
Pyrazolyl-Ureas as Interesting Scaffold in Medicinal Chemistry



Abstract
:1. Introduction
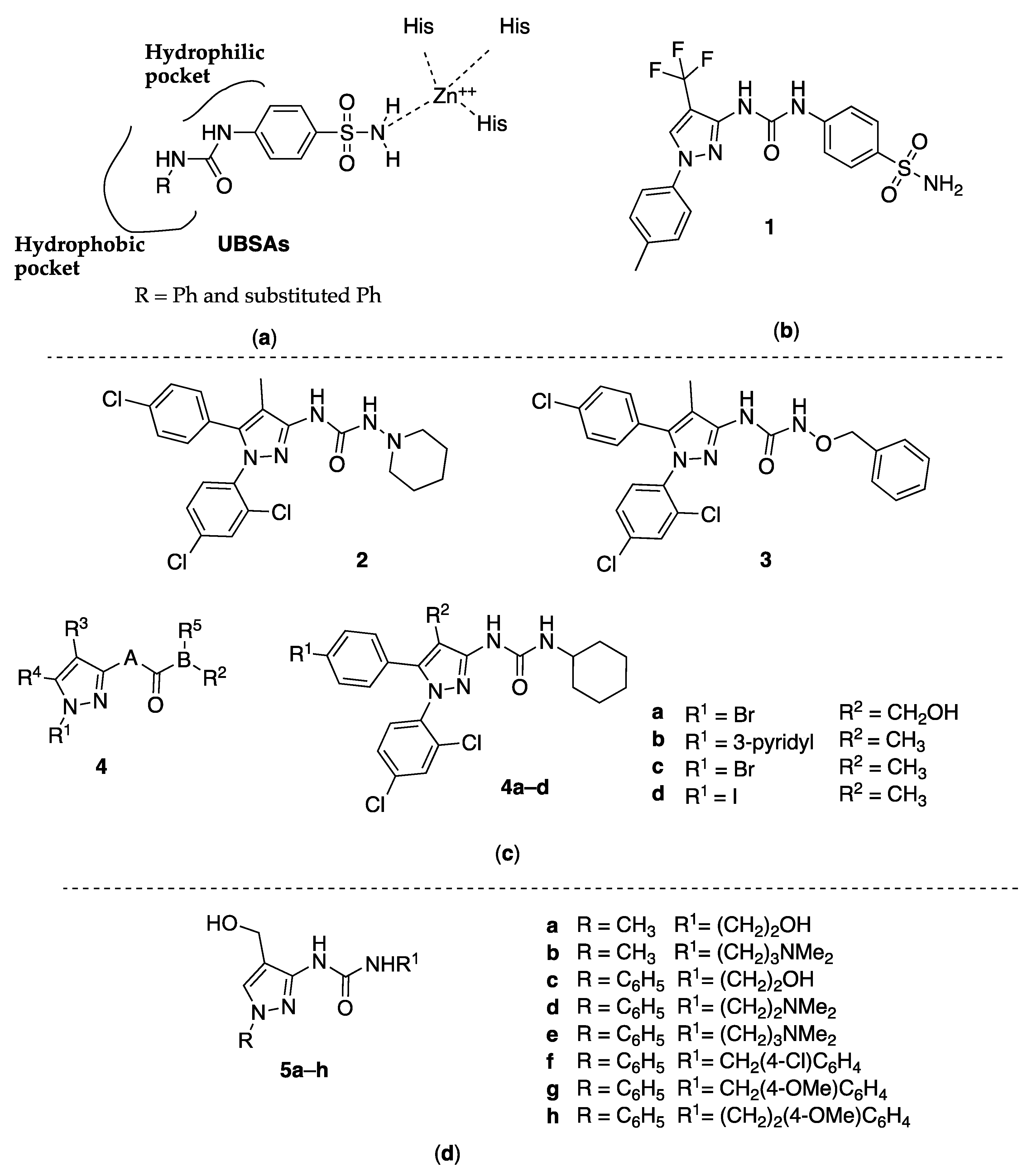
2. 3-Pyrazolyl-Ureas Derivatives
2.1. 3-Pyrazolyl-Ureas as Human Carbonic Anhydrase Inhibitors
2.2. 3-Pyrazolyl-Ureas as Cannabinoid Receptor Antagonist
2.3. 3-Pyrazolyl-Ureas as Antibacterial Agents
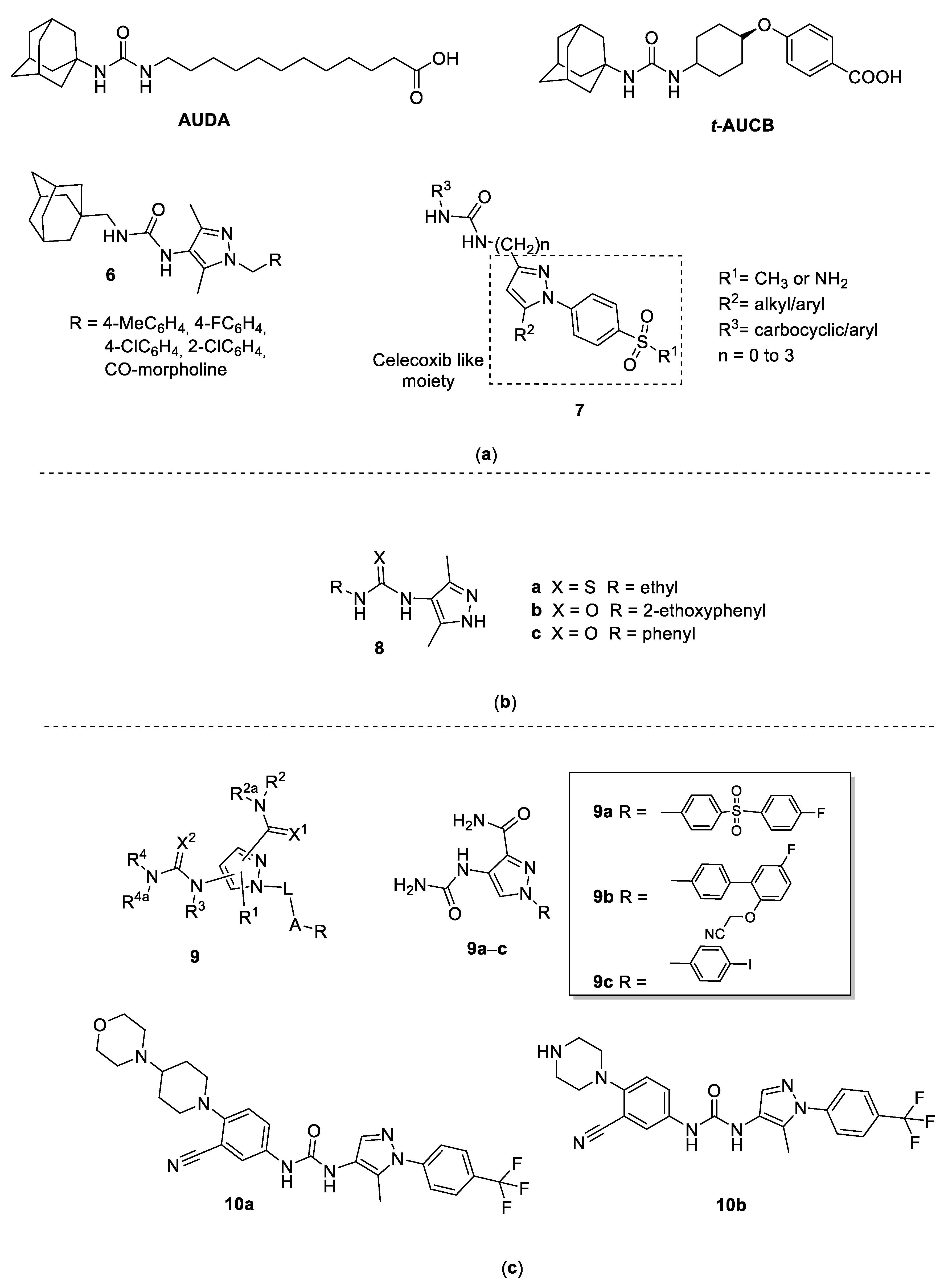
3. 4-Pyrazolyl-Ureas Derivatives
3.1. 4-Pyrazolyl-Ureas as Epoxide Hydrolase (sEH) Inhibitors
3.2. 4-Pyrazolyl-Ureas as Antiepileptic Drugs
3.3. 4-Pyrazolyl-Ureas as Anti-Inflammatory Agents
3.4. 4-Pyrazolyl-Ureas as Protein Kinases Inhibitors
3.4.1. CDK8 Inhibitors
3.4.2. RAS/RAF/MEK/ERK-MAPK Inhibitors
3.4.3. JNK Inhibitors
3.5. 4-Pyrazolyl-Ureas as Anticancer Agents
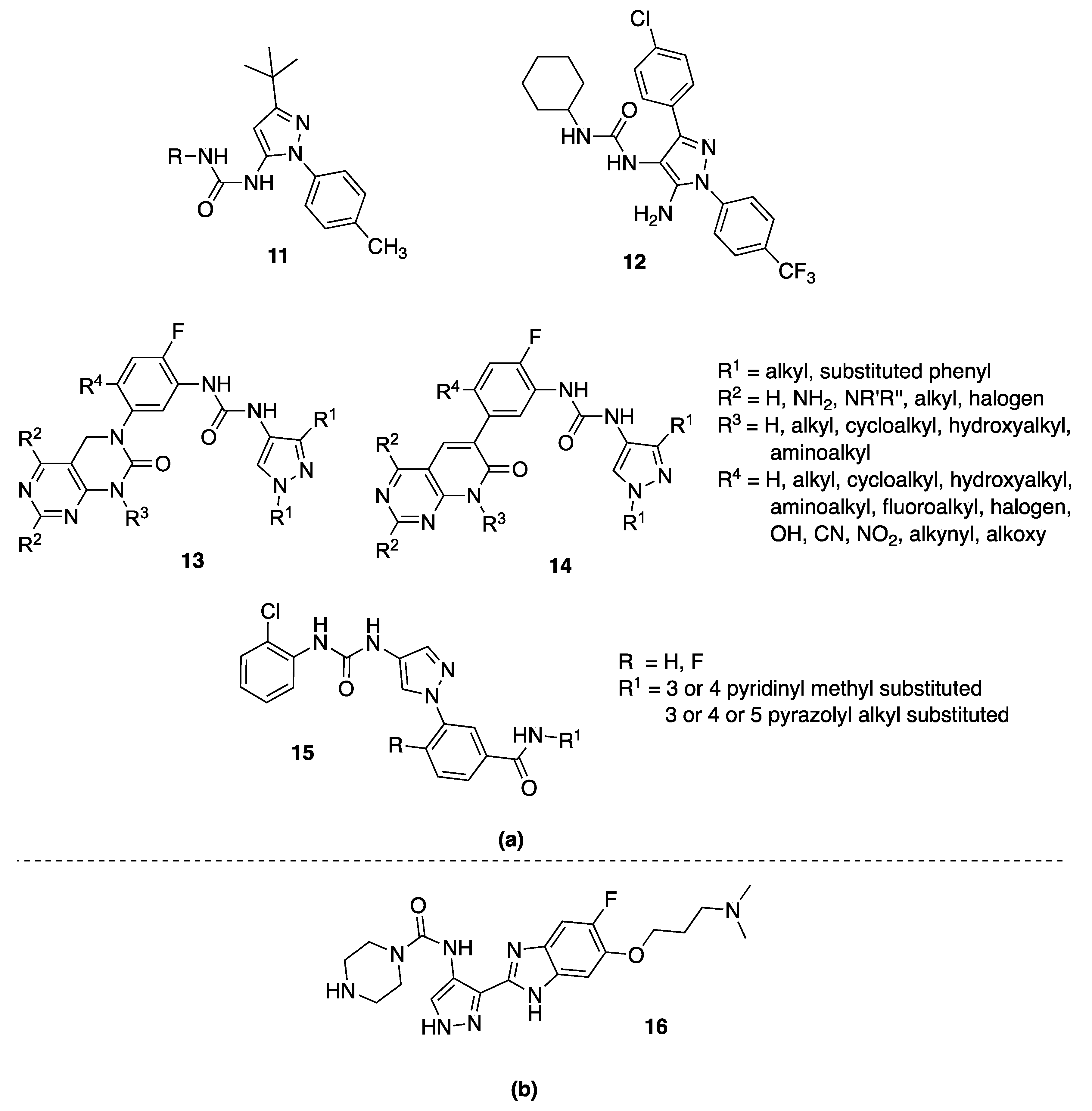
4. 5-Pyrazolyl-Ureas
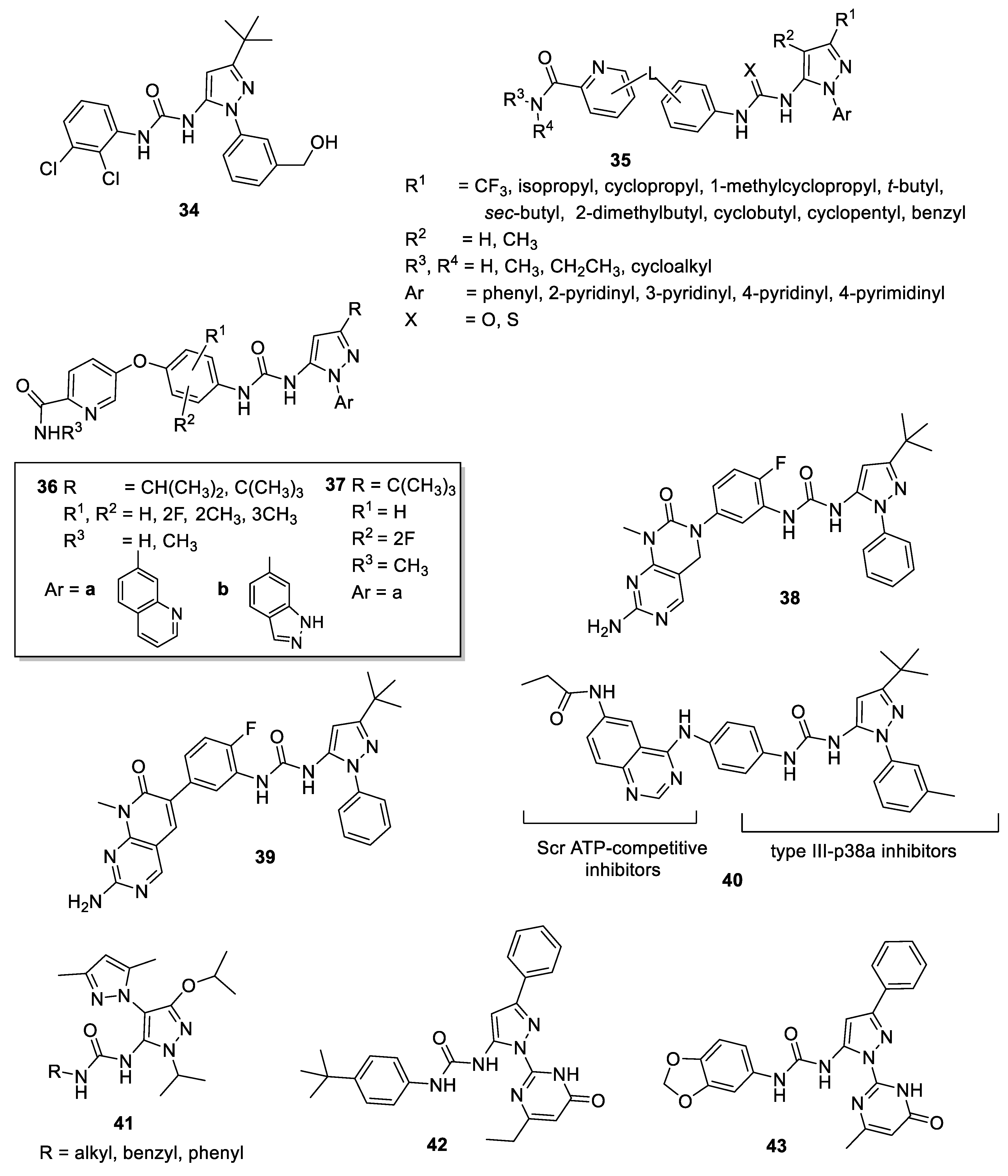
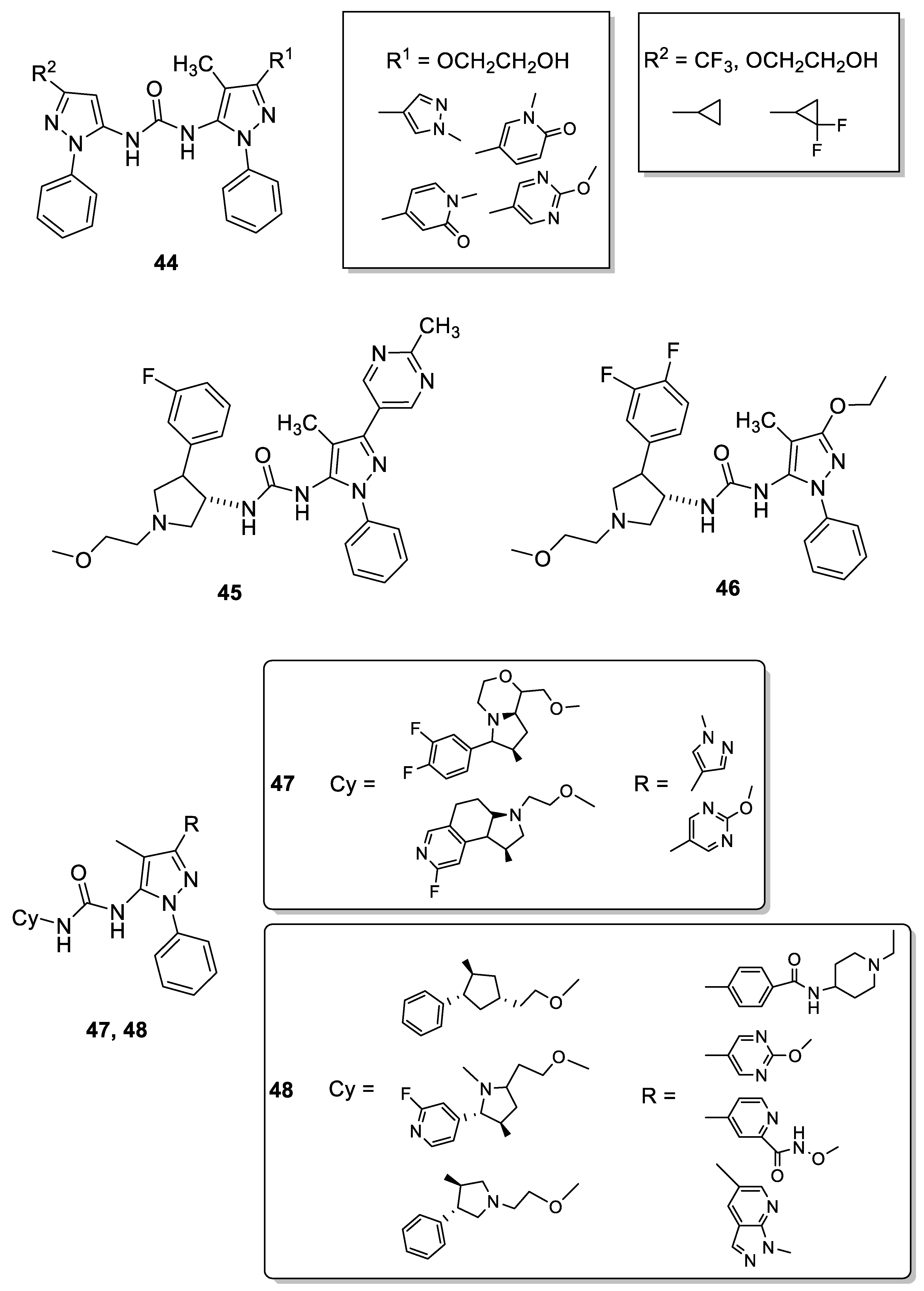
4.1. 5-Pyrazolyl-Ureas as p38MAPK Inhibitors
4.2. 5-Pyrazolyl-Ureas Interfering with Neutrophil Migration
4.3. 5-Pyrazolyl-Ureas as Antiangiogenic Agents
4.4. 5-Pyrazolyl-Ureas as Anticancer Agents
4.5. 5-Pyrazolyl-Ureas as TrKA Inhibitors
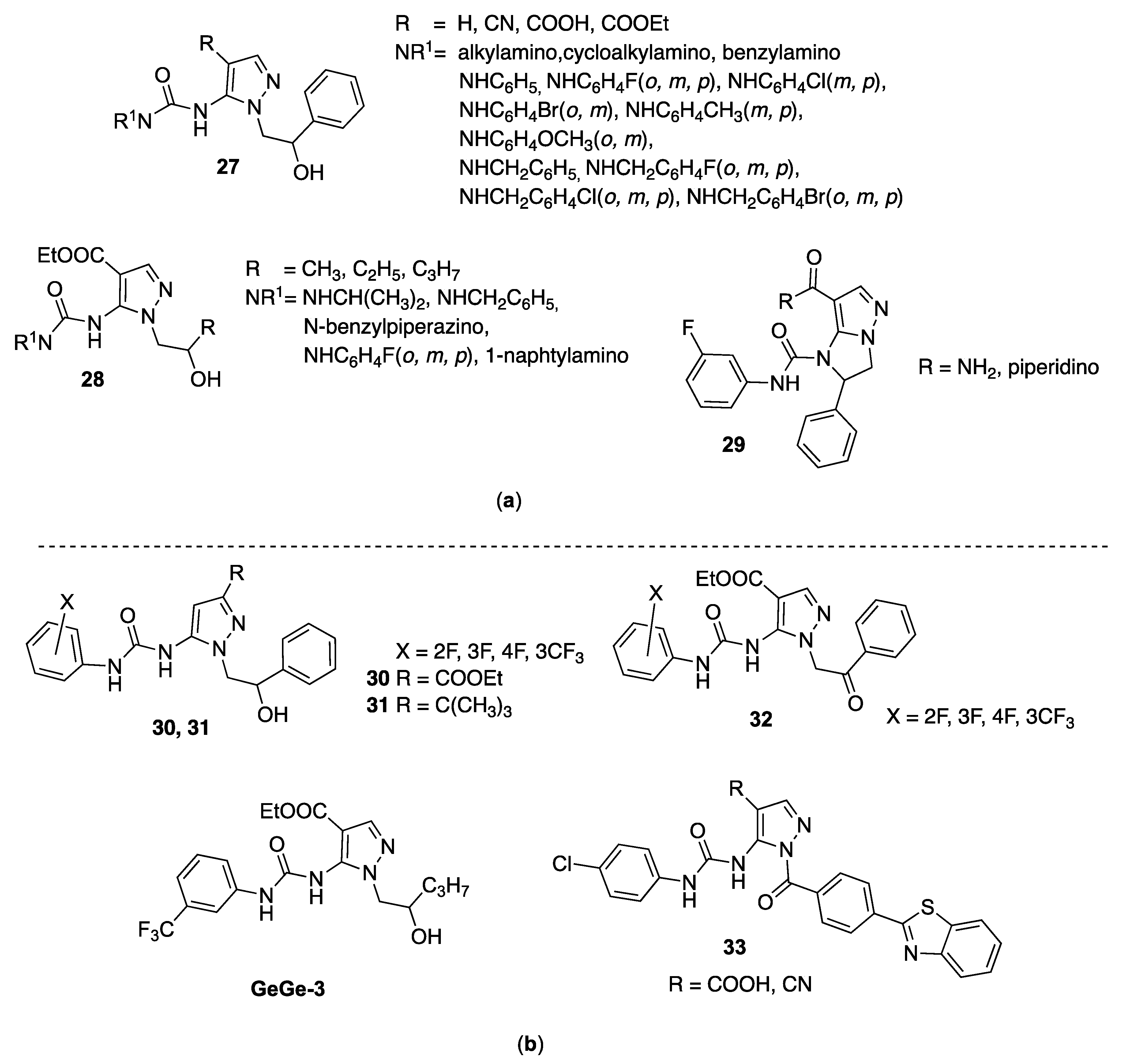
4.6. 5-Pyrazolyl-Ureas as Antimalarial Agents
4.6.1. Antimalarial Agents acting as Protein-Protein Interaction Inhibitors
4.6.2. Antimalarial Agents Acting as PfATP4 Inhibitors
4.6.3. Antimalarial Agents Acting as P. falciparum Prolyl-tRNA-synthetase Inhibitors
- in N1 is needed the methyl group; NH, or polar substituents are not tolerated;
- the trifluoromethyl substituent in position 3 of the pyrazole acts as vector to the solvent and its removal is not tolerated, while more polar groups (such an example 2-ethyl-morpholine) causes a potency decrease;
- removal of the p-fluorophenyl moiety in position 4 of the pyrazole led to inactive compounds, but substitution in the phenyl ring could be changed without affecting the potency.
4.7. 5-Pyrazolyl-Ureas as Anti-Toxoplasma Agents
4.8. 5-Pyrazolyl-Ureas as Antibacterial Agents
4.9. 5-Pyrazolyl-Ureas as Anti-Trypanosome Agents
4.10. 5-Pyrazolyl-Ureas as Antiviral Agents
4.11. 5-Pyrazolyl-Ureas as Potassium Channel Activators
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D-QSAR | Three-dimensional quantitative SAR |
| aaRS | Aminoacyl-tRNA synthase |
| Abl | Abelson kinase |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| AUDA | 12-(1-Adamantan-1-ylureido)- dodecanoic acid |
| CAMK | Ca2+/calmodulin- dependent protein kinase |
| CB1/2 | Cannabinoid receptors 1/2 |
| CDK8/CycC | Cyclin-dependent kinase 8 and cyclin C |
| COMFA | Comparative molecular field analysis |
| CoMSIA | Comparative molecular similarity |
| COPD | Chronic obstructive pulmonary disease |
| COX-2 | Cyclooxygenase-type 2 |
| CXCL8 | Interleukin 8 |
| CXCR1/R2 | CXC Chemokine receptors 1/2 |
| DFG (DMG) | Asp-Phe-Gly motif |
| DMPK1 | Dystrophia myotonica protein kinase 1 |
| ERK | extracellular-regulated protein kinase |
| Flt3 | Fms related tyrosine kinase 3 |
| fMLP | N-formyl-methionyl-leucyl-phenylalanine |
| GIRK | G protein-gated inwardly-rectifying potassium channels |
| GSK3α | Glycogen synthase kinases type 3α |
| hCA II | Human carbonic anhydrase II |
| hCAIs | Human carbonic anhydrase inhibitors |
| Hck | Hemopoietic Cell Kinase |
| HD | Huntington’s disease |
| HeLa | Henrietta Lacks immortal cells |
| hERG | Human Ether-à-go-go-Related Gene |
| HFF | Human foreskin fibroblasts |
| HSB | Hybrid structure based (virtual screening approach) |
| HTS | High-throughput screening |
| HUVEC | Human umbilical vein endothelial cells |
| I-κB | Inhibitor of kappa B kinase |
| IKK (1/2) | IkB kinases type 1/2 |
| IL-1-/6/-17/-18 | Interleukin type 1/6/17/18 |
| JM | Juxta-membrane |
| JNK | c-Jun NH2-terminal kinase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MBC | Minimum Bactericidal concentration |
| MES | Maximal electroshock seizure |
| MetRS | Methionyl-t-RNA synthetase |
| MFC | Minimum fungicidal concentration |
| MI | Myocardial infarction |
| MIC | Minimum inhibiting concentration |
| MLSCN | Molecular libraries screening center network |
| MLSMR | Molecular libraries small molecule repository |
| MS | Multiple sclerosis |
| MTIP | Myosin tail interacting protein |
| MyoA | Myosin A |
| NEK10 | NIMA (never in mitosis gene a)-related kinase 10 |
| NF-κΒ | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NGF | Nerve growth factor |
| ONIOM | N-Layered integrated molecular orbital & molecular mechanics |
| Panc-1 | Human pancreatic adenocarcinoma |
| PD | Parkinson’s disease |
| PDB | Protein data bank |
| PDGFR | Platelet derived growth factor |
| PfATP4 | Plasmodium falciparum p-type cation ATPase |
| PfProRS | Plasmodium falciparum prolyl-t-RNA-synthetase |
| PI3K | Protein inositol kinase 3 |
| PKC | Protein kinase C |
| PLK2, PLK3 | Polo-like kinase 2/3 |
| PPI | Protein-protein interaction |
| PTZ | Pentylenetetrazol-induced seizure |
| RAF/RAS | Rapidly accelerated Fibrosarcoma |
| RSV | Respiratory syncytial virus |
| SAR | Structure Activity Relationship |
| sEH | Soluble epoxide hydrolases |
| SUMO | Small ubiquitin-like modifier |
| Syk | Spleen tyrosine kinase |
| TNF-α | Tumor necrosis factor alpha |
| Trk | Tropomyosin receptor kinase |
| UBSAs | Ureido benzenesulfonamides |
| VEGF | Vascular endothelial growth factor |
| VEGFR1/2 | Vascular endothelial growth factor receptor |
| Zap-70 | Zeta Chain of T Cell Receptor Associated Protein Kinase 70 |
References
- Kucukguzel, S.G.; Senkardes, S. Recent advances in bioactive pyrazoles. Eur. J. Med. Chem. 2015, 97, 786–815. [Google Scholar] [CrossRef] [PubMed]
- Behr, L.C.; Fusco, R.; Jarboe, C.H. The Chemistry of Heterocyclic Compounds Pyrazoles, Pyrazoline, Pyrazolidines, Indazoles and Condensed Rings; Interscience Publisher: New York, NY, USA, 1967. [Google Scholar]
- Gilchrist, T.L. Heterocyclic Chemistry; Longman Scientific & Technical Publisher: New York, NY, USA, 1992. [Google Scholar]
- Bennania, F.E.; Doudachc, L.; Cherraha, Y.; Ramlib, Y.; Karrouchib, K.; Ansarb, M.; Abbes Faouzia, M.E. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorg. Chem. 2020, 97, 103470–103532. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Bao-Xiang, Z. Progress of the synthesis of condensed pyrazole derivatives (from 2010 to mid- 2013). Eur. J. Med. Chem. 2014, 85, 311–340. [Google Scholar] [CrossRef]
- Raffa, D.; Maggio, B.; Raimondi, M.V.; Cascioferro, S.; Plescia, F.; Cancemi, G.; Daidone, G. Recent advanced in bioactive systems containing pyrazole fused with a five membered heterocycle. Eur. J. Med. Chem. 2015, 97, 732–746. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, A.D.; Kondekar, N.B.; Sadani, A.A.; Chern, J.W. Ureas: Applications in Drug Design. Curr. Med. Chem. 2017, 24, 622–651. [Google Scholar] [CrossRef]
- Garuti, L.; Roberti, M.; Bottegoni, G.; Ferraro, M. Diaryl Urea: A Privileged Structure in Anticancer Agents. Curr. Med. Chem. 2016, 23, 1528–1548. [Google Scholar] [CrossRef]
- Supuran, C.T.; Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S. Ureido-Substituted Benzenesulfonamides Potently Inhibit Carbonic Anhydrase IX and Show Antimetastatic Activity in a Model of Breast Cancer Metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [Green Version]
- Sahu, C.; Sen, K.; Pakhira, S.; Mondal, B.; Das, A.K. Binding affinity of substituted ureido-benzenesulfonamide ligands to the carbonic anhydrase receptor: A theoretical study of enzyme inhibition. J. Comput. Chem. 2013, 34, 1907–1916. [Google Scholar] [CrossRef]
- Li, S.; Zheng, Z.; Tao, X.; Wang, L.; Zhou, X.; Chen, W.; Zhong, W.; Xiao, J.; Xie, Y.; Li, X.; et al. 4-Methyl-1H-diaryl Pyrazole Derivative as Cannabine Type I Receptor Inhibitor and Its Preparation. WO Patent Application No. 2016184310 A1 20161124, 24 November 2016. [Google Scholar]
- Makriyannis, A.; Liu, Q.; Thotapally, R. Preparation of Arylpyrazolecarboxamides as CB1 Cannabinoid Receptor Antagonists. U.S. Patent Application No. 20060100208 A1 20060511, 11 May 2006. [Google Scholar]
- Bratenko, M.K.; Barus, M.M.; Rotar, D.V.; Vovk, M.V. Polyfunctional Pyrazoles. 9*. Synthesis of 1-Alkyl(Aryl)-3-[4-(Hydroxymethyl)-1H-Pyrazol-3-Yl]ureas. Chem. Heterocyc. Compd. 2014, 50, 1252–1258. [Google Scholar] [CrossRef]
- Morisseau, C.; Goodrow, M.H.; Dowdy, D.; Zheng, J.; Greene, J.F.; Sanborn, J.R.; Hammock, B.D. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc. Natl. Acad. Sci. USA 1999, 96, 8849–8854. [Google Scholar] [CrossRef] [Green Version]
- Kodani, S.D.; Hammock, B.D. The 2014 Bernard B. Brodie award lecture-epoxide hydrolases: Drug metabolism to therapeutics for chronic pain. Drug Metab. Dispos. 2015, 43, 788–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammock, B.D.; Kim, I.-H.; Morisseau, C.; Watanabe, T.; Newmann, J.W. Improved Inhibitors for the Soluble Epoxide Hydrolase. WO Patent Application No. 2004/089296 A2, 21 October 2004. [Google Scholar]
- D’yachenko, V.S.; Danilov, D.V.; Shkineva, T.K.; Vatsadze, I.A.; Burmistrov, V.V.; Butov, G.M. Synthesis and properties of 1-[(adamantan-1-yl)methyl]-3-pyrazolyl ureas. Chem. Heterocycl. Compd. 2019, 55, 129–134. [Google Scholar] [CrossRef]
- Hwang, S.H.; Wagner, K.M.; Morisseau, C.; Hua Dong, J.-Y.L.; Wecksler, A.T.; Hammock, B.D. Synthesis and Structure Activity Relationship Studies of Urea-Containing Pyrazoles as Dual Inhibitors of Cyclooxygenase-2 and Soluble Epoxide Hydrolase. J. Med. Chem. 2011, 54, 3037–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinisch, G.; Matuszczak, B.; Rakowitz, D.; Tantisina, B. Synthesis of N-aryl-N′-heteroaryl-substituted urea and thiourea derivatives and evaluation of their anticonvulsant activity. Arch. Pharm. (Weinheim) 1997, 330, 207–210. [Google Scholar] [CrossRef]
- Pandeya, S.N.; Mishra, V.; Sign, P.N.; Rupainwar, D.C. Anticonvulsant activity of thioureido derivatives of acetophenone semicarbazone. Pharmacol. Res. 1998, 37, 17–22. [Google Scholar] [CrossRef]
- Dimmock, J.R.; Vashishtha, S.C.; Stables, J.P. Ureylene anticonvulsants and related compounds. Pharmazie 2000, 55, 490–494. [Google Scholar]
- Kaymakcioglu, B.K.; Rollas, S.; Koercegez, E.; Aricioglu, F. Synthesis and biological evaluation of new N-substituted-N′-(3,5-di/1,3,5-trimethylpyrazole-4-yl)thiourea/urea derivatives. Eur. J. Pharmac. Sci. 2005, 26, 97–103. [Google Scholar] [CrossRef]
- Clare, M.; Fletcher, T.R.; Hamper, B.C.; Hanson, G.A.; Heier, R.F.; Huang, H.; Lennon, P.J.; Oburn, D.S.; Reding, M.T.; Stealey, M.A.; et al. Preparation of Substituted Pyrazole Ureas for the Treatment of Inflammation. WO Patent Application No. 2005037797 A1 20050428, 28 April 2005. [Google Scholar]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-kappa B system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef]
- Baxter, A.; Brough, S.; Faull, A.; Johnstone, C.; Mcinally, T. Heteroaromatic Carboxamide Derivatives and their Use as Inhibitors of the Enzyme IKK-2. WO Patent Application No. 01/58890 Al PCTISE01/00248, 7 February 2001. [Google Scholar]
- Sharma, R.; Jain, A.; Sahu, B.; Singh, D.; Mali, S. Preparation of Heterocyclyl Compounds as Inhibitors of IL-17 and TNF-α for the Treatment of Inflammatory Diseases and Autoimmune Disorders. WO Patent Application No. 2014181287 A1 20141113, 8 May 2014. [Google Scholar]
- Schneider, E.V.; Bottcher, J.; Huber, R.; Maskos, K.; Neumann, L. Structure–kinetic relationship study of CDK8/CycC specific compounds. Proc. Natl. Acad. Sci. USA 2013, 110, 8081–8086. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Ren, X.; Chang, C.A. Discovery of CDK8/CycC Ligands with a New Virtual Screening Tool. ChemMedChem 2019, 14, 107–118. [Google Scholar] [CrossRef]
- Flynn, D.L.; Petillo, P.A.; Kaufman, M.D.; Patt, W.C. Preparation of Heterocyclic Ureas as Kinase Inhibitors Useful for the Treatment of Proliferative Diseases. WO Patent Application No. 2008033999 A2 20080320, 20 March 2008. [Google Scholar]
- Flynn, D.L.; Petillo, P.A.; Kaufman, M.D.; Patt, W.C. Preparation of Heterocyclic Ureas as Kinase Inhibitors Useful for the Treatment of Proliferative Diseases. U.S. Patent Application No. 20090099190 A1 20090416, 16 April 2009. [Google Scholar]
- Flynn, D.L.; Kaufman, M.D.; Patt, W.C.; Petillo, P.A. Preparation of Heterocyclic Ureas as Kinase Inhibitors Useful for the Treatment of Proliferative and Inflammatory Diseases. WO Patent Application No. 2008034008 A2 20080320, 20 March 2008. [Google Scholar]
- Flynn, D.L.; Petillo, P.A.; Kaufman, M.D.; Patt, W.C. Preparation of Dihydropyridopyrimidinyl, Dihydronaphthyidinyl and Related Compounds Useful as Kinase Inhibitors for the Treatment of Proliferative Diseases. U.S. Patent Application No. 8188113 B2 20120529, 29 May 2012. [Google Scholar]
- Eshraghi, A.A.; Wang, J.; Adil, E.; He, J.; Zine, A.; Bublik, M.; Bonny, C.; Puel, J.; Balkany, T.J.; Van De Water, T.R. Blocking c-Jun-N-terminal kinase signaling can prevent hearing loss induced by both electrode insertion trauma and neomycin ototoxicity. Hear Res. 2007, 226, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Van De Water, T.R.; Bonny, C.; de Ribaupierre, F.; Puel, J.L.; Zine, A. A Peptide Inhibitor of c-Jun N-Terminal Kinase Protects against Both Aminoglycoside and Acoustic Trauma-Induced Auditory Hair Cell Death and Hearing Loss. J. Neurosci. 2003, 23, 8596–8607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; LoGrasso, P.; Zheng, K.; Park, C.M. Preparation of Substituted Pyrazolylbenzamides as JNK Kinase Inhibitors. WO Patent Application No. 2015084936 A1 20150611, 11 June 2015. [Google Scholar]
- Cherrier, M.P.; Parmantier, E.; Minoux, H.; Clerc, F.; Angouillant-Boniface, O.; Brollo, M.; Schio, L. Pyrazolylbenzimidazole Derivative, their Preparation, Compositions Containing them and their Use for Treating Diseases, Especially Cancer. WO Patent Application No. 2008003857 A1 20080110, 10 January 2008. [Google Scholar]
- Swapna, D.; Pooja, V.P.; Sucharitha, D.; Sunitha, K.; Swetha, M. A review of p38 kinase inhibitors as anti-inflammatory drugs targets. Int. J. Pharm. Technol. (IJPT) 2010, 2, 86–101. [Google Scholar]
- Barun, O.; Advait, N.; Francisco, J.A. A general strategy for creating “inactive-conformation’’ abl inhibitors. Chem. Biol. 2006, 13, 779–786. [Google Scholar] [CrossRef] [Green Version]
- Dumas, J.; Sibley, R.; Riedl, B.; Monahan, M.K.; Lee, W.; Lowinger, T.B.; Redman, A.M.; Johnson, J.S.; Kingery-Wood, J.; Scott, W.J.; et al. Discovery of a new class of p38 kinase inhibitors. Bioorg. Med. Chem. Lett. 2000, 10, 2047–2050. [Google Scholar] [CrossRef]
- Dumas, J.; Hatoum-Mokdad, H.; Sibley, R.; Riedl, B.; Scott, W.J.; Monahan, M.K.; Lowinger, T.B.; Brennan, C.; Natero, R.; Turner, T.; et al. 1-Phenyl-5-pyrazolyl ureas: Potent and selective p38 kinase inhibitors. Bioorg. Med. Chem. Lett. 2000, 10, 2051–2054. [Google Scholar] [CrossRef]
- Redman, A.M.; Johnson, J.S.; Dally, R.; Swartz, S.; Wild, H.; Paulsen, H.; Caringal, Y.; Gunn, D.; Renick, J.; Osterhout, M.; et al. p38 Kinase Inhibitors for the Treatment of Arthritis and Osteoporosis: Thienyl, Furyl, and Pyrrolyl Ureas. Bioorg. Med. Chem. Lett. 2001, 11, 9–12. [Google Scholar] [CrossRef]
- Dumas, J.; Hatoum-Mokdad, H.; Sibley, R.N.; Smith, R.A.; Scott, W.J.; Khire, U.; Lee, W.; Wood, J.; Wolanin, D.; Cooley, J.; et al. Synthesis and pharmacological characterization of a potent, orally active p38 kinase inhibitor. Bioorg. Med. Chem. Lett. 2002, 12, 1559–1562. [Google Scholar] [CrossRef]
- Cirillo, P.F.; Gilmore, T.A.; Hickey, E.R.; Regan, J.R.; Zhang, L.-H. Preparation of Aromatic Heterocyclic Ureas as Antiinflammatory Agents. WO Patent Application No. 2000043384 A1 20000727, 27 July 2000. [Google Scholar]
- Zhang, L.-H.; Zhu, L. Novel Process for Synthesis of Heteroaryl-Substituted Ureas. WO Patent Application No. 2001004115 A2 20010118, 18 January 2001. [Google Scholar]
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Grob, P.M.; Hickey, E.R.; Moss, N.; Pav, S.; et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002, 9, 268–272. [Google Scholar] [CrossRef]
- Regan, J.; Breitfelder, S.; Cirillo, P.; Gilmore, T.; Graham, A.G.; Hickey, E.; Klaus, B.; Madwed, J.; Moriak, M.; Moss, N.; et al. Pyrazole urea-based inhibitors of p38 MAP kinase: From lead compound to clinical candidate. J. Med. Chem. 2002, 45, 2994–3008. [Google Scholar] [CrossRef]
- Soyoon, R.; Jida, C.; Jaemyung, K.; Suyoung, B.; Jaewoo, H.; Seunghyun, J.; Soohyun, K.; Youngmin, L. BIRB 796 has Distinctive Anti-inflammatory Effects on Different Cell Types. Immune Netw. 2013, 13, 283–288. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.clinicaltrials.gov/ (accessed on 5 February 2020).
- Bagley, M.C.; Davis, T.; Dix, M.C.; Widdowson, C.S.; Kipling, D. Microwave-assisted Synthesis of N-pyrazole Ureas and the p38alpha Inhibitor BIRB 796 for Study into Accelerated Cell Ageing. Org. Biomol. Chem. 2006, 4, 4158–4164. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.G.; Srivani, P.; Achaiah, G.; Sastry, G.N. Strategies to design pyrazolyl urea derivatives for p38 kinase inhibition: A molecular modeling study. J. Comput.-Aided Mol. Des. 2007, 21, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Toru, A.; Hitoshi, Y.; Chiyoshi, K. Identification, synthesis, and biological evaluation of 6-[(6R)-2-(4-fluorophenyl)-6-(hydroxymethyl)-4,5,6,7-tetrahydropyrazolo [1,5-a]pyrimidin-3-yl]-2-(2-methylphenyl)pyridazin-3(2H)-one (AS1940477), a potent p38 MAP kinase inhibitor. J. Med. Chem. 2012, 55, 7772–7785. [Google Scholar] [CrossRef]
- Shunsuke, I.; Yoshiji, A.; Hideo, A. A possible mechanism for hepatotoxicity induced by BIRB-796, an orally active p38 mitogen-activated protein kinase inhibitor. J. Appl. Toxicol. 2011, 31, 671–677. [Google Scholar] [CrossRef]
- Li, X.; Zhou, X.; Zhang, J.; Wang, L.; Long, L.; Zheng, Z.; Li, S.; Zhong, W. Synthesis and biological evaluation of chromenylurea and chromanylurea derivatives as anti-TNF-α agents that target the p38 MAPK pathway. Molecules 2014, 19, 2004–2028. [Google Scholar] [CrossRef]
- Li, X.; Zhou, X.; Zheng, Z.; Zhong, W.; Xiao, J.; Li, S. Short Synthesis of 1-(3-tert-Butyl-1-phenyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl) Urea Derivatives. Synth. Commun. 2009, 39, 3999–4009. [Google Scholar] [CrossRef]
- Flynn, D.L.; Petillo, P.A. Preparation of 1-pyrazolyl-3-phenylurea p38 MAP Kinase Inhibitors as Antiinflammatory Medicaments. U.S. Patent Application No. 20050288286 A1 20051229, 29 December 2005. [Google Scholar]
- Flynn, D.L.; Petillo, P.A. Preparation of Pyrazolyl aryl Ureas as Modulators of the Protein Kinase Activation State for Treatment of Inflammation and Hyperproliferative Diseases. WO Patent Application No. 2006081034 A2 20060803, 3 August 2006. [Google Scholar]
- Flynn, D.L.; Petillo, P.A. Preparation of 1-pyrazolyl-3-phenylurea p38 MAP Kinase Inhibitors as Antiinflammatory Medicaments. U.S. Patent Application No. 20070191336 A1 20070816, 16 August 2007. [Google Scholar]
- Arai, T.; Ohno, M.; Inoue, H.; Hayashi, S.; Aoki, T.; Hirokawa, H.; Meguro, H.; Koga, Y.; Oshida, K.; Kainoh, M.; et al. Design and synthesis of novel p38α MAP kinase inhibitors: Discovery of pyrazole-benzyl ureas bearing 2-molpholinopyrimidine moiety. Bioorg. Med. Chem. Lett. 2012, 22, 5118–5122. [Google Scholar] [CrossRef]
- Longshaw, A.I.; Fordyce, E.A.F.; Onions, S.T.; King-Underwood, J.; Venable, J.D.; Walters, I. Preparation of Pyrazolyl-Urea Compounds as p38 MAP Kinase Inhibitors. WO Patent Application No. 2015121660, 20 August 2015. [Google Scholar]
- Charron, C.E.; Ito, K.; Rapeport, W.G. Ureido-pyrazole Derivatives for Use in the Treatment of Respiratory Disorders. WO Patent Application No. 2011158042, 22 December 2011. [Google Scholar]
- Charron, C.E.; Fenton, R.; Crowe, S.; Ito, K.; Strong, P.; Rapeport, G.; Ray, K. Preparation of Pyrazolylurea Compounds as Hemopoietic Cell Kinase (p59-HCK) Inhibitors for Treatment of Influenza Infection. WO Patent Application No. 2011/070369, 16 June 2011. [Google Scholar]
- Longshaw, A.I.; Fordyce, E.A.F.; Onions, S.T.; King-Underwood, J.; Venable, J.D. Preparation of Aromatic Heterocyclic Compounds as p38 MAP Kinase Inhibitors with Antiinflammatory Activity. WO Patent Application No. 2015121444, 20 August 2015. [Google Scholar]
- King-Underwood, J.; Murray, P.J.; Williams, J.G.; Buck, I.; Onions, S.T. Preparation of Pyrazolyl Naphthyl Ureas as p38 MAP Kinase Inhibitors. WO Patent Application No. 2011124930, 13 October 2011. [Google Scholar]
- King-Underwood, J.; Ito, K.; Strong, P.; Rapeport, W.G.; Charron, C.E.; Murray, P.J.; Williams, J.G.; Onions, S.T. 1-(5-Pyrazolyl)-3-(1-naphthyl)ureas as Enzyme Inhibitors and their Preparation, Pharmaceutical Compositions and Use in the Treatment of Inflammatory Disorders. WO Patent Application No. 2011124923, 13 October 2011. [Google Scholar]
- Ito, K.; Strong, P.; Rapeport, W.G.; Murray, P.J.; King-Underwood, J.; Williams, J.G.; Onions, S.T.; Joly, K.; Charron, C.E. Preparation of Pyrazolyl Ureas as p38 MAP Kinase Inhibitors. WO Patent Application No. 2010067130, 21 April 2011. [Google Scholar]
- Ito, K.; Charron, C.E.; King-Underwood, J.; Onions, S.T.; Longshaw, A.I.; Broeckx, R.; Filliers, W.; Copmans, A. Preparation of 1-pyrazolyl-3-{4-[(2-anilinopyrimidin-4-yl)oxy]naphththalen-1-yl}ureas as p38 MAP Kinase Inhibitors. WO Patent Application No. 2013050757, 11 April 2013. [Google Scholar]
- Duffy, L.A.; King-Underwood, J.; Longshaw, A.I.; Murray, P.J.; Onions, S.T.; Taddei, D.M.A.; Williams, J.G.; Ito, K.; Charron, C.E. Preparation of Pyrazole Ureas as p38 MAP Inhibitors. WO Patent Application No. 2014033448 A1 20140306, 6 March 2014. [Google Scholar]
- Cariou, C.A.M.; Charron, C.E.; Fordyce, E.A.F.; Hamza, D.; Fyfe, M.C.T.; Ito, K.; King-Underwood, J.; Murray, P.J.; Onions, S.T.; Thom, S.M.; et al. Preparation of Pyrazolyl-Urea Compounds as Kinase Inhibitors for Treatment of Inflammatory Diseases. WO Patent Application No. 2014027209, 20 February 2014. [Google Scholar]
- Bruno, O.; Brullo, C.; Bondavalli, F.; Schenone, S.; Ranise, A.; Arduino, N.; Bertolotto, M.B.; Montecucco, F.; Ottonello, L.; Dallegri, F.; et al. Synthesis and biological evaluation of N-pyrazolyl-N′-alkyl/benzyl/phenylureas: A new class of potent inhibitors of interleukin 8-induced neutrophil chemotaxis. J. Med. Chem. 2007, 50, 3618–3626. [Google Scholar] [CrossRef]
- Bruno, O.; Brullo, C.; Bondavalli, F.; Schenone, S.; Spisani, S.; Falzarano, M.S.; Varani, K.; Barocelli, E.; Ballabeni, V.; Giorgio, C.; et al. 1-Methyl and 1-(2-hydroxyalkyl)-5-(3-alkyl/cycloalkyl/phenyl/naphthylureido)-1H-pyrazole-4-carboxylic acid ethyl esters as potent human neutrophil chemotaxis inhibitors. Bioorg. Med. Chem. 2009, 17, 3379–3387. [Google Scholar] [CrossRef]
- Bertolotto, M.; Brullo, C.; Ottonello, L.; Montecucco, F.; Dallegri, F.; Bruno, O. Treatment with 1-(2-hydroxyalkyl)-5-(3-alkyl/phenyl/naphthylureido)-1Hpyrazole-4-carboxylic acid ethyl esters abrogates neutrophil migration towards fMLP and CXCL8 via the inhibition of defined signalling pathways. Eur. J. Clin. Invest. 2014, 44, 39. [Google Scholar]
- Bruno, O.; Bondavalli, F.; Brullo, C.; Schenone, S. Ureas Derivatives of 1H-pyrazol-4-carboxylic Acid with Neutrophil Chemotaxis Inhibiting Activity. EP Patent Application No. 2033955 A1, 11 March 2009. [Google Scholar]
- Brullo, C.; Spisani, S.; Selvatici, R.; Bruno, O. N-Aryl-2-phenyl-2,3-dihydro-imidazo[1,2-b]pyrazole-1-carboxamides 7-substituted strongly inhibiting both fMLP-Ome- and IL-8-induced human neutrophil chemotaxis. Eur. J. Med. Chem. 2012, 47, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Selvatici, R.; Brullo, C.; Bruno, O.; Spisani, S. Differential inhibition of signaling pathways by two new imidazo-pyrazoles molecules in fMLF-Ome- and IL8-stimulated human neutrophil. Eur. J. Pharmacol. 2013, 718, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Meta, E.; Brullo, C.; Sidibé, A.; Imhof, B.A.; Bruno, O. Design, synthesis and biological evaluation of new pyrazolyl-ureas and imidazopyrazolecarboxamides able to interfere with MAPK and PI3K upstream signaling involved in the angiogenesis. Eur. J. Med. Chem. 2017, 133, 24–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meta, E.; Imhof, B.A.; Roprazb, P.; Fish, R.J.; Brullo, C.; Bruno, O.; Sidibé, A. The pyrazolyl-urea GeGe3 inhibits tumor angiogenesis and reveals dystrophia myotonica protein kinase (DMPK)1 as a novel angiogenesis target. Oncotarget 2017, 8, 108195–108212. [Google Scholar] [CrossRef] [Green Version]
- Abd El-Meguid, E.A.; Ali, M.M. Synthesis of some novel 4-benzothiazol-2-ylbenzoyl-1H-pyrazoles, and evaluation as antiangiogenic agents. Res. Chem. Intermediat. 2016, 42, 1521–1536. [Google Scholar] [CrossRef]
- Flynn, D.L.; Petillo, P.A. Preparation of Pyrazolyl Phenyl Ureas as Enzyme Modulators. WO Patent Apllication No. 2006071940 A2 20060706, 6 July 2006. [Google Scholar]
- Flynn, D.L.; Petillo, P.A. Preparation of Pyrazolyl Phenyl Ureas as Enzyme Modulators. U.S. Patent Application No. 20080113967 A1 20080515, 15 May 2008. [Google Scholar]
- Smith, R.; Hatoum-Mokdad, H.N.; Cantin, L.D.; Bierer, D.E.; Fu, W.; Nagarathnam, D.; Ladouceur, G.; Wang, Y.; Ogutu, H.; Wilhelm, S.; et al. Pyrazole Urea Compounds Useful in the Treatment of Cancer and their Preparation. WO Patent Application No. 2007064872, 7 June 2007. [Google Scholar]
- Flynn, D.L.; Petillo, P.A.; Kaufman, M.D. Preparation of Urea Derivatives as Kinase Inhibitors Useful for the Treatment of Myeloproliferative Diseases and Other Proliferative Diseases. U.S. Patent Application No. 7790756 B2 20100907, 7 September 2010. [Google Scholar]
- Flynn, D.L.; Petillo, P.A.; Kaufman, M.D. Preparation of Heterocyclic Urea Derivatives as Kinase Inhibitors Useful for the Treatment of Myeloproliferative Diseases and Other Proliferative Diseases. WO Patent Application No. 2013036232 A2 20130314, 14 March 2013. [Google Scholar]
- Getlik, M.; Gruetter, C.; Simard, J.R.; Kluter, S.; Rabiller, M.; Rode, H.B.; Robubi, A.; Rauh, D. Hybrid Compound Design to Overcome the Gatekeeper T338M Mutation in cSrc. J. Med. Chem. 2009, 52, 3915–3926. [Google Scholar] [CrossRef]
- Indrasena, R.K.; Aruna, C.; Manisha, M.; Srihari, K.; Sudhakar, B.K.; Vijayakumar, V.; Sarveswari, S.; Priya, R.; Amrita, A.; Siva, R. Synthesis, DNA binding and in-vitro cytotoxicity studies on novel bis-pyrazoles. J. Photochem. Photobiol. B Biol. 2017, 168, 89–97. [Google Scholar] [CrossRef]
- Kessler, J.D.; Kahle, K.T.; Sun, T.; Meerbrey, K.L.; Schlabach, M.R.; Schmitt, E.M.; Skinner, S.O.; Xu, Q.; Li, M.Z.; Hartman, Z.C.; et al. A SUMOylation-dependent Transcriptional Subprogram Is Required for Myc-driven Tumorigenesis. Science 2012, 335, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Ito, A.; Hirohama, M.; Yoshida, M.; Zhang, K.Y.J. Identification of new SUMO activating enzyme 1 inhibitors using virtual screening and scaffold hopping. Bioorg. Med. Chem. Lett. 2016, 26, 1218–1223. [Google Scholar] [CrossRef] [Green Version]
- Benito-Gutiérrez, È.; Garcia-Fernàndez, J.; Comella, J.X. Origin and Evolution of the Trk Family of Neurotrophic Receptors. Mol. Cell. Neurosci. 2006, 31, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Brandhuber, B.J.; Jiang, Y.; Kolakowski, G.R.; Winski, S.L. Pyrazolyl Urea, Thiourea, Guanidine and Cyanoguanidine Compounds as TrkA Kinase Inhibitors and their Preparation. WO Patent Application No. 2014078417 A1 20140522, 22 May 2014. [Google Scholar]
- Allen, S.; Andrews, S.W.; Baer, B.; Crane, Z.; Liu, W.; Watson, D.J. Preparation of 1-((3S,4R)-4-(3-fluorophenyl)-1-(2-methoxyethyl)pyrrolidin-3-yl)-3-(4-methyl-3-(2-methylpyrimidin-5-yl)-1-phenyl-1H-pyrazol-5-yl)urea as a TRKA Kinase Inhibitor. WO Patent Application No. 2015175788 A1 20151119, 19 November 2015. [Google Scholar]
- Subramanian, G.; Johnson, P.D.; Zachary, T.; Roush, N.; Zhu, Y.; Bowen, S.J.; Janssen, A.; Duclos, B.A.; Williams, T.; Javens, C.; et al. Deciphering the Allosteric Binding Mechanism of the Human Tropomyosin Receptor Kinase A (hTrkA) Inhibitors. ACS Chem. Biol. 2019, 14, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Furuya, N.; Momose, T.; Katsuno, K.; Fushimi, N.; Muranaka, H.; Handa, C.; Ozawa, T.; Kinoshita, T. The juxtamembrane region of TrkA kinase is critical for inhibitor selectivity. Bioorg. Med. Chem. Lett. 2017, 27, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Shikano, K.; Yamaguchi, H.; Horiguchi, T.; Yasuo, K.; Hata, K. Pain Therapeutic or Preventive Containing Fused Heterocycles and Heterocycle-Fused Carbocycles Having TrkA Inhibitory Activity. JP Patent Application No. 2019189573, 31 October 2019. [Google Scholar]
- Yukimasa, A.; Shikano, K.; Horiguchi, T.; Nakamura, K.; Inoue, T.; Fujo, M.; Yamaguchi, H. Pain Treatment and/or Preventive Agent Containing Nitrogen-Containing Heterocycles Having Tropomyosin Receptor Kinase A (TrkA)-Inhibitory Activity. JP Patent Application No. 2019026646, 21 February 2019. [Google Scholar]
- Vaidya, A.B.; Welsh, W.J.; Kortagere, S.; Bergman, L.W. Mechanism-Based Small Heterocyclic Molecule Parasite Inhibitors. WO Patent Application No. 2009065096 A1 20090522, 22 May 2009. [Google Scholar]
- Welsh, W.J.; Kortagere, S.; Bergman, L.W.; Vaidya, A.B. Mechanism-Based Small-Molecule Parasite Inhibitors. U.S. Patent Application No. 8486987 B2 20130716, 16 July 2013. [Google Scholar]
- Kortagere, S.; Welsh, W.J. Development and application of hybrid structure-based method for efficient screening of ligands binding to G-protein coupled receptors. J. Comput.-Aided. Mol. Des. 2006, 20, 789–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortagere, S.; Welsh, W.J.; Morrisey, J.M.; Daly, T.; Ejigiri, I.; Sinnis, P.; Vaidya, A.B.; Bergman, L.W. Structure-based design of novel small-molecule inhibitors of Plasmodium falciparum. J. Chem. Inf. Model. 2010, 50, 840–849. [Google Scholar] [CrossRef] [Green Version]
- Kuhlbrandt, W. Biology, structure, and mechanism of P-type ATPases. Nat. Rev. Mol. Cell Biol. 2004, 5, 282–295. [Google Scholar] [CrossRef]
- Yatime, L.; Buch-Pedersen, M.J.; Musgaard, M.; Morth, J.P.; Lund Winther, A.M.; Pedersen, B.P.; Olesen, C.; Andersen, J.P.; Vilsen, B.; Schiott, B.; et al. P-type ATPases as drug targets: Tools for medicine and science. Biochim. Biophys. Acta 2009, 1787, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Navarro, A.; Benito, B. Sodium or potassium efflux ATPase a fungal, bryophyte, and protozoal ATPase. Biochim. Biophys. Acta 2010, 1798, 1841–1853. [Google Scholar] [CrossRef] [Green Version]
- Spillman, N.J.; Allen, R.J.; McNamara, C.W.; Yeung, B.K.; Winzeler, E.A.; Diagana, T.T.; Kirk, K. Na(+) regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe 2013, 13, 227–237. [Google Scholar] [CrossRef] [Green Version]
- White, N.J.; Pukrittayakamee, S.; Phyo, A.P.; Rueangweerayut, R.; Nosten, F.; Jittamala, P.; Jeeyapant, A.; Jain, J.P.; Lefevre, G.; Li, R.; et al. Spiroindolone KAE609 for falciparum and vivax malaria. N. Engl. J. Med. 2014, 371, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Flannery, E.L.; Fidock, D.A.; Winzeler, E.A. Using genetic methods to define the targets of compounds with antimalarial activity. J. Med. Chem. 2013, 56, 7761–7771. [Google Scholar] [CrossRef] [Green Version]
- Flannery, E.L.; McNamara, C.W.; Kim, S.W.; Kato, T.S.; Li, F.; Teng, C.H.; Gagaring, K.; Manary, M.J.; Barboa, R.; Meister, S.; et al. Mutations in the P-Type Cation-Transporter ATPase 4, PfATP4, Mediate Resistance to Both Aminopyrazole and Spiroindolone Antimalarials. ACS Chem. Biol. 2015, 10, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.N.; Dranow, D.M.; Horst, B.G.; Abendroth, J.A.; Forte, B.; Hallyburton, I.; Jansen, C.; Baragana, B.; Choi, R.; Rivas, K.L.; et al. Biochemical and Structural Characterization of Selective Allosteric Inhibitors of the Plasmodium falciparum Drug Target, Prolyl-tRNA-synthetase. ACS Infect. Dis. 2017, 3, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortagere, S.; Mui, E.; McLeod, R.; Welsh, W.J. Rapid discovery of inhibitors of Toxoplasma gondii using hybrid structure-based computational approach. J. Comput.-Aided Mol. Des. 2011, 25, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.L.; Hirth, B.H.; Liang, B.; Gourlie, B.B.; Nahill, S.; Barsomian, G. Ureas of 5-aminopyrazole and 2-aminothiazole inhibit growth of gram-positive bacteria. Bioorg. Med. Chem. Lett. 2003, 13, 4463–4466. [Google Scholar] [CrossRef]
- Pedro-Rosa, L.; Buckner, F.S.; Ranade, R.M.; Eberhart, C.; Madoux, F.; Gillespie, J.R.; Koh, C.Y.; Brown, S.; Lohse, J.; Verlinde, C.L.M.; et al. Identification of potent inhibitors of the Trypanosoma brucei methionyl-tRNA synthetase via high-throughput orthogonal screening. J. Biomol. Screen. 2015, 20, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Onions, S.T.; Williams, J.G.; Joly, K. Preparation of Pyrido[2,3-b]pyrazine Compounds for Use in Drug Formulations for Treating Inflammation, Respiratory Disorders, and Viral Infections. WO Patent Application No. 2011158044 A2 20111222, 22 December 2011. [Google Scholar]
- Luscher, C.; Slesinger, P. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 2010, 11, 301–315. [Google Scholar] [CrossRef]
- Niswender, C.M.; Johnson, K.A.; Luo, Q.; Ayala, J.E.; Kim, C.; Conn, P.J.; Weaver, C.D. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 2008, 73, 1213–1224. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, K.; Romaine, I.; Days, E.; Pascual, C.; Malik, A.; Yang, L.; Zou, B.; Du, Y.; Sliwoski, G.; Morrison, R.D.; et al. ML297 (VU0456810), the first potent and selective activator of the GIRK potassium channel, displays antiepileptic properties in mice. ACS Chem. Neurosci. 2013, 4, 1278–1286. [Google Scholar] [CrossRef] [Green Version]
- Wieting, J.M.; Vadukoot, A.K.; Sharma, S.; Abney, K.K.; Bridges, T.M.; Daniels, J.S.; Morrison, R.D.; Wickman, K.; Weaver, C.D.; Hopkins, C.R. Discovery and Characterization of 1H-Pyrazol-5-yl-2-phenylacetamides as Novel, Non-Urea-Containing GIRK1/2 Potassium Channel Activators. ACS Chem. Neurosci. 2017, 8, 1873–1879. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Affinity Ki (nM) | Selectivity | ||
|---|---|---|---|
| Comp. 4 | CB1 | CB2 | CB1 |
| a | 9 | 4920 | 546.7 |
| b | 29 | 10863 | 374.6 |
| c | 6.8 | 4319 | 635.2 |
| d | 26 | 21791 | 838.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brullo, C.; Rapetti, F.; Bruno, O. Pyrazolyl-Ureas as Interesting Scaffold in Medicinal Chemistry. Molecules 2020, 25, 3457. https://doi.org/10.3390/molecules25153457
Brullo C, Rapetti F, Bruno O. Pyrazolyl-Ureas as Interesting Scaffold in Medicinal Chemistry. Molecules. 2020; 25(15):3457. https://doi.org/10.3390/molecules25153457
Chicago/Turabian StyleBrullo, Chiara, Federica Rapetti, and Olga Bruno. 2020. "Pyrazolyl-Ureas as Interesting Scaffold in Medicinal Chemistry" Molecules 25, no. 15: 3457. https://doi.org/10.3390/molecules25153457
APA StyleBrullo, C., Rapetti, F., & Bruno, O. (2020). Pyrazolyl-Ureas as Interesting Scaffold in Medicinal Chemistry. Molecules, 25(15), 3457. https://doi.org/10.3390/molecules25153457