472. [Google Scholar] [CrossRef] [PubMed]
- Boblak, K.N.; Gasonoo, M.; Zhang, Y.; Klumpp, D.A. Intramolecular conjugate addition with heterocylic olefins. J. Org. Chem. 2015, 80, 11948–11952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sheets, M.R.; Raja, E.; Boblak, K.; Klumpp, D.A. Superacid-promoted additions involving vinyl-substituted pyrimidines, quinoxalines, and quinazolines: Mechanisms correlated to charges. J. Am. Chem. Soc. 2011, 133, 8467–8469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
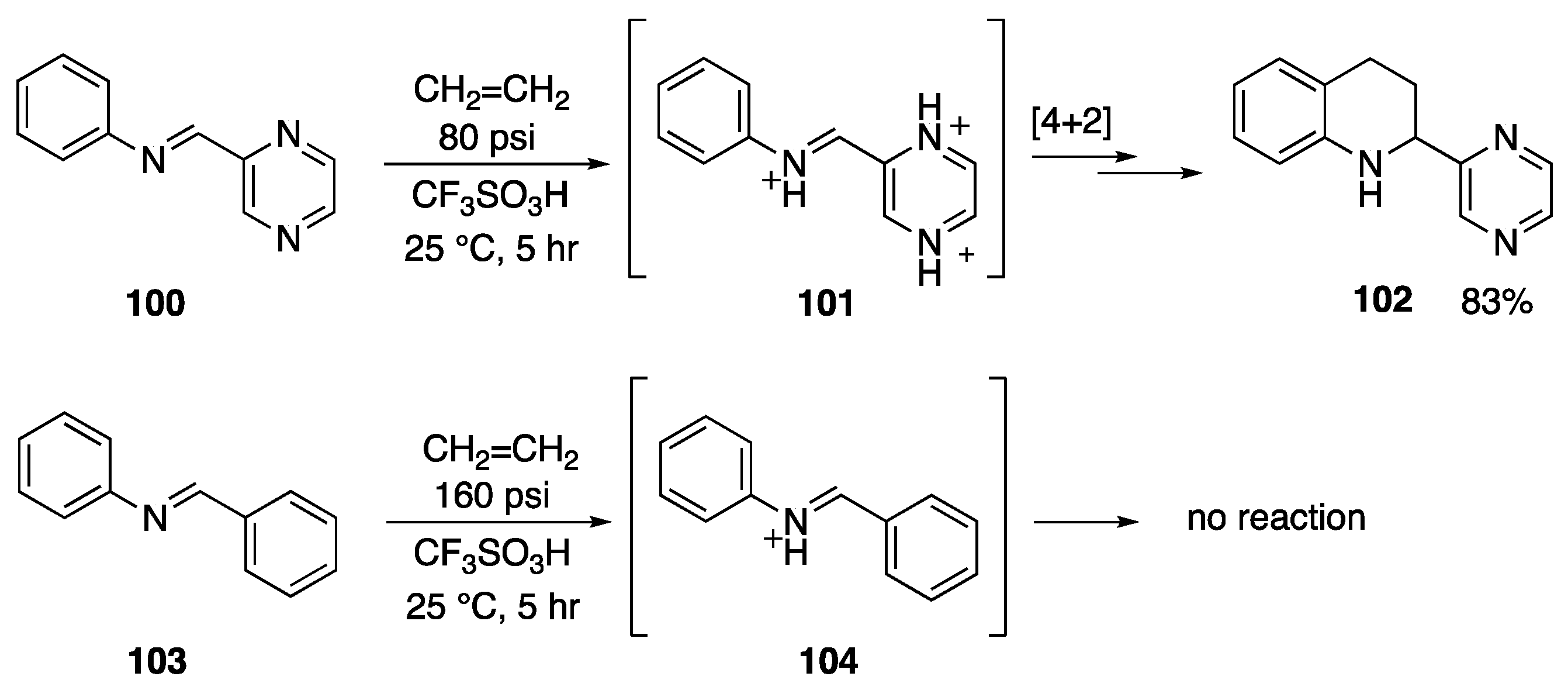
- Zhang, Y.; Briski, J.; Zhang, Y.; Rendy, R.; Klumpp, D.A. Superacid-catalyzed reactions of olefinic pyrazines: An example of anti-Markovnikov addition involving superelectrophiles. Org. Lett. 2005, 7, 2505–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohwada, T.; Suzuki, T.; Shudo, K. Superacid-catalyzed electrocyclization of diphenylmethyl cations to fluorenes. kinetic and theoretical revisit supporting the involvement of ethylene dications. J. Am. Chem. Soc. 1998, 120, 4629–4637. [Google Scholar] [CrossRef]
- Ohwada, T.; Shudo, K. Reaction of diphenylmethyl cations in a strong acid. participation of carbodications with positive charge substantially delocalized over the aromatic rings. J. Am. Chem. Soc. 1988, 110, 1862–1870. [Google Scholar] [CrossRef]
- Levy, A.; Rakowitz, A.; Mills, N.S. Dications of fluorenylidenes. The effect of substituent electronegativity and position on the antiaromaticity of substituted tetrabenzo[5.5]fulvalene dications. J. Org. Chem. 2003, 68, 3990–3998. [Google Scholar] [CrossRef]
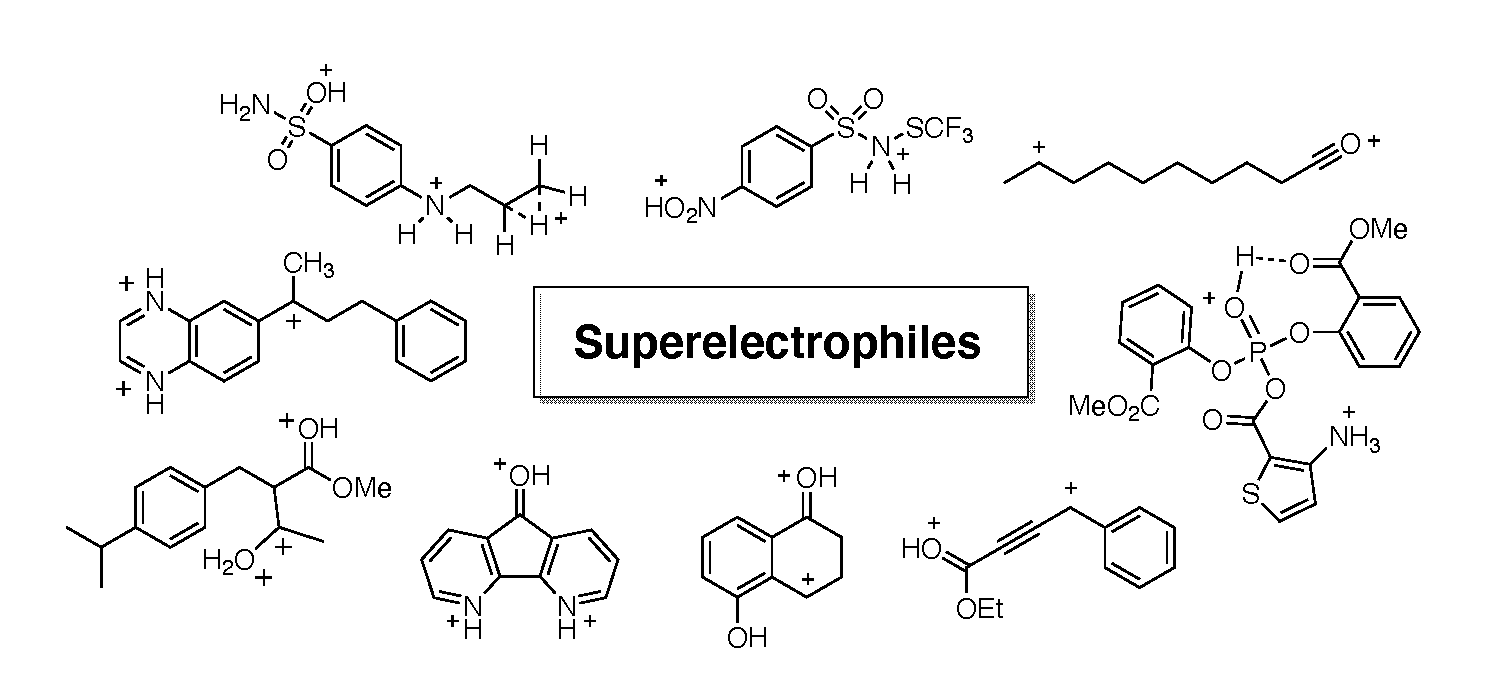
- Sai, K.K.S.; Esteves, P.M.; Tanoue da Penha, E.T.; Klumpp, D.A. Superacid-promoted reactions of α-keto amides and related systems. J. Org. Chem. 2008, 73, 6506–6512. [Google Scholar] [CrossRef]
- Yoshida, N.; Ohwada, T. Regioselective superacid-catalyzed electrocyclization of diphenylmethyl cations to fluorenes, phenanthrols and benzofurans. Synthesis 2001, 10, 1487–1494. [Google Scholar] [CrossRef]
- Hoefnagel, A.J.; van Bekkum, H. New zeolite-catalyzed ring-closure reaction of benzilic acid. Micropor. Mesopor. Mat 2000, 35–36, 155–161. [Google Scholar] [CrossRef]
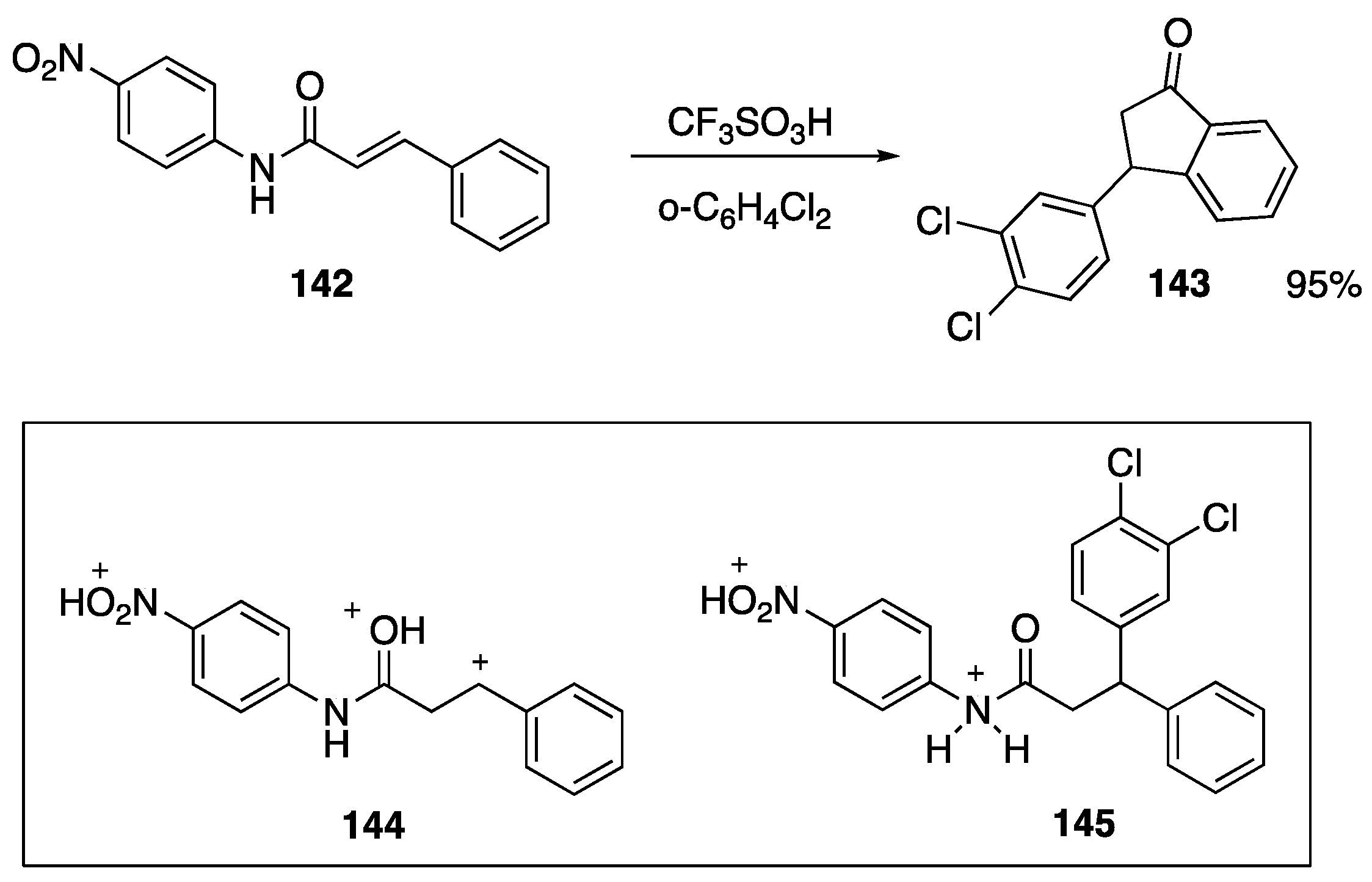
- Suzuki, T.; Ohwada, T.; Shudo, K. Superacid-catalyzed electrocyclization of 1-Phenyl-2-propen-1-ones to 1-indanones. Kinetic and theoretical studies of electrocyclization of oxonium-carbenium dications. J. Am. Chem. Soc. 1997, 119, 6774–6780. [Google Scholar] [CrossRef]
- Klumpp, D.A.; Zhang, Y.; O’Connor, M.J.; Esteves, P.M.; de Almeida, L.S. Aza-Nazarov reaction and the role of superelectrophiles. Org. Lett. 2007, 9, 3085–3088. [Google Scholar] [CrossRef] [PubMed]
- Sai, K.K.S.; O’Connor, M.J.; Klumpp, D.A. Aza-Nazarov cyclization cascades. Tetrahedron Lett. 2011, 52, 2195–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, R.; Daniliuc, C.-G.; Würthwein, E.-U. Preparation of NH-Pyrroles under superelectrophilic conditions by an Aza-Nazarov reaction cascade with indole as neutral leaving group: Experiment and theory. Eur. J. Org. Chem. 2012, 2012, 6021–6032. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Q.; Zhang, D.; Xin, X.; Zhang, R.; Zhou, F.; Dong, D. PPA-mediated C-C-C bond formation: A synthetic route to substituted indeno[2,1-c]quinoline-6(7h)-ones. Org. Lett. 2013, 15, 776–779. [Google Scholar] [CrossRef]
- Liu, X.; Xin, X.; Xiang, D.; Zhang, R.; Kumar, S.; Zhou, F.; Dong, D. Facile and efficient synthesis of quinolin-2(1H)-ones via cyclization of Penta-2,4-Dienamides mediated by H2SO4. Org. Biomol. Chem. 2012, 10, 5643–5646. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, S.H.; Qurah, A.; Kethe, A.; Klumpp, D.A. Superacid-promoted synthesis of indolizidine derivatives. Tetrahedron Lett. 2018, 59, 1932–1935. [Google Scholar] [CrossRef]
- Qurah, A.; Klumpp, D.A. Cyclizations of N-carbamoyl and N-thiocarbamoyl iminium ions leading to ring-fused heterocycles. Tetrahedron Lett. 2016, 57, 4788–4790. [Google Scholar] [CrossRef] [Green Version]
- Hart, H.; Sulzberg, T.; Rafos, R.R. dicarbonium ions of the triarylmethyl type. ions from o-, m- and p-xylene glycols. J. Am. Chem. Soc. 1963, 85, 1800–1806. [Google Scholar] [CrossRef]
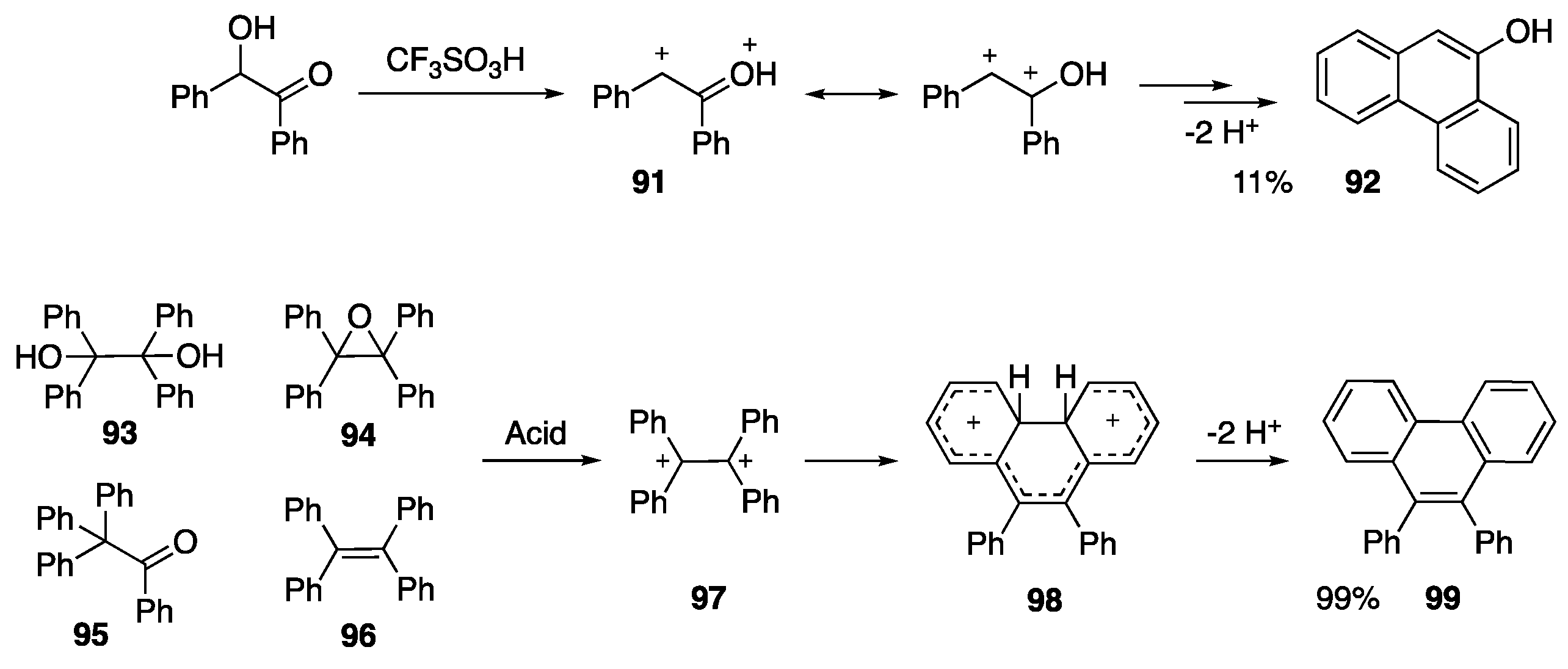
- Klumpp, D.A.; Baek, D.N.; Prakash, G.K.S.; Olah, G.A. The preparation of condensed aromatics by the superacidic dehydration cyclization of aryl pinacols and epoxides. J. Org. Chem. 1997, 62, 6666–6671. [Google Scholar] [CrossRef]
- Vuong, H.; Dash, B.P.; Nilsson, L.S.O.; Klumpp, D.A. Diels-Alder reactions with ethylene and superelectrophiles. Org. Lett. 2018, 20, 1849–1852. [Google Scholar] [CrossRef]
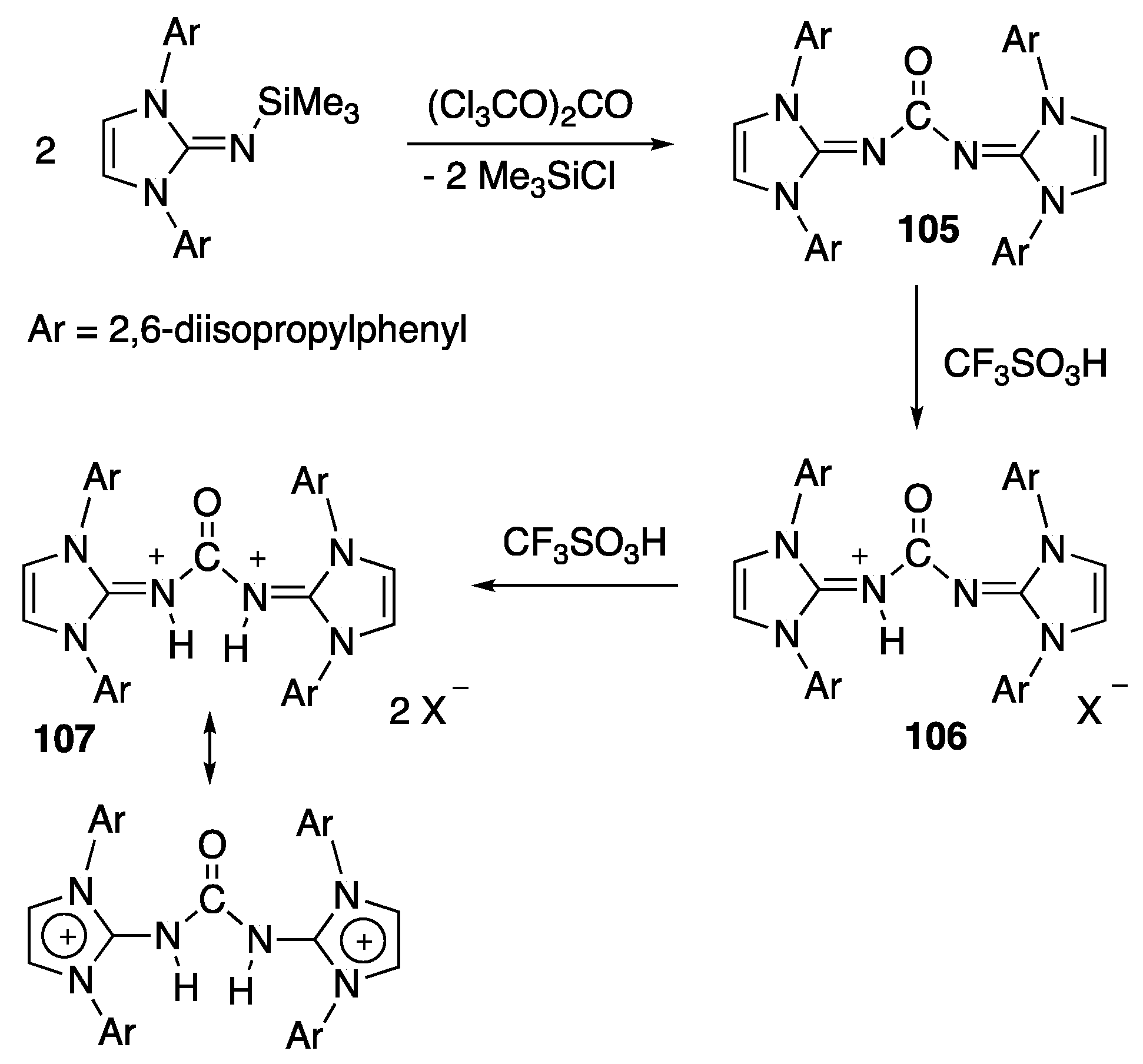
- Loh, Y.K.; Fuentes, M.A.; Vasko, P.; Aldridge, S. Successive protonation of an N-Heteroyclic imine derived carbonyl: superrelectrophilic dication versus nasked acylium ion. Angew. Chem. Int. Edit. 2018, 57, 16559–16563. [Google Scholar] [CrossRef] [PubMed]
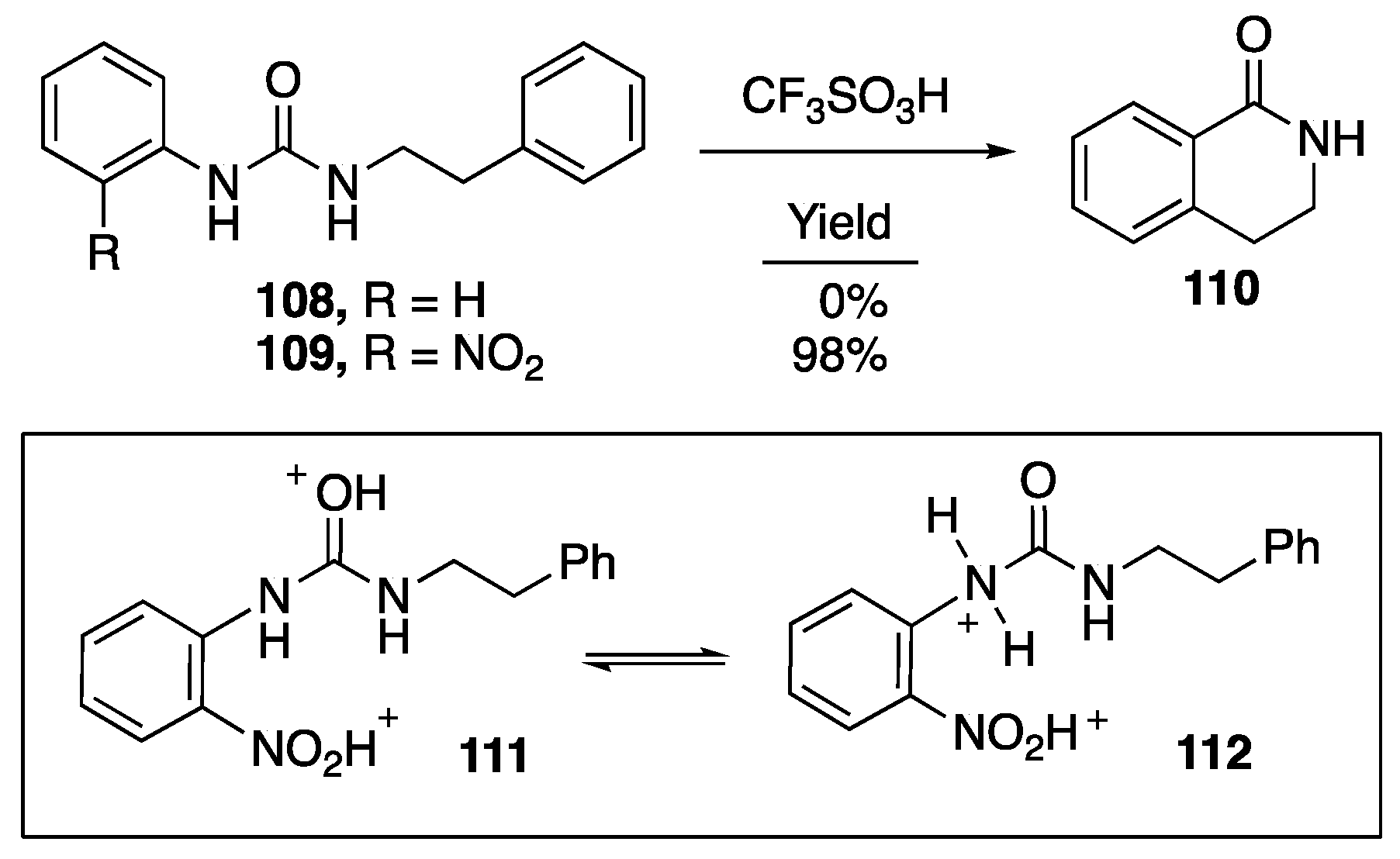
- Raja, E.K.; Nilsson, L.S.O.; Klumpp, D.A. Friedel-Crafts-type reactions with ureas and thioureas. Chem. Commun. 2012, 48, 8141–8143. [Google Scholar] [CrossRef] [PubMed]
- Kurouchi, H.; Sumita, A.; Otani, Y.; Ohwada, T. Protonation swithcing to the least-basic heteroatom of carbamate trough cationic hydrogen bonding promotes the formation of isocyanate cations. Chem. Eur. J. 2014, 20, 8682–8690. [Google Scholar] [CrossRef] [PubMed]
- Kurouchi, H.; Kawamoto, K.; Sugimoto, H.; Nakamura, S.; Otani, Y.; Ohwada, T. activation of electrophility of stable Y-delocalized carbamate cations in intramolecular aromatic substitution reaction: evidence for formation of diprotonated carbamates leading to generation of isocyanates. J. Org. Chem. 2012, 77, 9313–9328. [Google Scholar] [CrossRef]
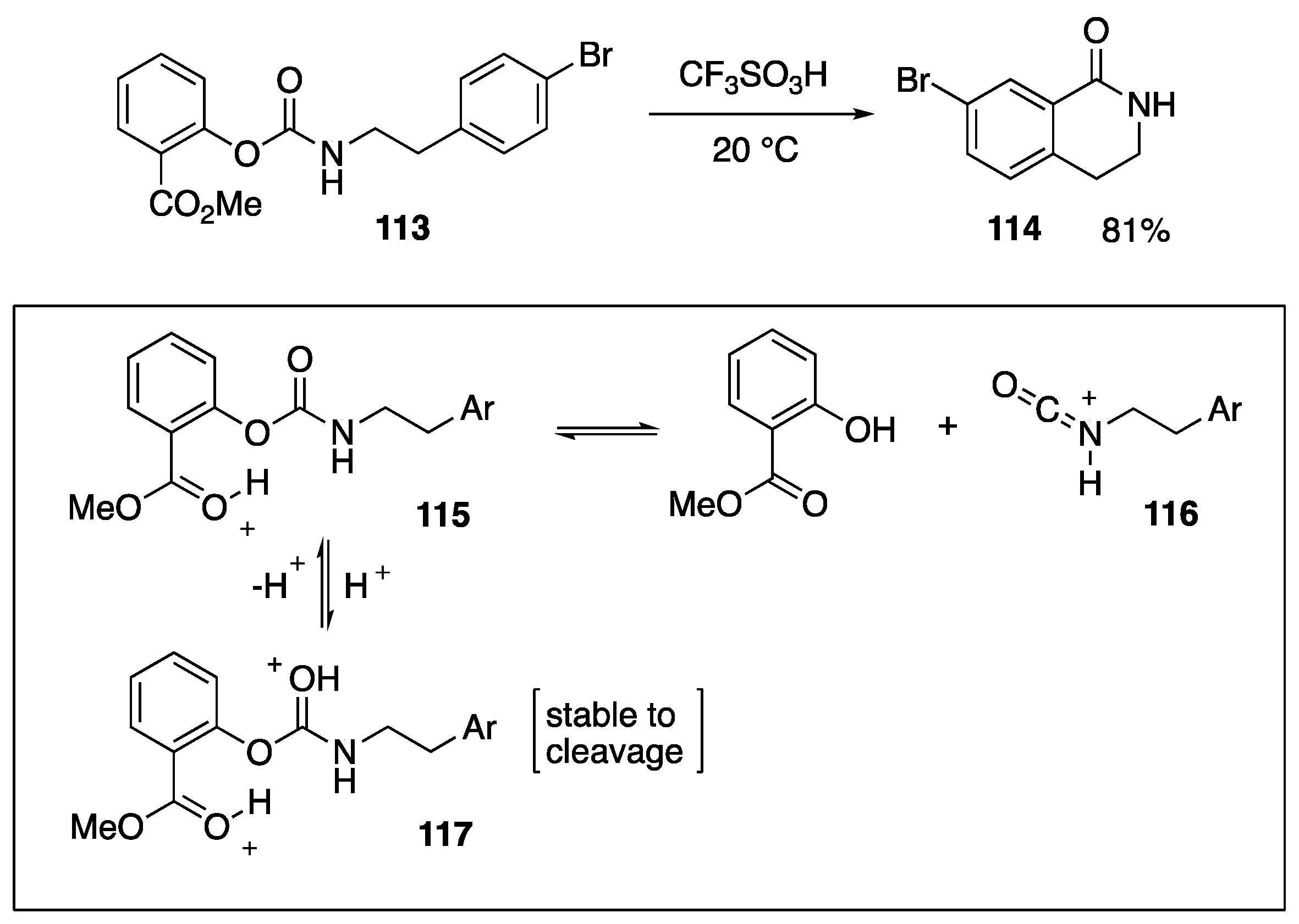
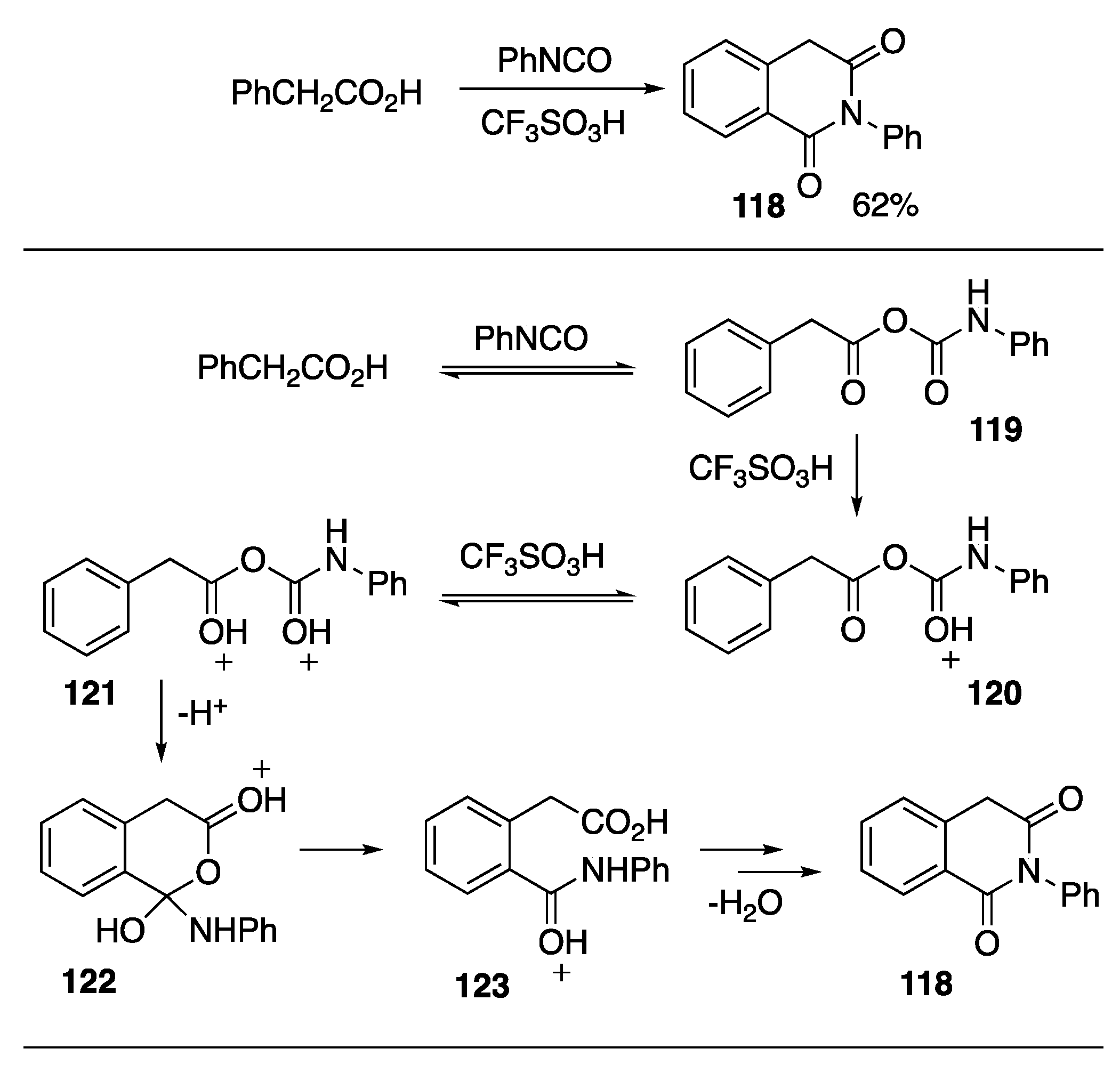
- Kennedy, S.H.; Schaeff, M.N.; Klumpp, D.A. Superelectrophiles in synthesis: Preparation of aromatic imides. J. Org. Chem. 2019, 84, 14133–14140. [Google Scholar] [CrossRef]
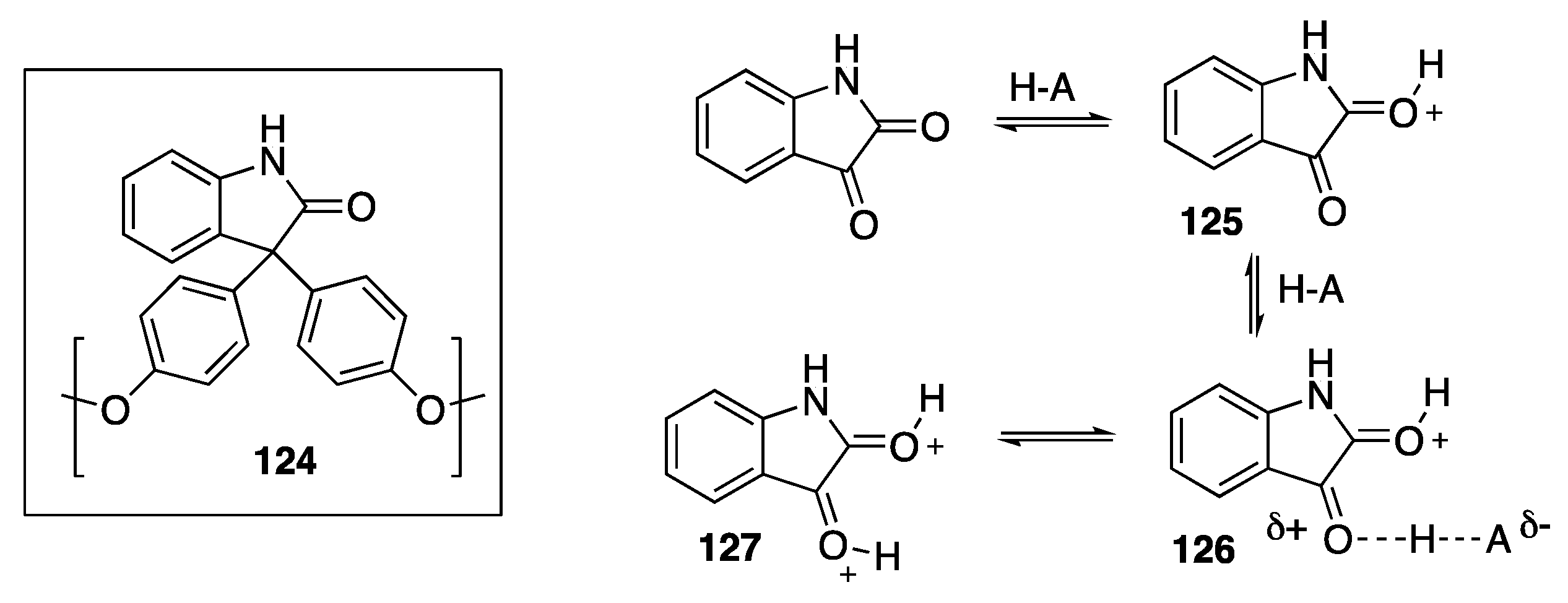
- Soultan, A.H.; Verheyen, T.; Smet, M.; de Borggraeve, W.M.; Patterson, J. Synthesis of functionalization of hyperbranched poly(arylene oxindole) towards versatile biomaterials. Polym. Chem. 2018, 9, 2775–2784. [Google Scholar] [CrossRef]
- Mancilla, E.C.; Hernández-Martínez, H.; Zolotukhin, M.G.; Ruiz-Treviño, A.F.; Gonzaález-Díaz, M.O.; Cardenas, J.; Scherf, U. POXINAR membrane family for gas separation. Ind. Eng. Chem. Res. 2019, 58, 15280–15287. [Google Scholar] [CrossRef]
- Hernandez, M.C.G.; Zolotukhin, M.G.; Fomine, S.; Cedillo, G.; Morales, S.L.; Fröhlich, N.; Preis, E.; Scherf, U.; Salmon, M.; Chavez, M.I.; et al. Novel, metal-free, superacid-catalyzed “click” reactions of isatins with linear, nonactivated, multiring aromatic hydrocarbons. Macromolecules 2010, 43, 6968–6979. [Google Scholar] [CrossRef]
- Zolotukhin, M.G.; Fomina, L.; Salcedo, R.; Sansores, L.E.; Colquhoun, H.M.; Khalilov, L.M. Superelectrophiles in polymer chemistry. A novel, one-pot synthesis of high-Tg, high-temperature polymers. Macromolecules 2004, 37, 5140–5141. [Google Scholar] [CrossRef]
- Colquhoun, H.M.; Zolotukhin, M.G.; Khalilov, L.M.; Dzhemilev, U.M. Superelectrophiles in aromatic polymer chemistry. Macromolecules 2001, 34, 1122–1124. [Google Scholar] [CrossRef]
- Smet, M.; Fu, Y.; Zhang, X.; Schacht, E.H.; Dehaen, W. A convenient A2 + B3 approach to hyperbranched poly(arylene oxindoles). Macromol. Rapid Commun. 2005, 26, 1458–1463. [Google Scholar] [CrossRef]
- Klumpp, D.A.; Yeung, K.Y.; Prakash, G.K.S.; Olah, G.A. Preparation of 3,3-Diaryloxindoles by superacid-induced condensations of isatins and aromatics with a combinatorial approach. J. Org. Chem. 1998, 63, 4481–4484. [Google Scholar] [CrossRef]
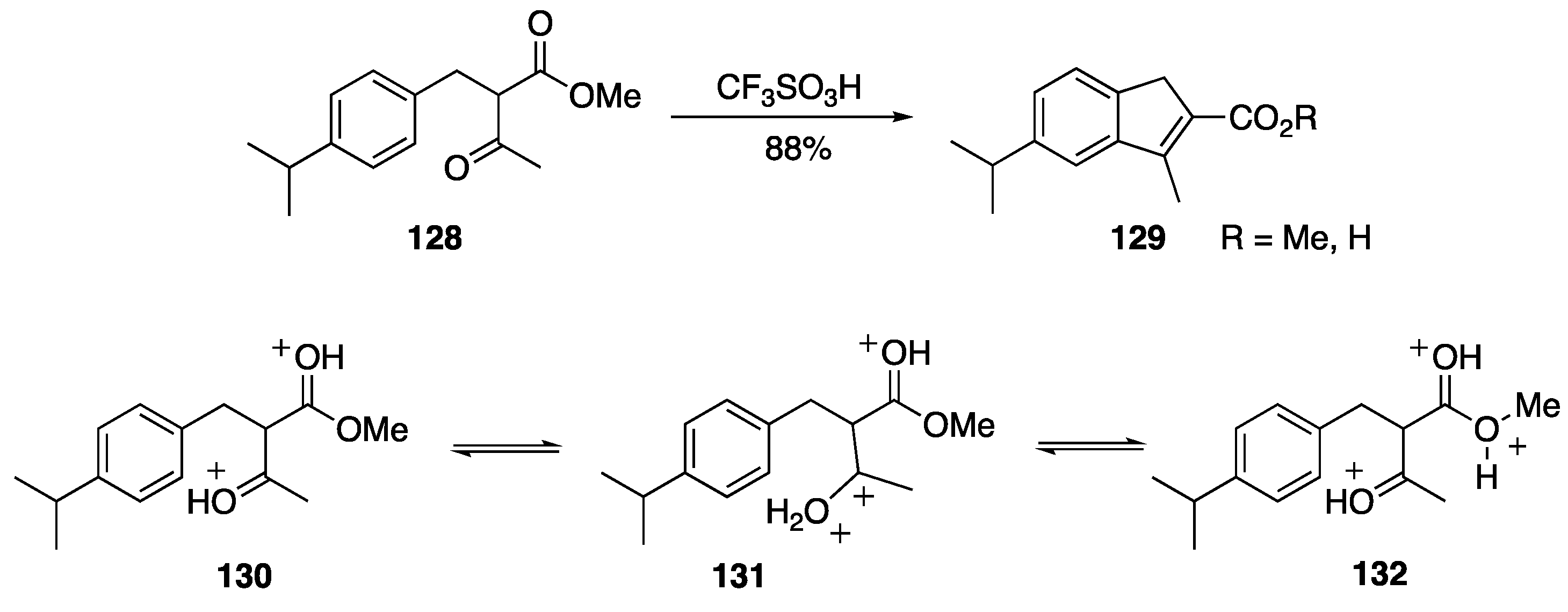
- Kuouchi, H.; Sugimoto, H.; Otani, Y.; Ohwada, T. Cyclization of arylacetoacetates to indene and duhydronaphtalene derivatives in strong acid. evidence for involment of further protonation of O,O-Diprotonated β-Ketoester, leading to enhancement. J. Am. Chem. Soc. 2010, 132, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Nirmalchandar, A.; Pakina, F.; Rasul, G.; Mathew, T.; Prakash, G.K.S. Superelectrophilic activation of phenylglyoxamides: Efficient synthesis of triarylacetamides and fruorenecarboxamides by superacid catalysis. Top. Catal. 2018, 61, 652–663. [Google Scholar] [CrossRef]
- Brouwer, D.M.; Kiffen, A.A. Hydride transfer reactions III: Rates of hydride transfer from isobutane to hydroxycarbonium ions. Recl. Trav. Chim. Pays-Bas. 1973, 92, 809–813. [Google Scholar] [CrossRef]
- Olah, G.A.; Mathew, T.; Marinez, E.R.; Esteves, P.M.; Etzkorn, M.; Rasul, G.; Prakash, G.K.S. Acid-catalyzed isomerization of pivalaldehyde to methyl isopropyl ketone via a reactive protosolvated ion intermediate. J. Am. Chem. Soc. 2001, 123, 11556–11561. [Google Scholar] [CrossRef]
- Olah, G.A.; Ramaiah, P.; Prakash, G.K.S. electrophilic nitration of alkanes with nitronium hexafluorophosphate. Proc. Natl. Acad. Sci. USA 1997, 94, 11783–11785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vol’pin, M.; Akhrem, I.; Orlinkov, A. Aprotic organic superacids—Efficient reagents and catalysts for transformations of alkanes and cycloalkanes under mild conditions. New J. Chem. 1989, 13, 771–789. [Google Scholar] [CrossRef]
- Akhrem, I.; Orlinkov, A. Polyhalomethanes combined with lewis acids in alkane chemistry. Chem. Rev. 2007, 107, 2037–2079. [Google Scholar] [CrossRef] [PubMed]
- Akhrem, I.S.; Christyakov, A.L.; Gambaryan, N.P.; Stankevich, I.V.; Vol’pin, M.E. Polyhalomethanes combined with aluminum halides as generators of superelectrophiles of a novel type. J. Organomet. Chem. 1997, 536–537, 489–495. [Google Scholar] [CrossRef]
- Sommer, J.; Bukala, J. Selective electrophilic activation of alkanes. Acc. Chem. Res. 1993, 26, 370–376. [Google Scholar] [CrossRef]
- Olah, G.A.; Rasul, G.A.; Yudin, A.K.; Burrichter, A.; Prakash, G.K.S.; Christyakov, A.L.; Stankevich, I.V.; Akhrem, I.S.; Gambaryan, N.P.; Vol’pin, M.E.J. Trihalomethyl Cations and Their Superelectrophilic Activation. Am. Chem. Soc. 1996, 118, 1446–1451. [Google Scholar] [CrossRef]
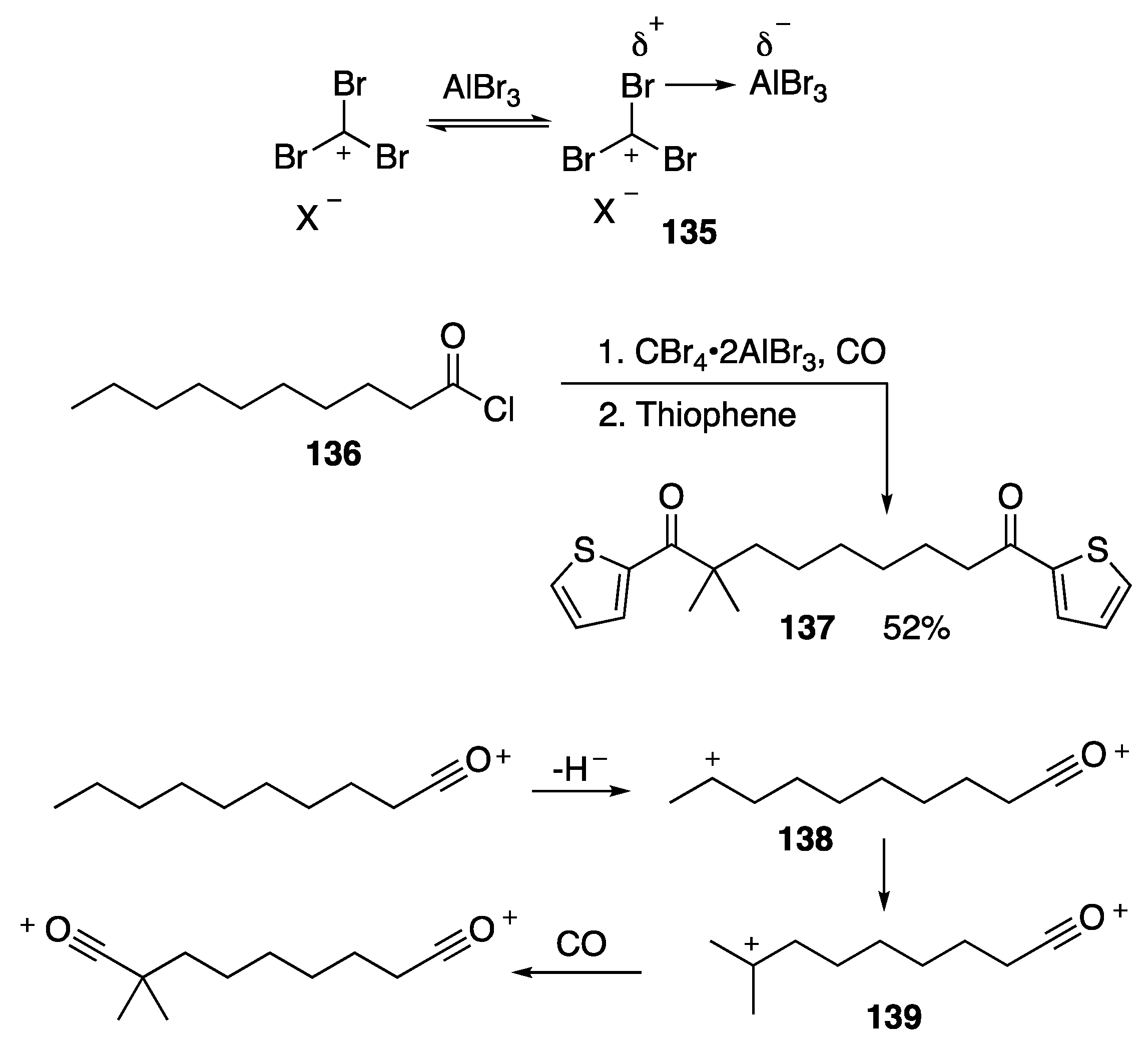
- Akhrem, I.S.; Avetisyan, D.V.; Afanas’eva, L.V.; Artushin, O.I.; Kargamanov, N.D.; Sigan, A.L. A new method for one-pot synthesis of dicarboxylic acids derivatives and diketones containing quaternary carbon atom and remote functional groups from linear and cyclic halides. Tetrahedron Lett. 2017, 58, 4014–4019. [Google Scholar] [CrossRef]
- Akhrem, I.S.; Avetisyan, D.V.; Afanas’eva, L.V.; Tyutyunov, A.A.; Artyushin, O.I.; Kagramanov, N.D. Regioselective functionalization of gem-bis(trifluoromethylated) alkanols. Mendeleev Commun. 2018, 28, 651–652. [Google Scholar] [CrossRef]
- Artault, M.; Mokhtari, N.; Cantin, T.; Martin-Mingot, A.; Thibaudeau, S. Superelectrophilic Csp 3 –H bond fluorination of aliphatic amines in superacid: the striking role of ammonium–carbenium dications. Commun. 2020, 56, 5905–5908. [Google Scholar]
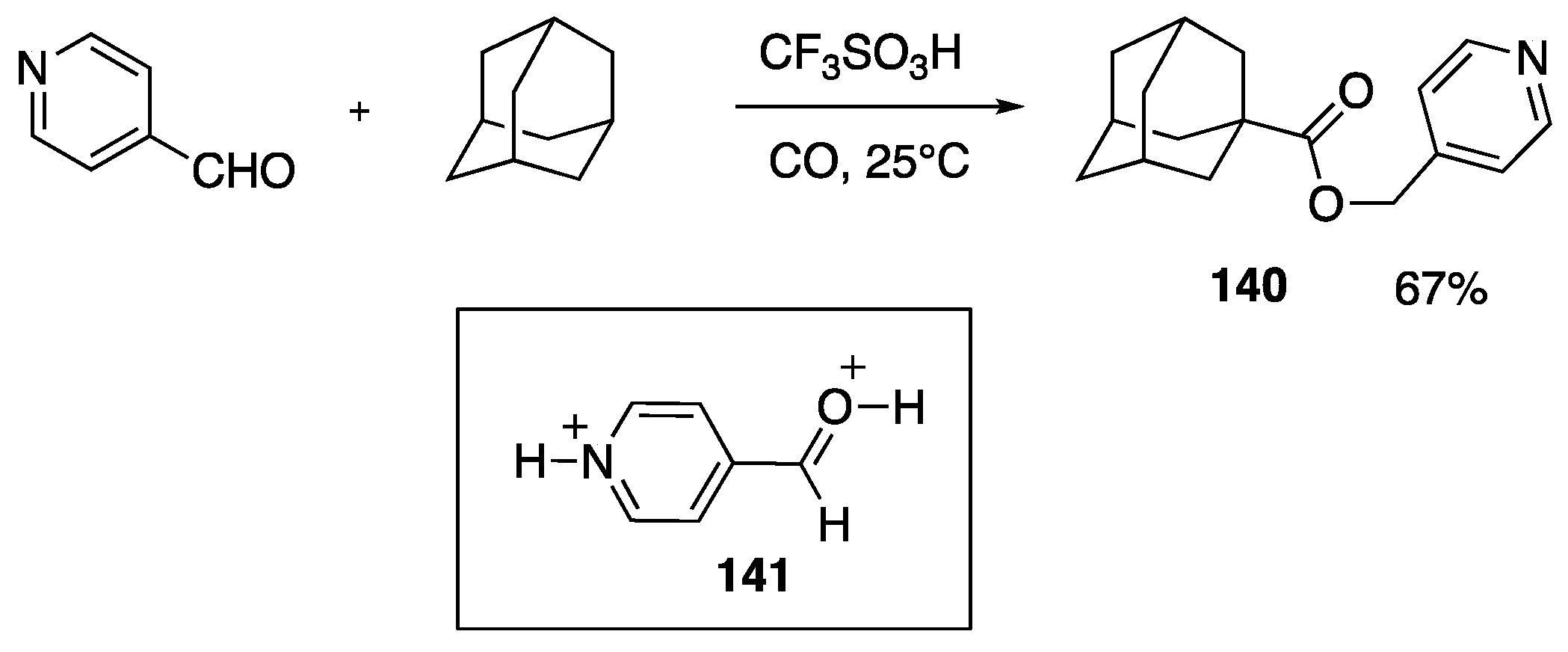
- Klumpp, D.A.; Zhang, Y.; Kindelin, P.J.; Lau, S. Superacid-catalyzed reactions of pyridinecarboxaldehydes. Tetrahedron 2006, 62, 5915–5921. [Google Scholar] [CrossRef]
- Shudo, K.; Ohwada, T. Stable Carbocation Chemistry; Prakash, G.K.S., Schleyer, P.V.R., Eds.; Wiley: New York, NY, USA, 1997; pp. 529–531. [Google Scholar]
- Raja, E.K.; DeSchepper, D.J.; Nilsson, L.S.O.; Klumpp, D.A. Friedel-Crafts acylation with amides. J. Org. Chem. 2012, 77, 5788–5793. [Google Scholar] [CrossRef] [Green Version]
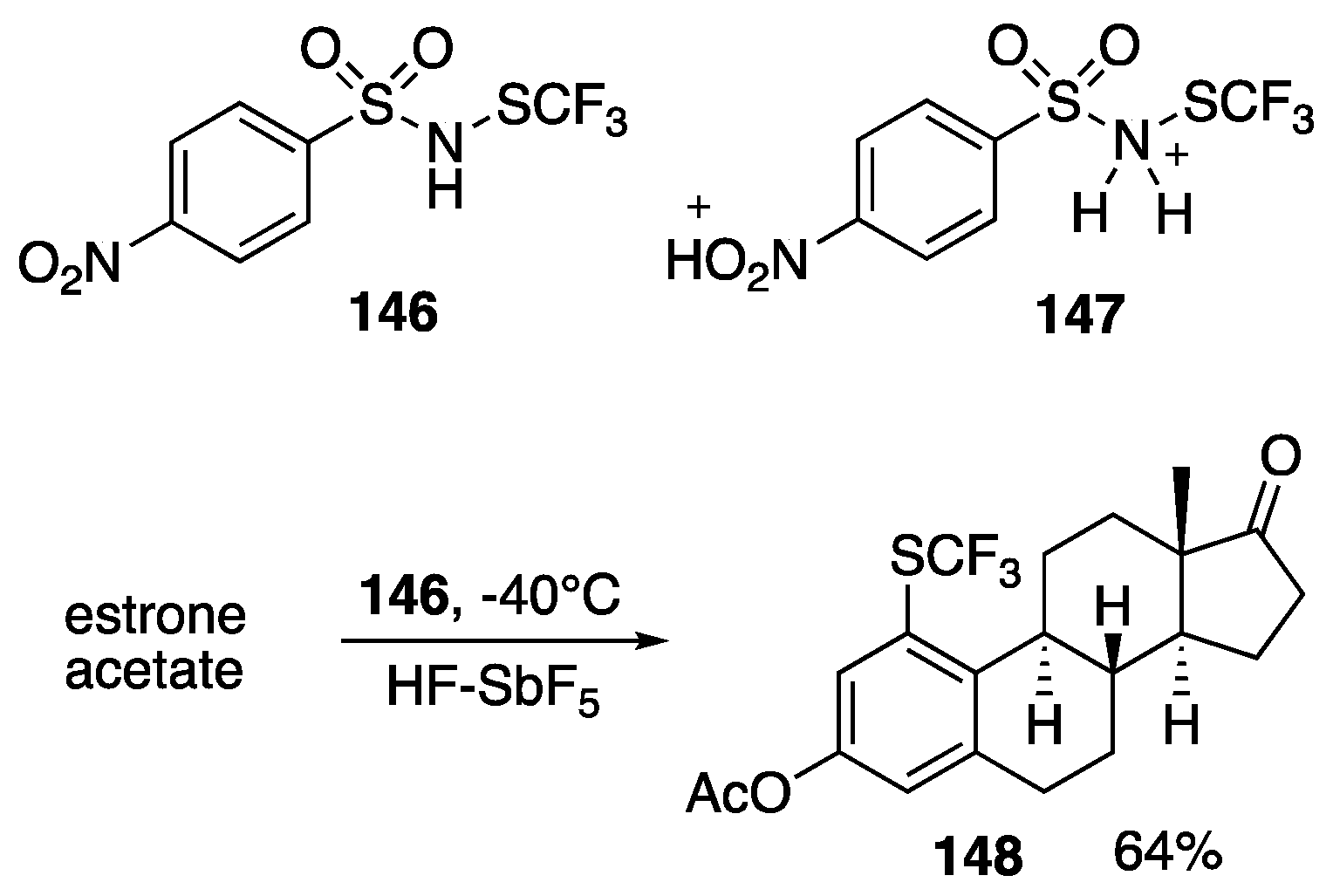
- Milandou, L.J.C.B.; Carreyre, H.; Alazet, S.; Greco, G.; Martin-Mingot, A.; Loumpangou, C.N.; Ouamba, J.-M.; Bouazza, F.; Billard, T.; Thibaudeau, S. Superacid-catalyzed trifluoromethylation of aromatic amines. Angew. Chem. Int. Ed. 2017, 56, 169–172. [Google Scholar] [CrossRef]
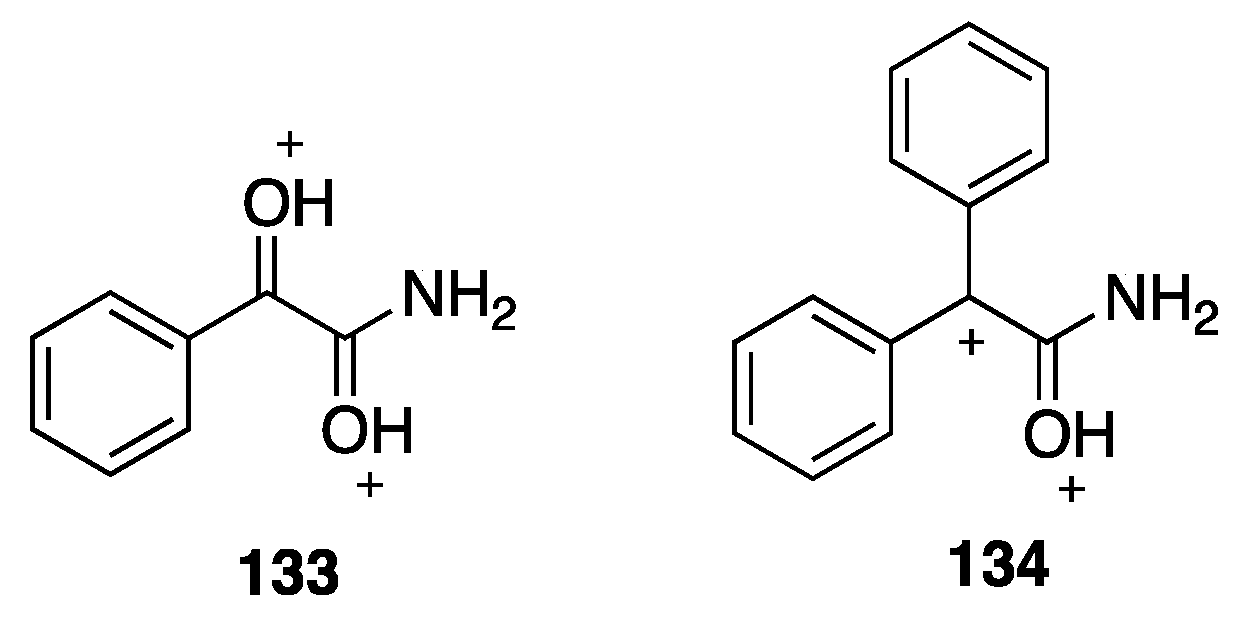
- Zhu, Z.; Genaev, A.M.; Salnikov, G.E.; Koltunov, K.Y. Superelectrophilic activation of 1-nitronaphthalene in the presence of aluminum chloride. Reactions with benzene and cyclohexane. Org. Biomol. Chem 2018, 16, 9129–9132. [Google Scholar] [CrossRef]
- Ohta, T.; Shudo, K.; Okamoto, T. Pheylation of Nitroso-, Azo-, Azosy-, and Nitrobenzene, A Variety of Onium-Benzenium Ions. Tetrahedron Lett. 1977, 101–104. [Google Scholar] [CrossRef]



































{kind=link}
{kind=link}
{kind=link}
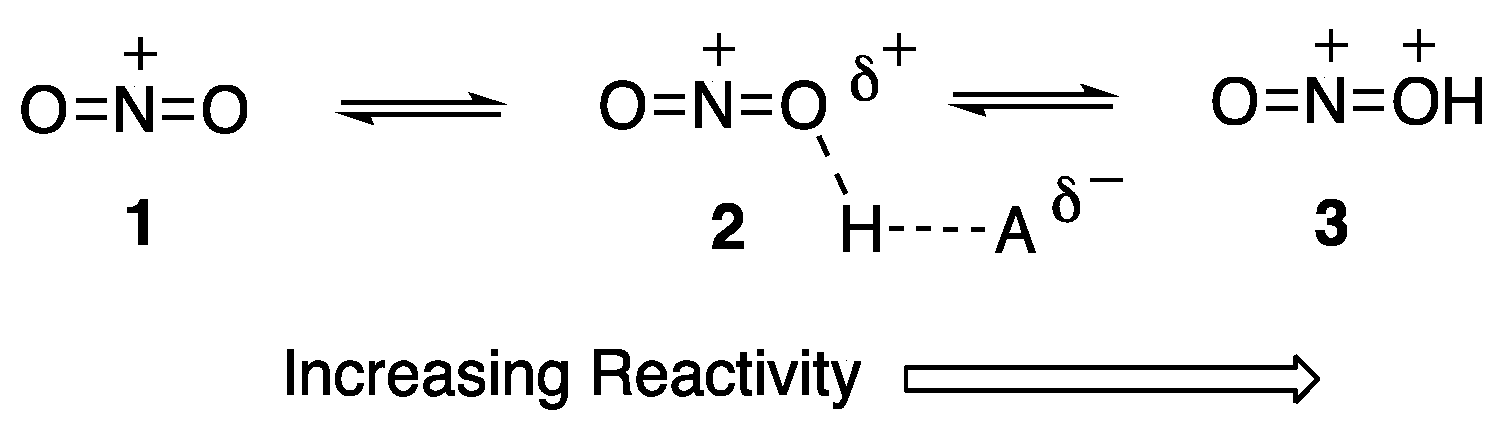{kind=link}
{kind=link}
{kind=link}
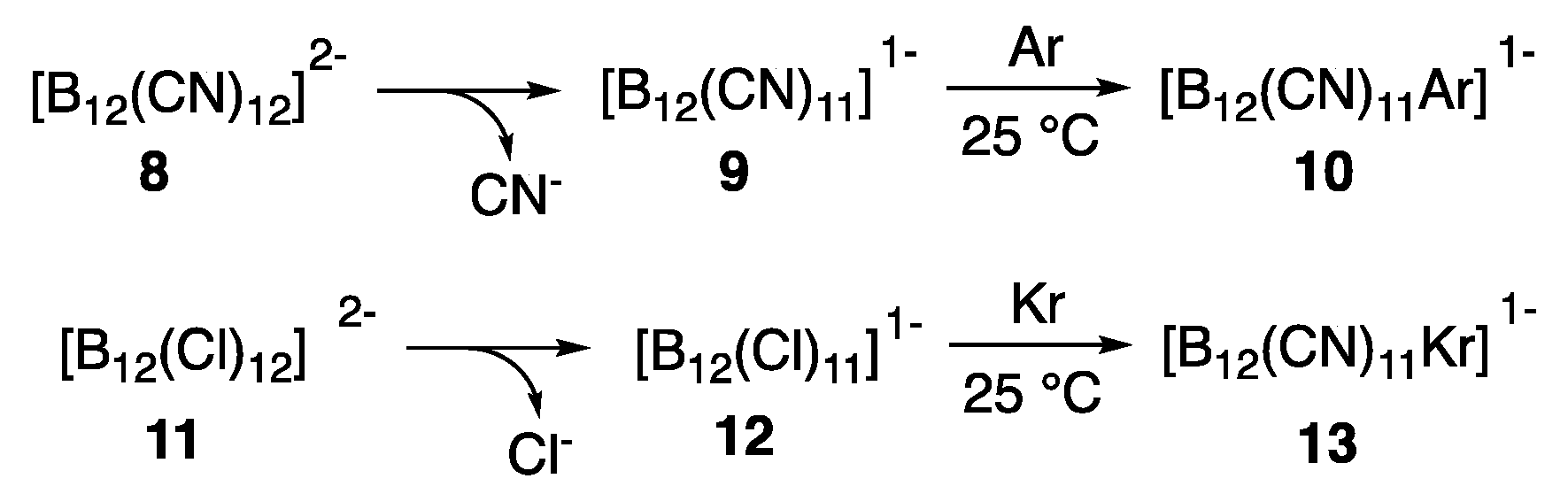{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
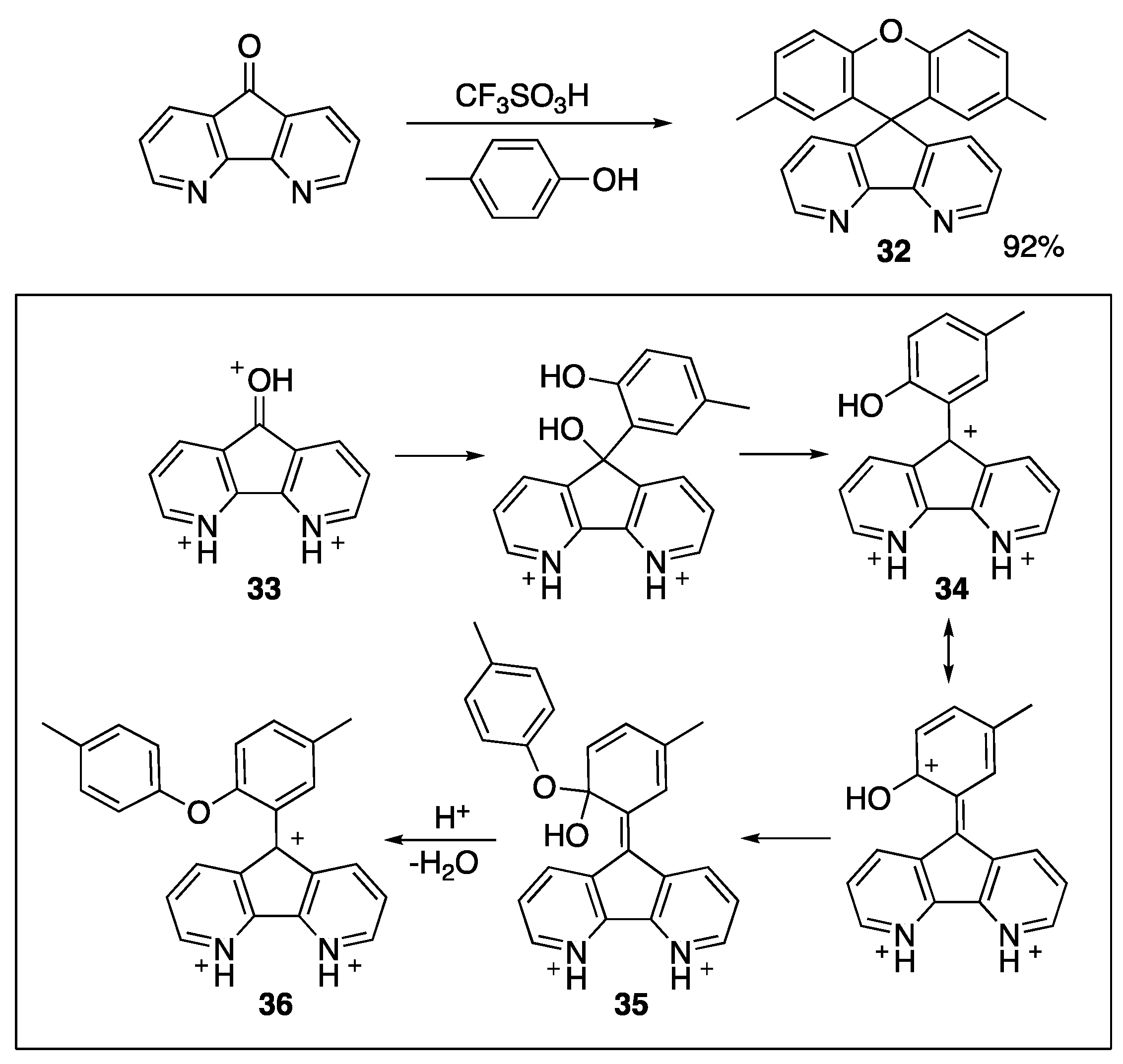{kind=link}
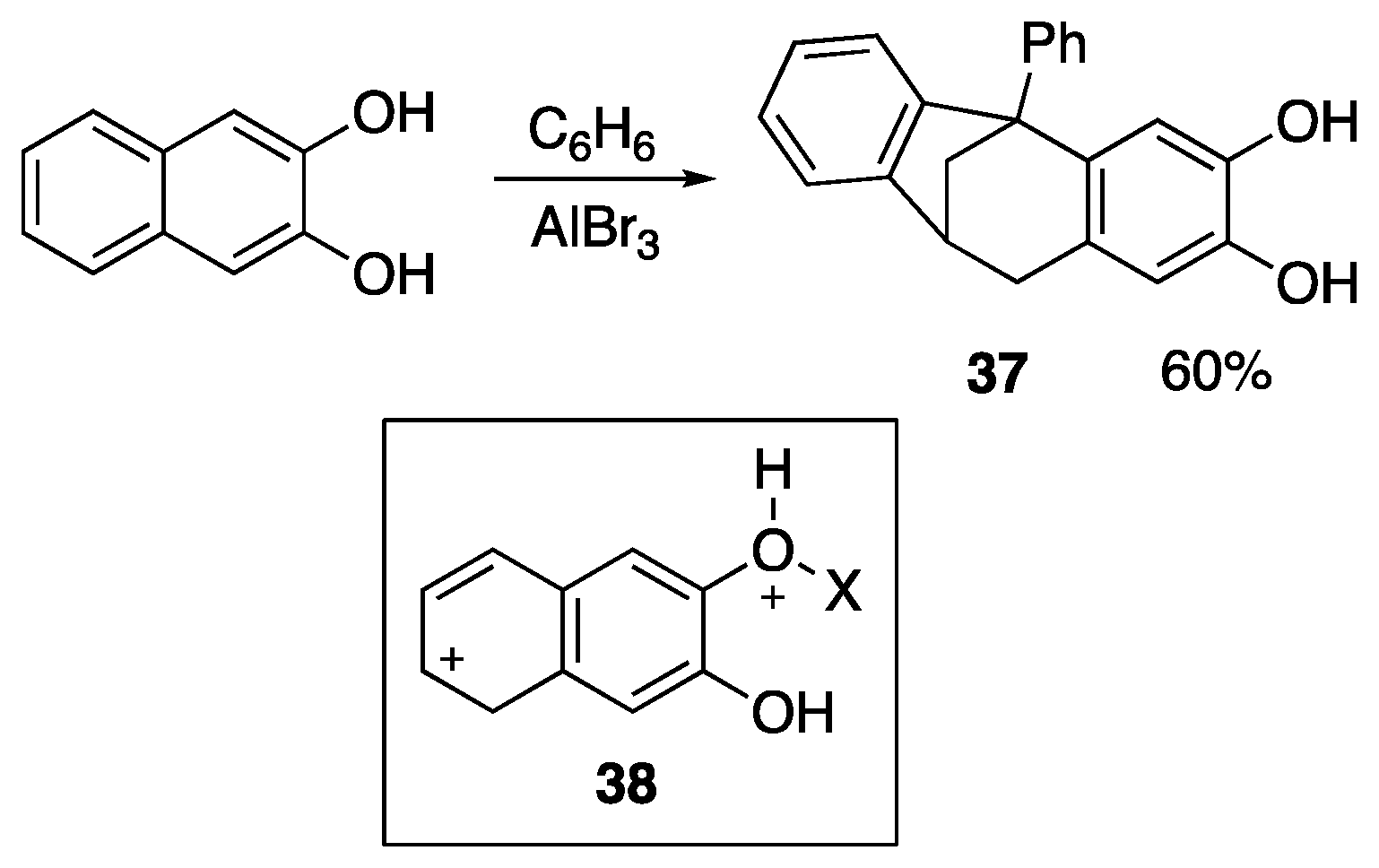{kind=link}
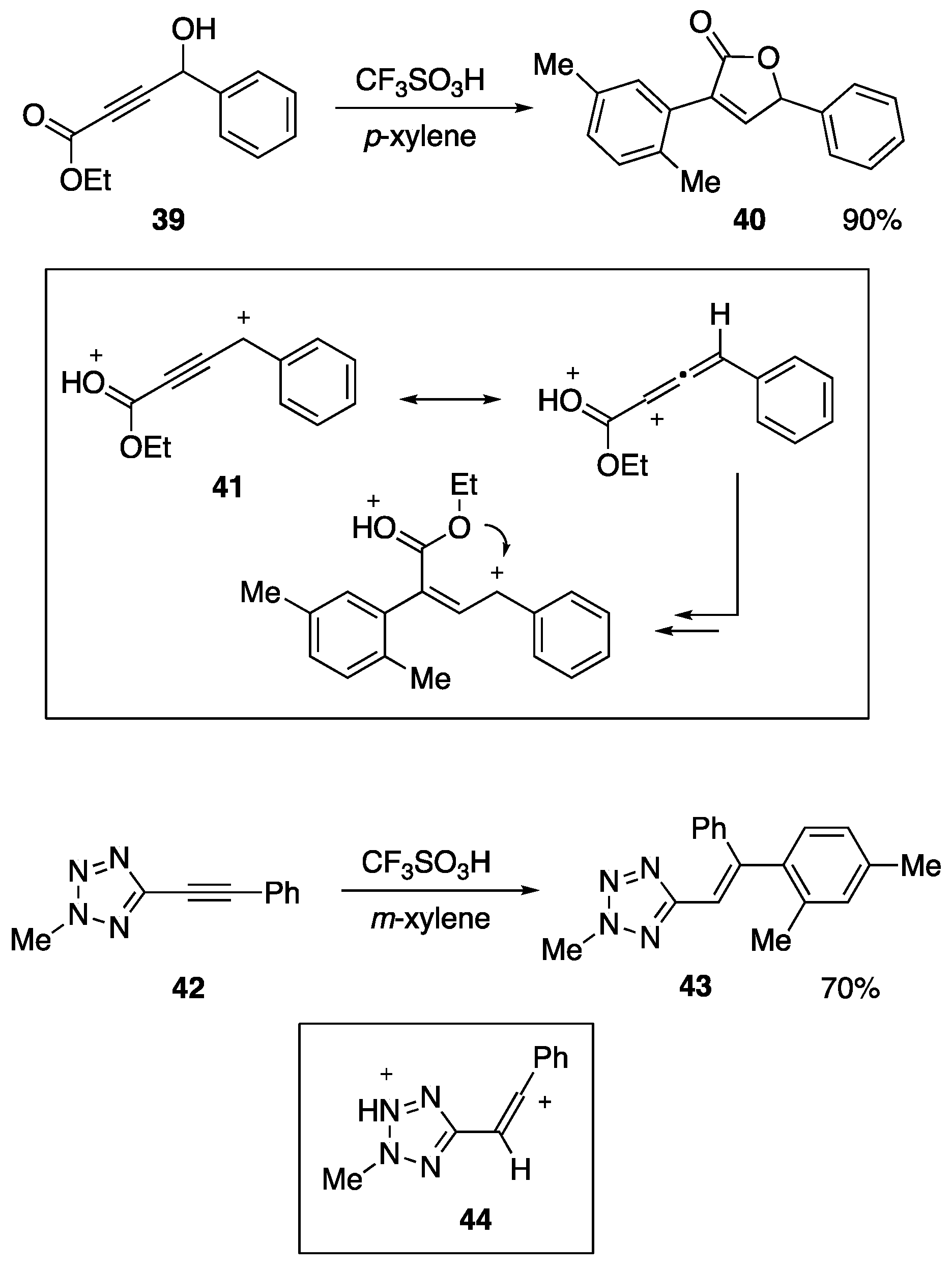{kind=link}
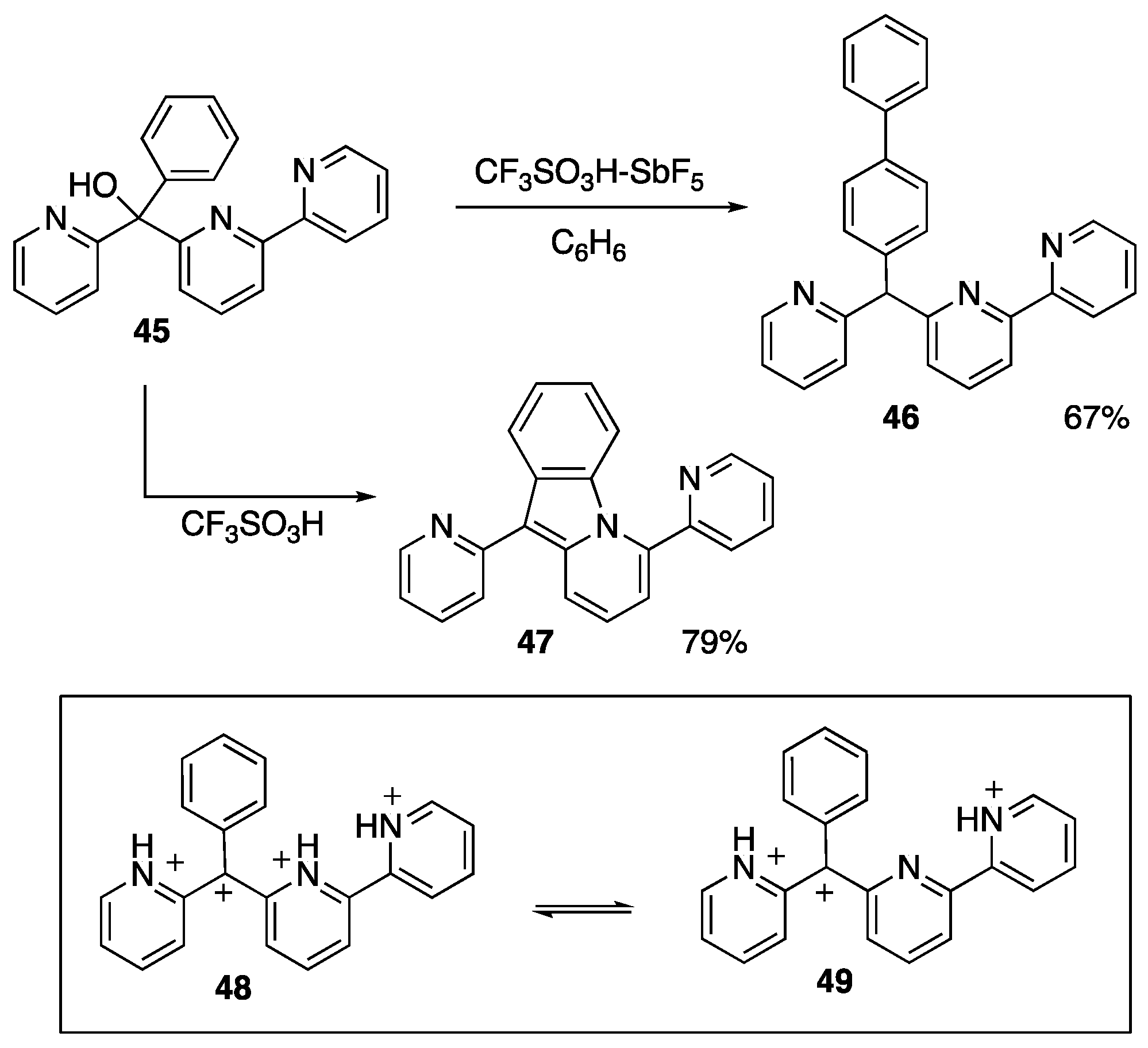{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
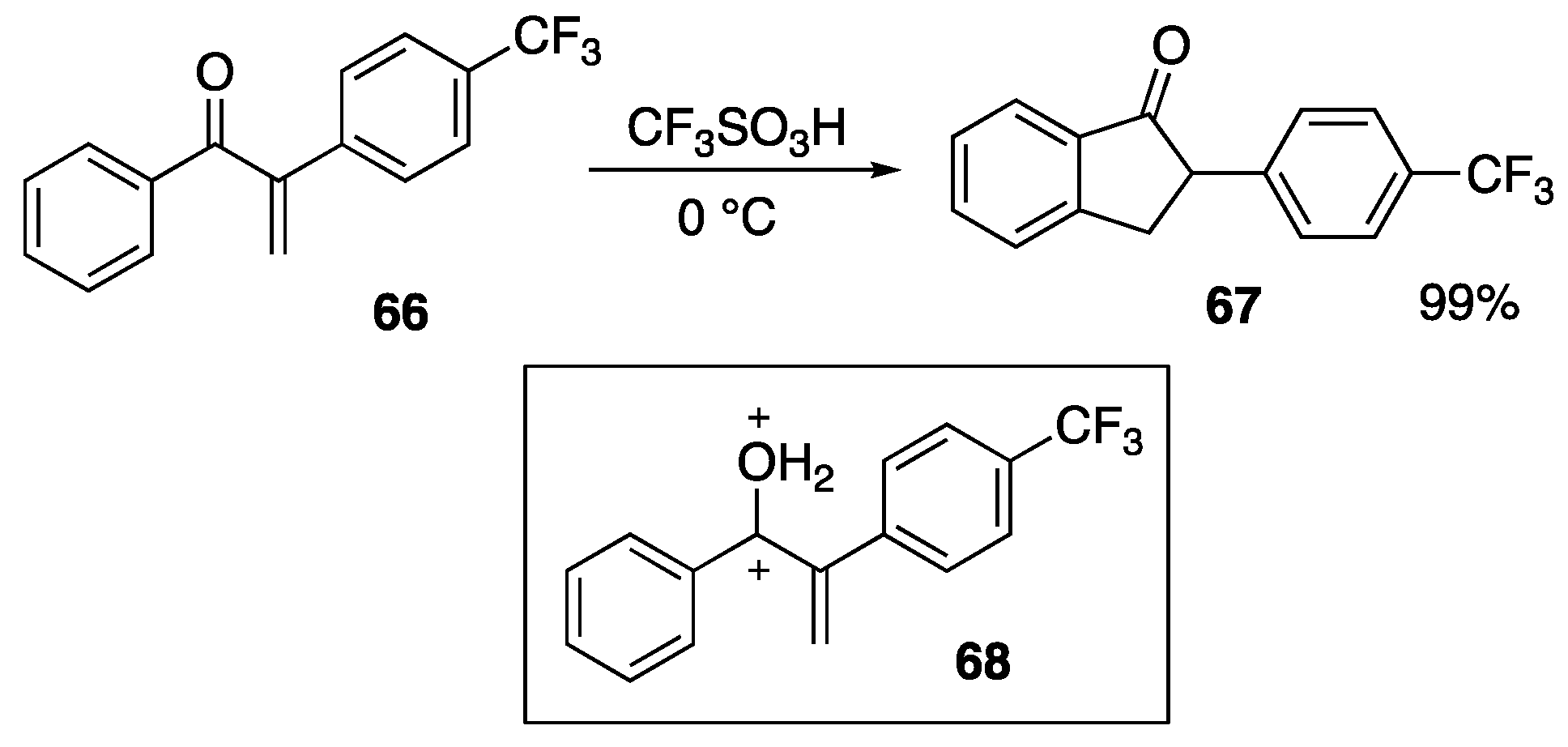{kind=link}
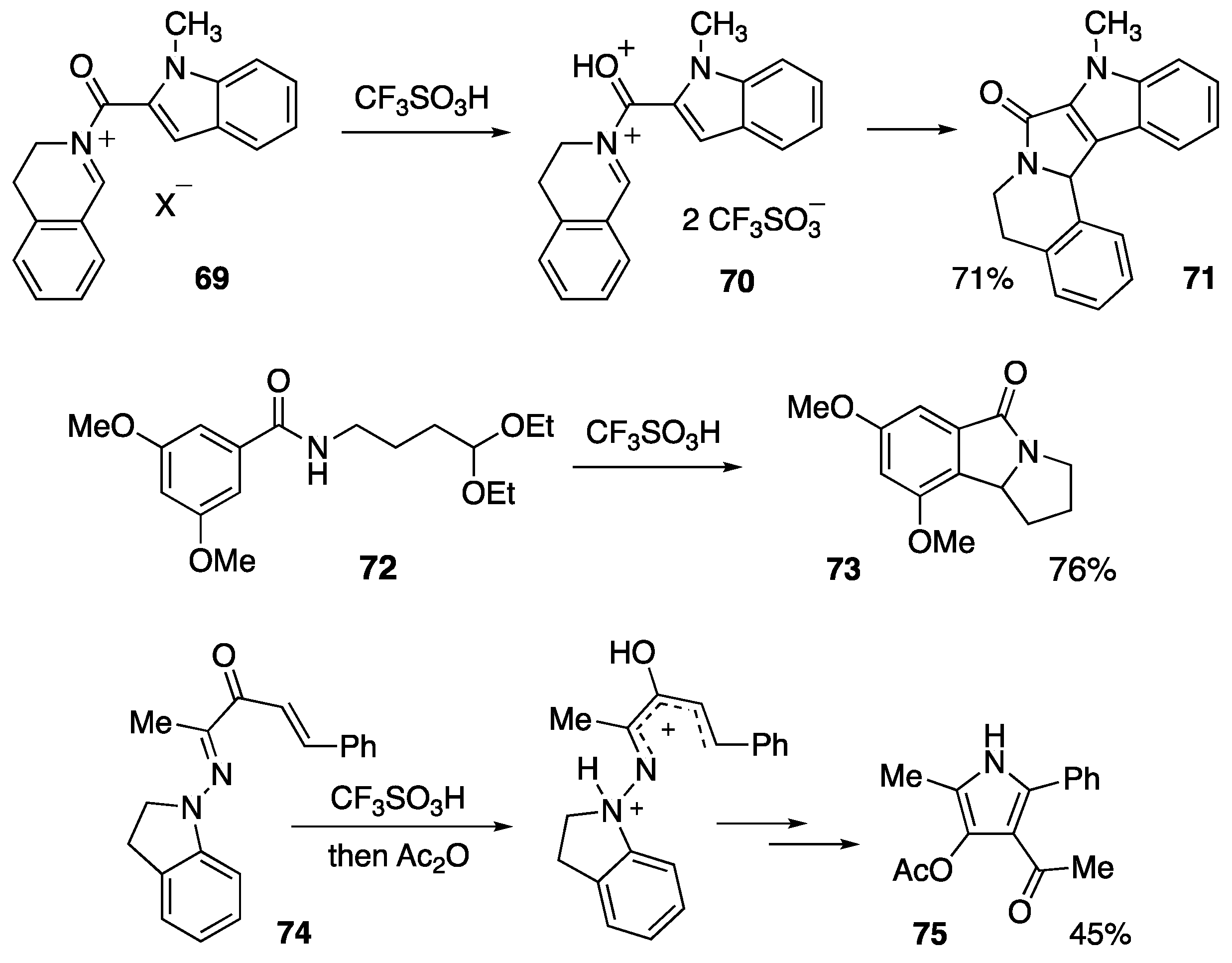{kind=link}
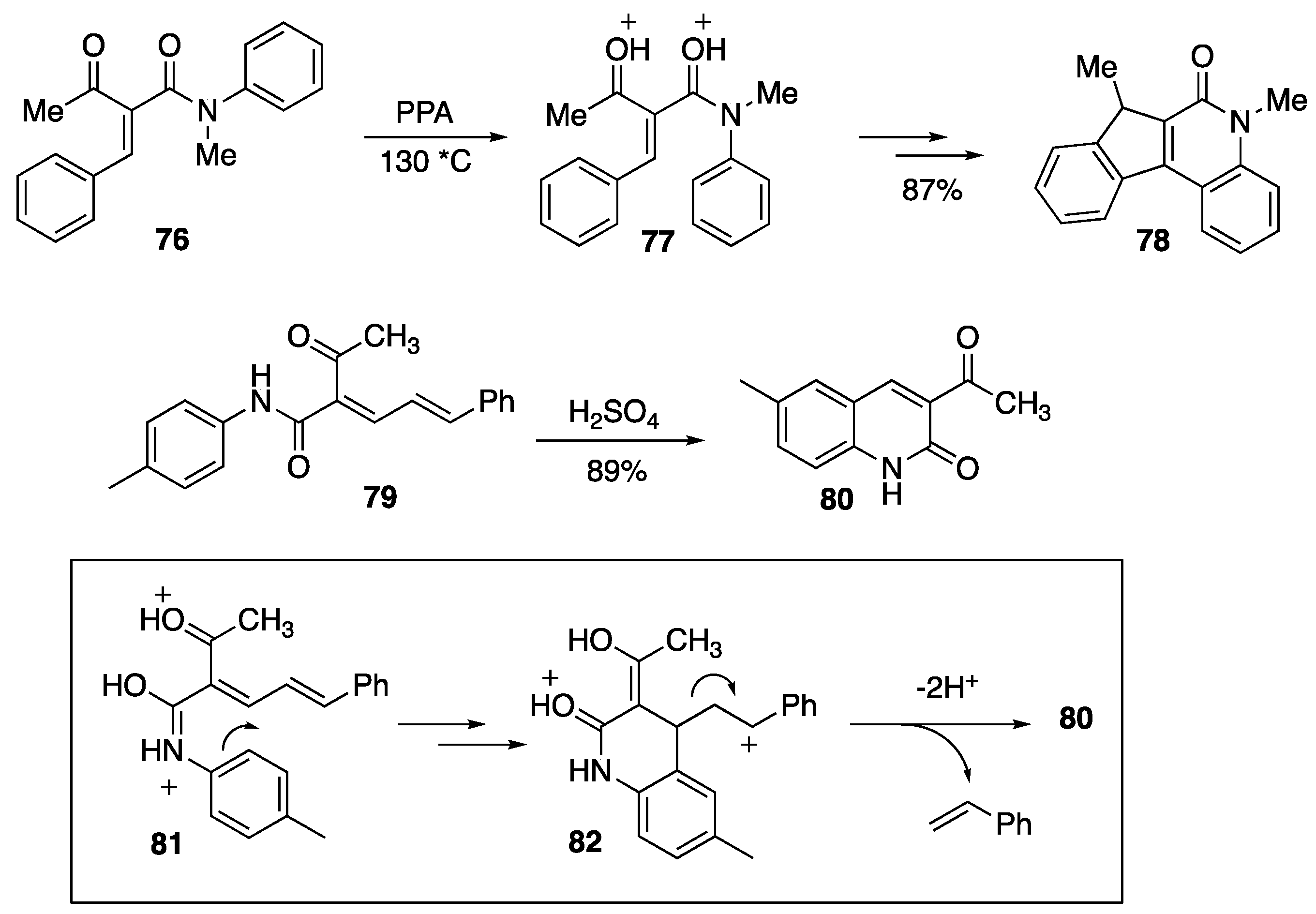{kind=link}
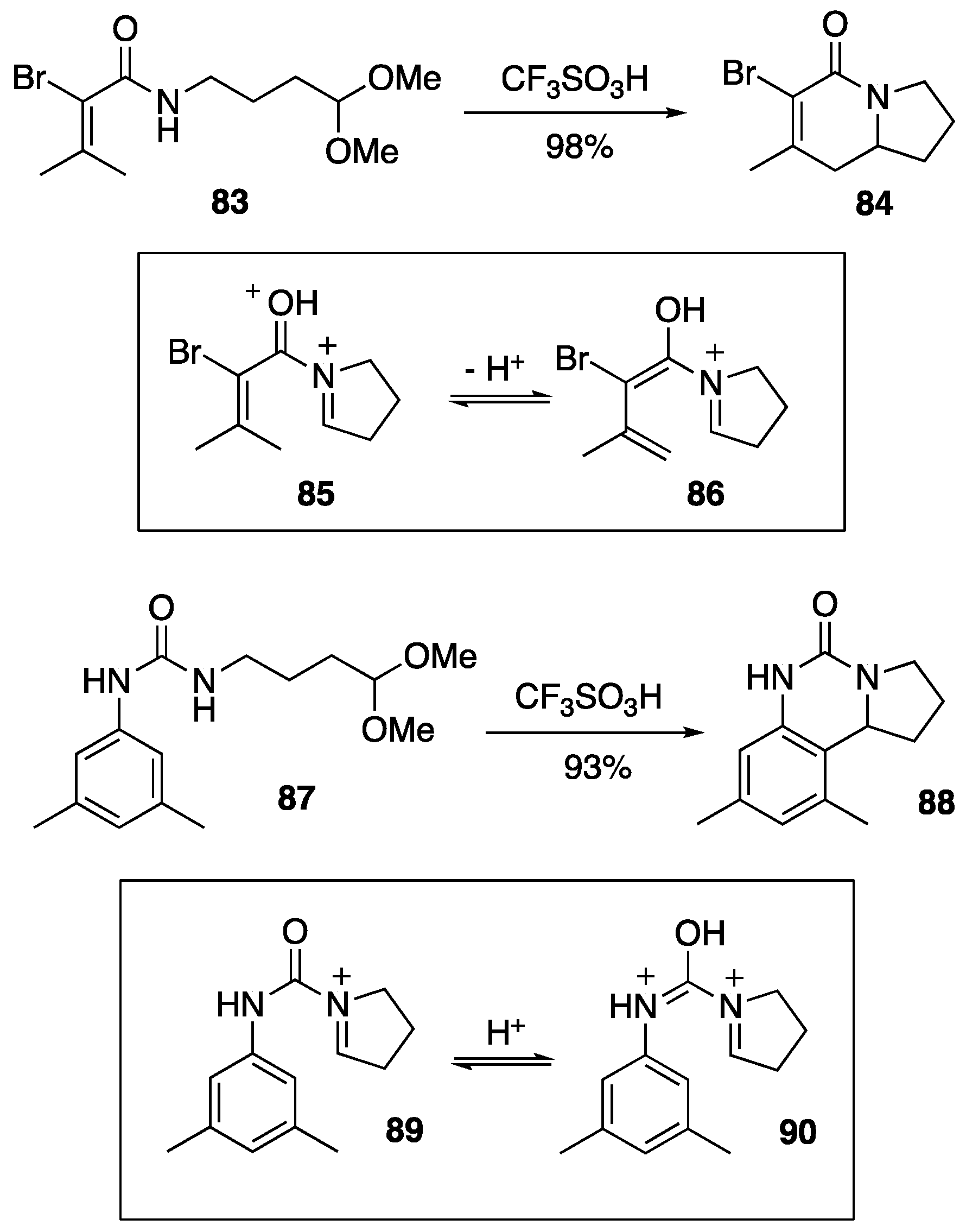{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Superelectrophile |
|---|---|
 |  |
 |  |
 |  |
 |  |
 |  |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klumpp, D.A.; Anokhin, M.V. Superelectrophiles: Recent Advances. Molecules 2020, 25, 3281. https://doi.org/10.3390/molecules25143281
Klumpp DA, Anokhin MV. Superelectrophiles: Recent Advances. Molecules. 2020; 25(14):3281. https://doi.org/10.3390/molecules25143281
Chicago/Turabian StyleKlumpp, Douglas A., and Maksim V. Anokhin. 2020. "Superelectrophiles: Recent Advances" Molecules 25, no. 14: 3281. https://doi.org/10.3390/molecules25143281
APA StyleKlumpp, D. A., & Anokhin, M. V. (2020). Superelectrophiles: Recent Advances. Molecules, 25(14), 3281. https://doi.org/10.3390/molecules25143281