Structure-Based Virtual Screening, Synthesis and Biological Evaluation of Potential FAK-FAT Domain Inhibitors for Treatment of Metastatic Cancer


Abstract
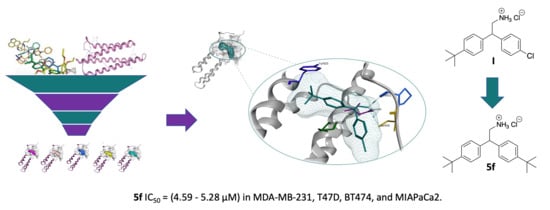
:
1. Introduction
2. Results and Discussion
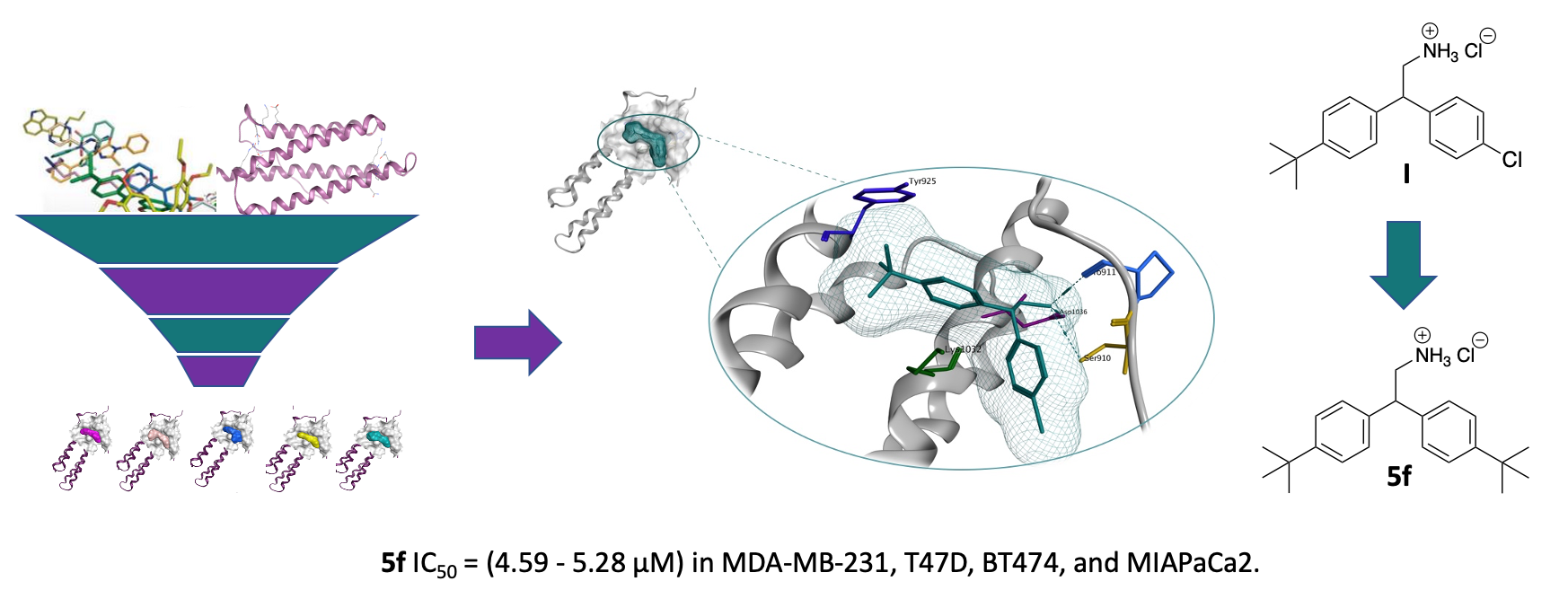
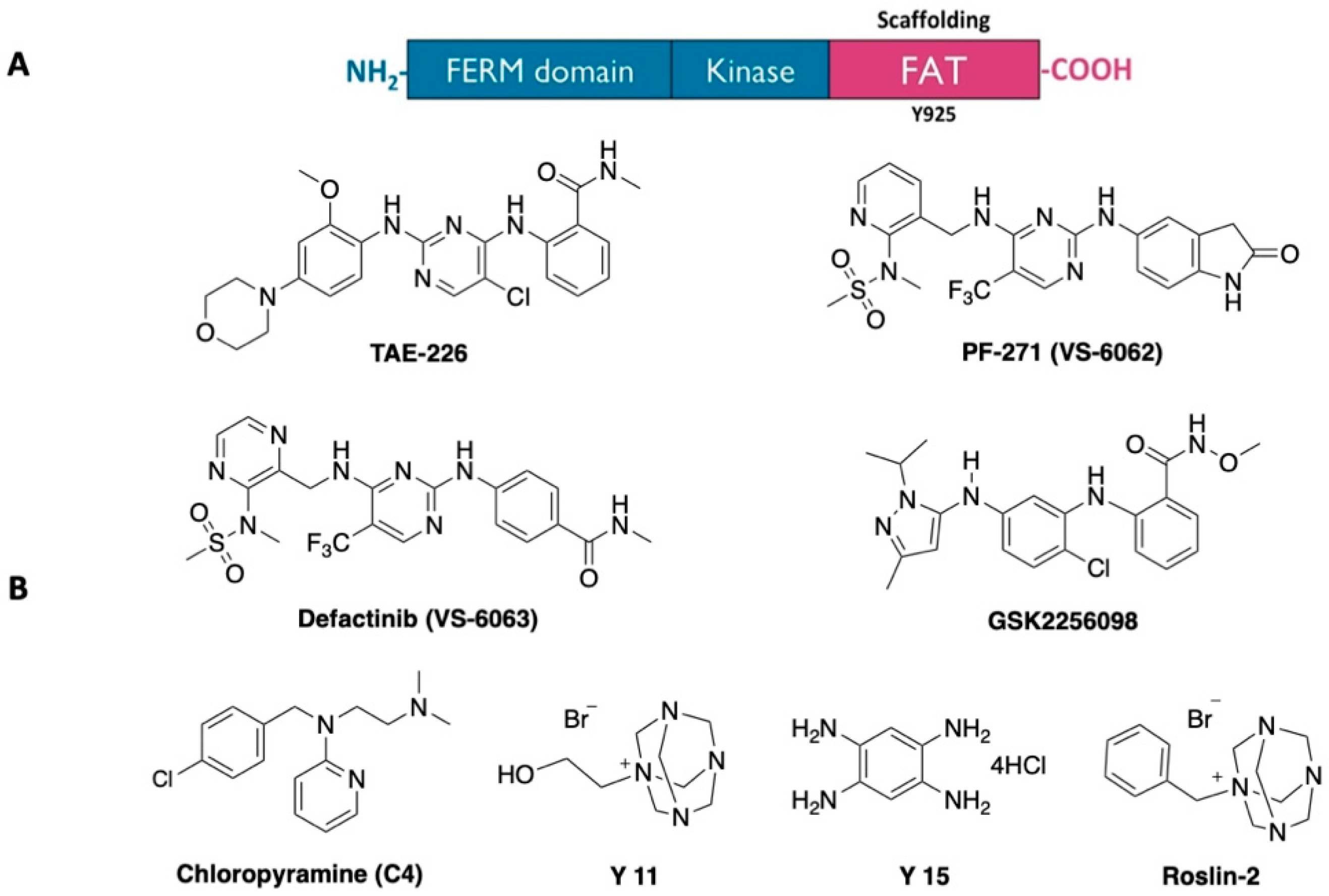
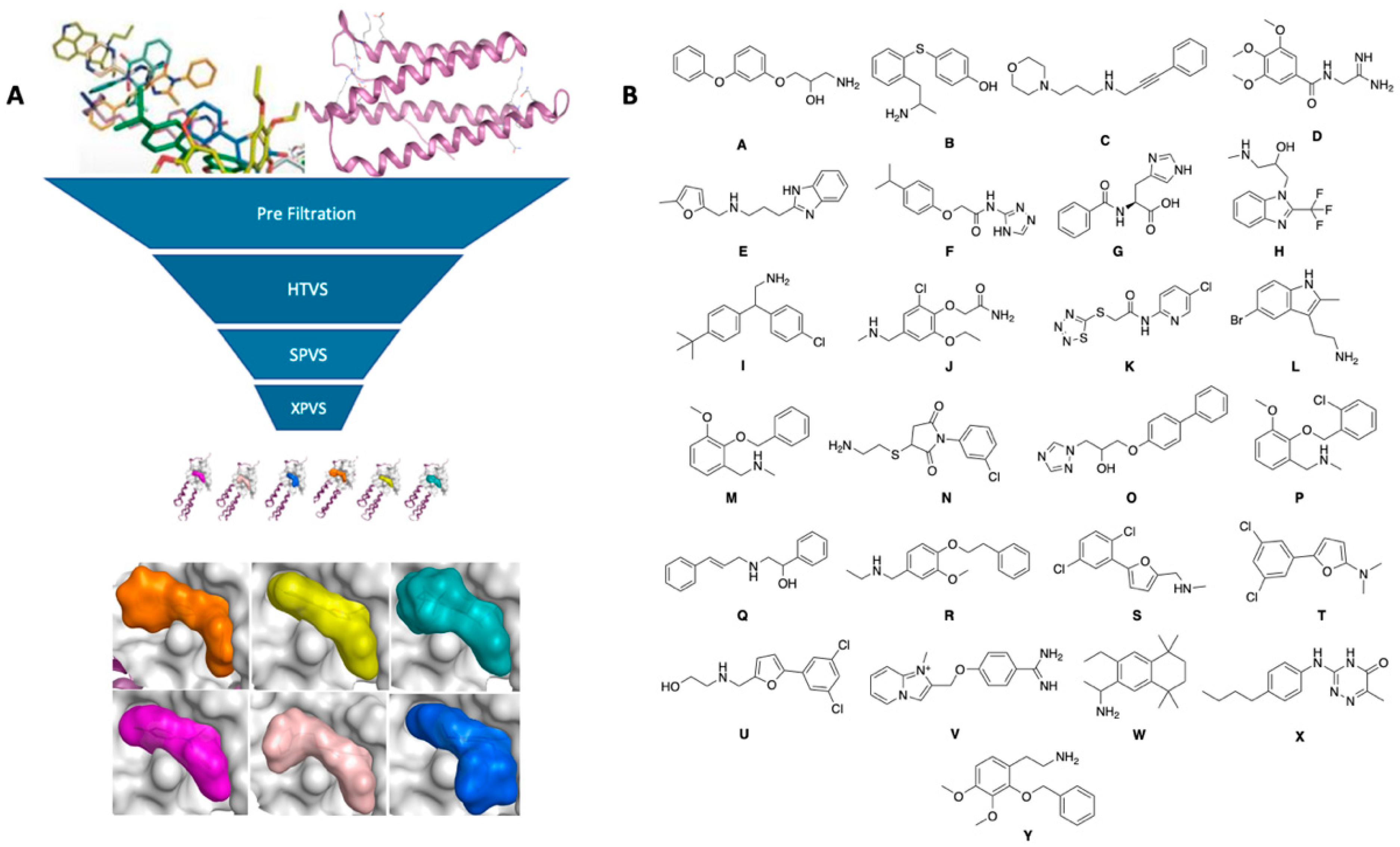
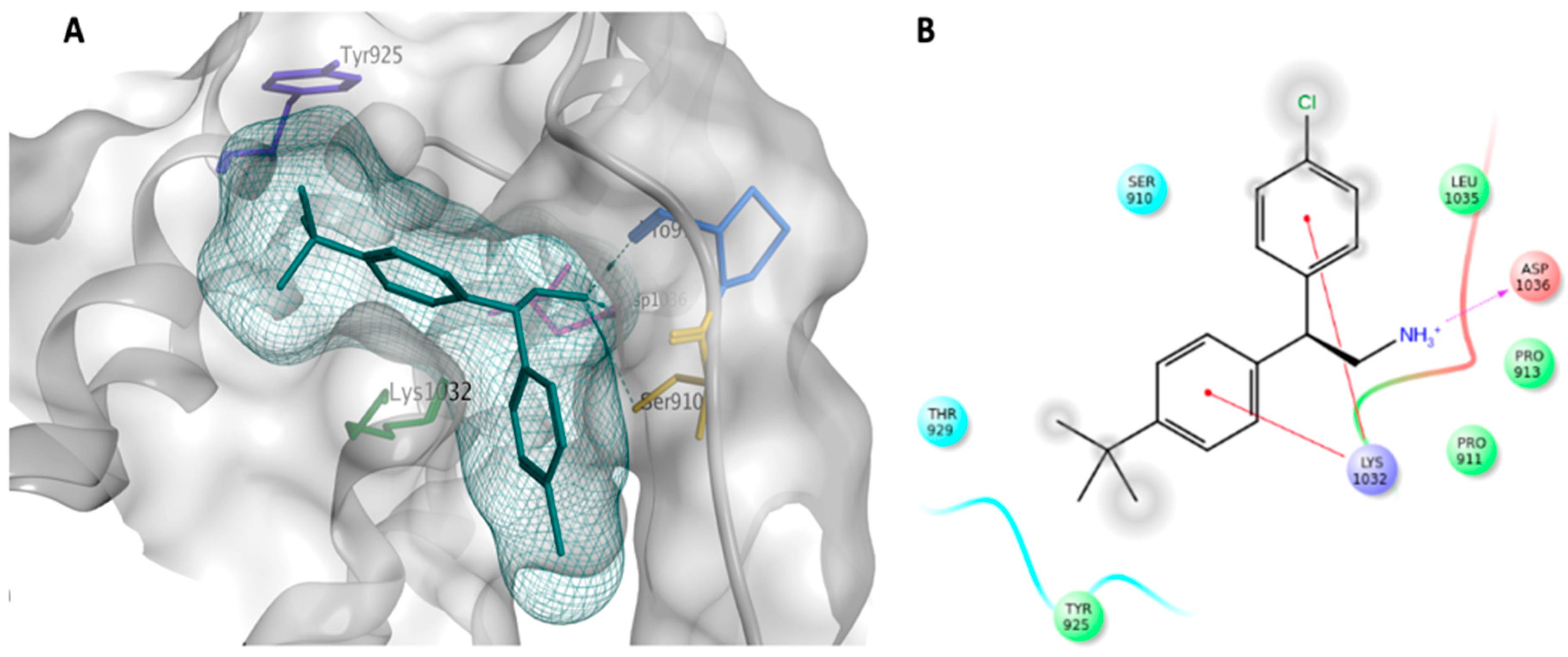
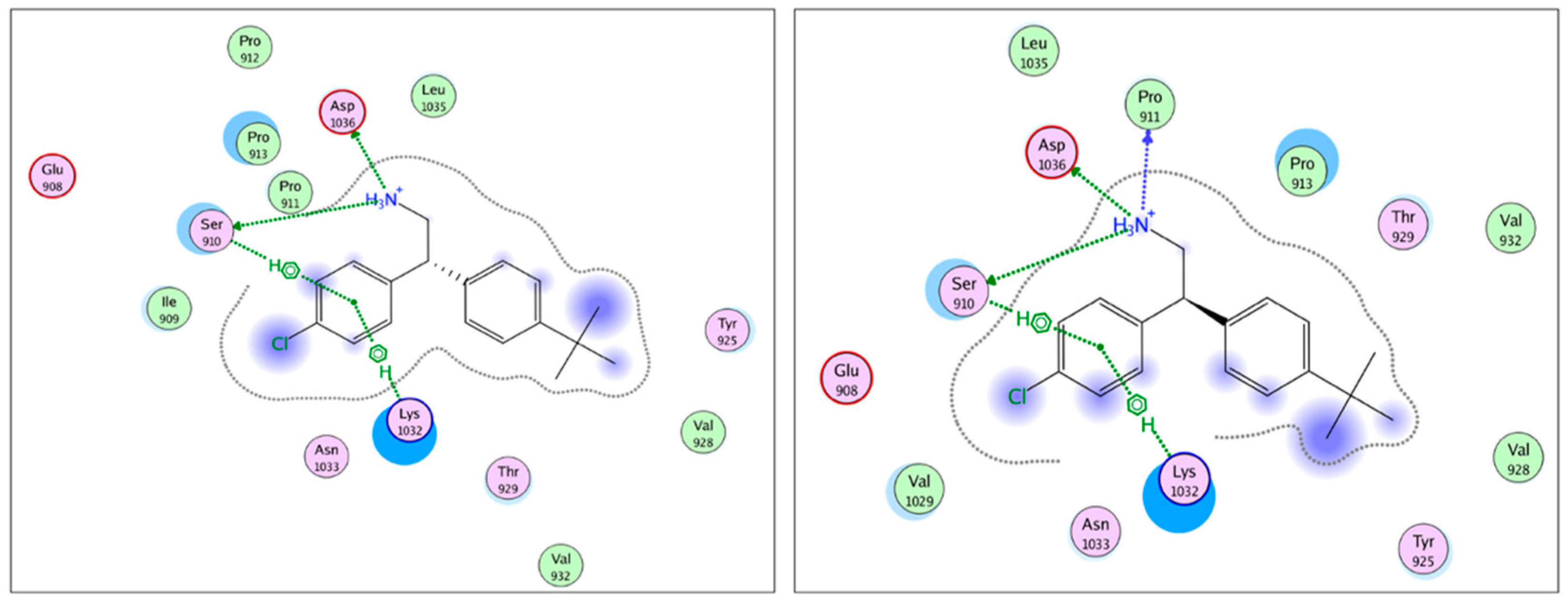
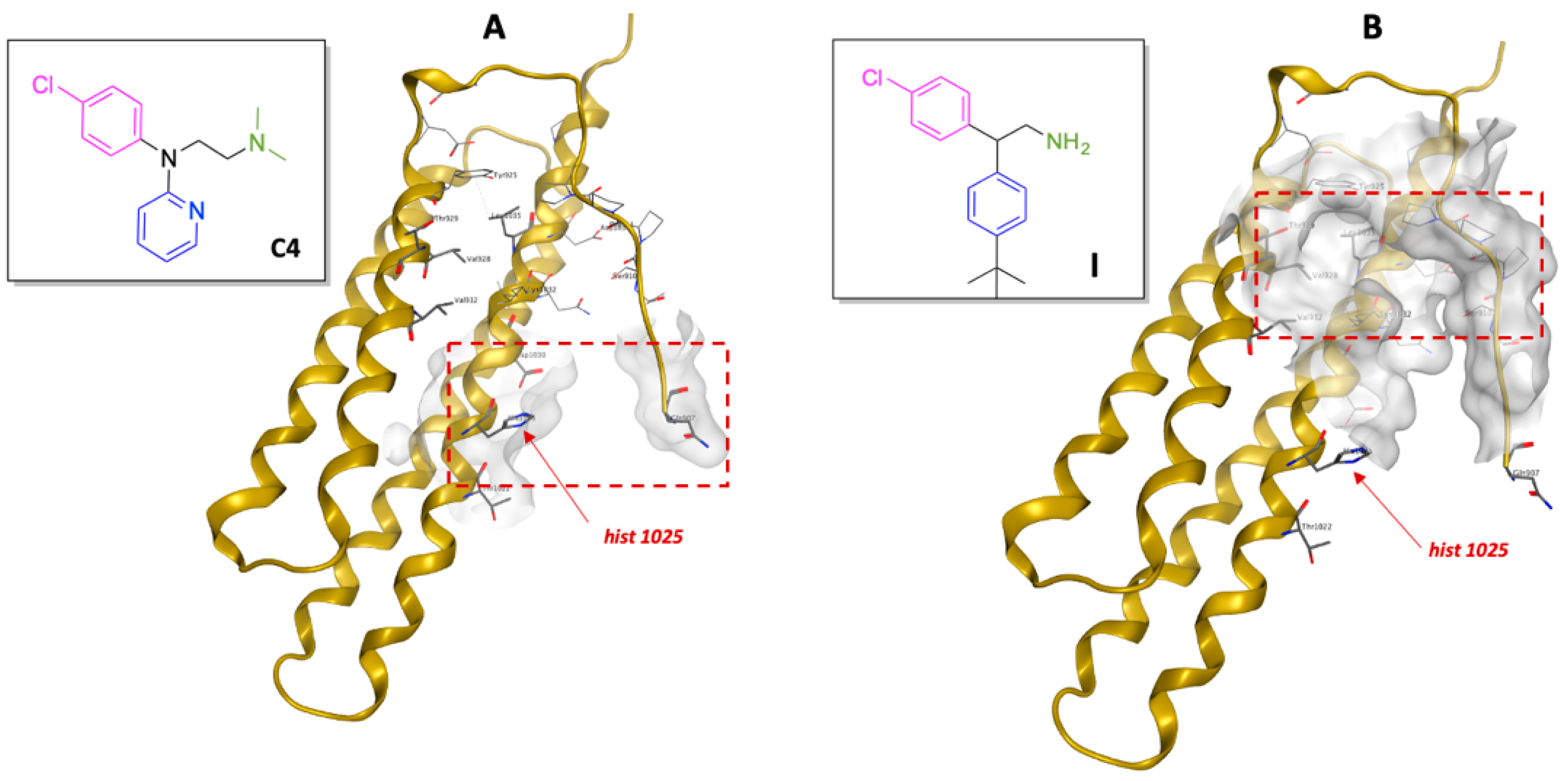
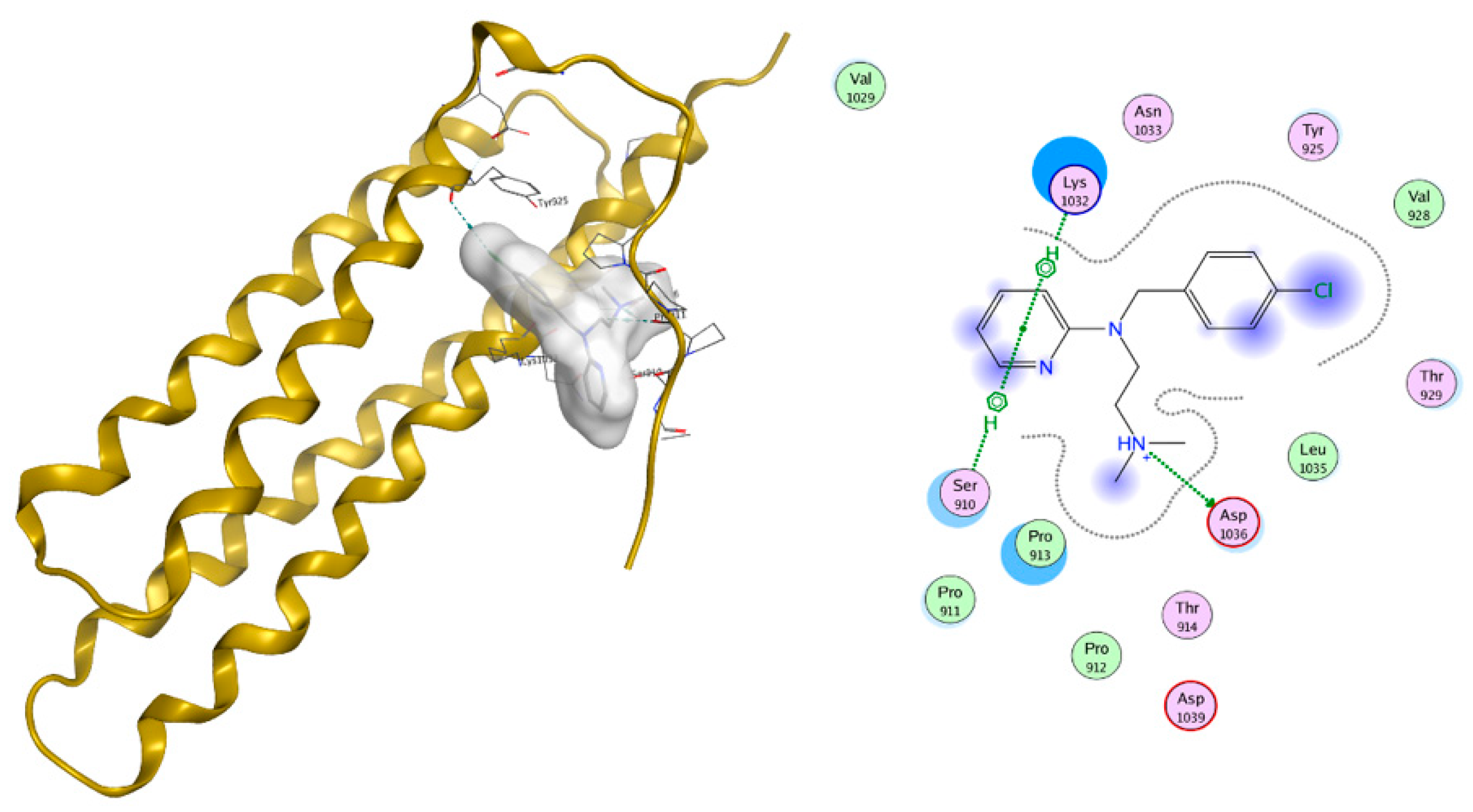
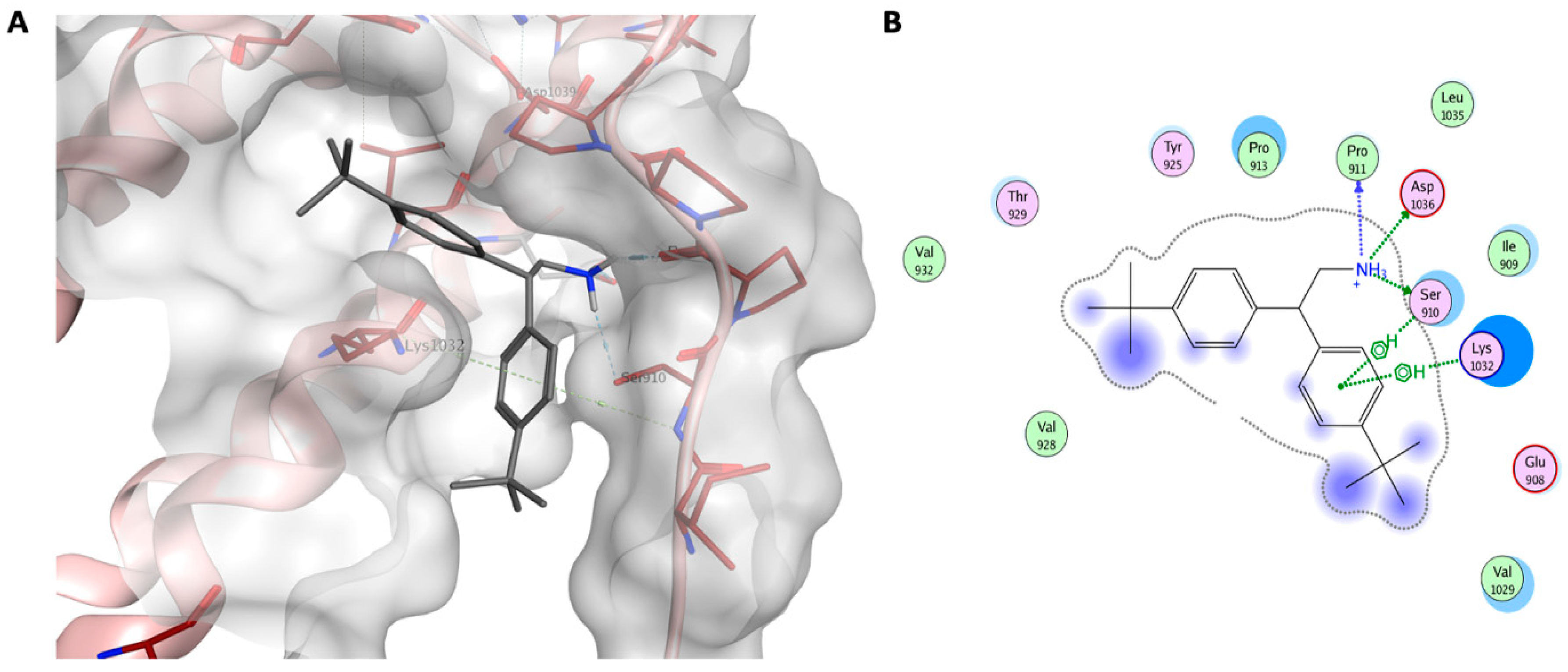
2.1. Virtual Screening (Molecular Modelling)
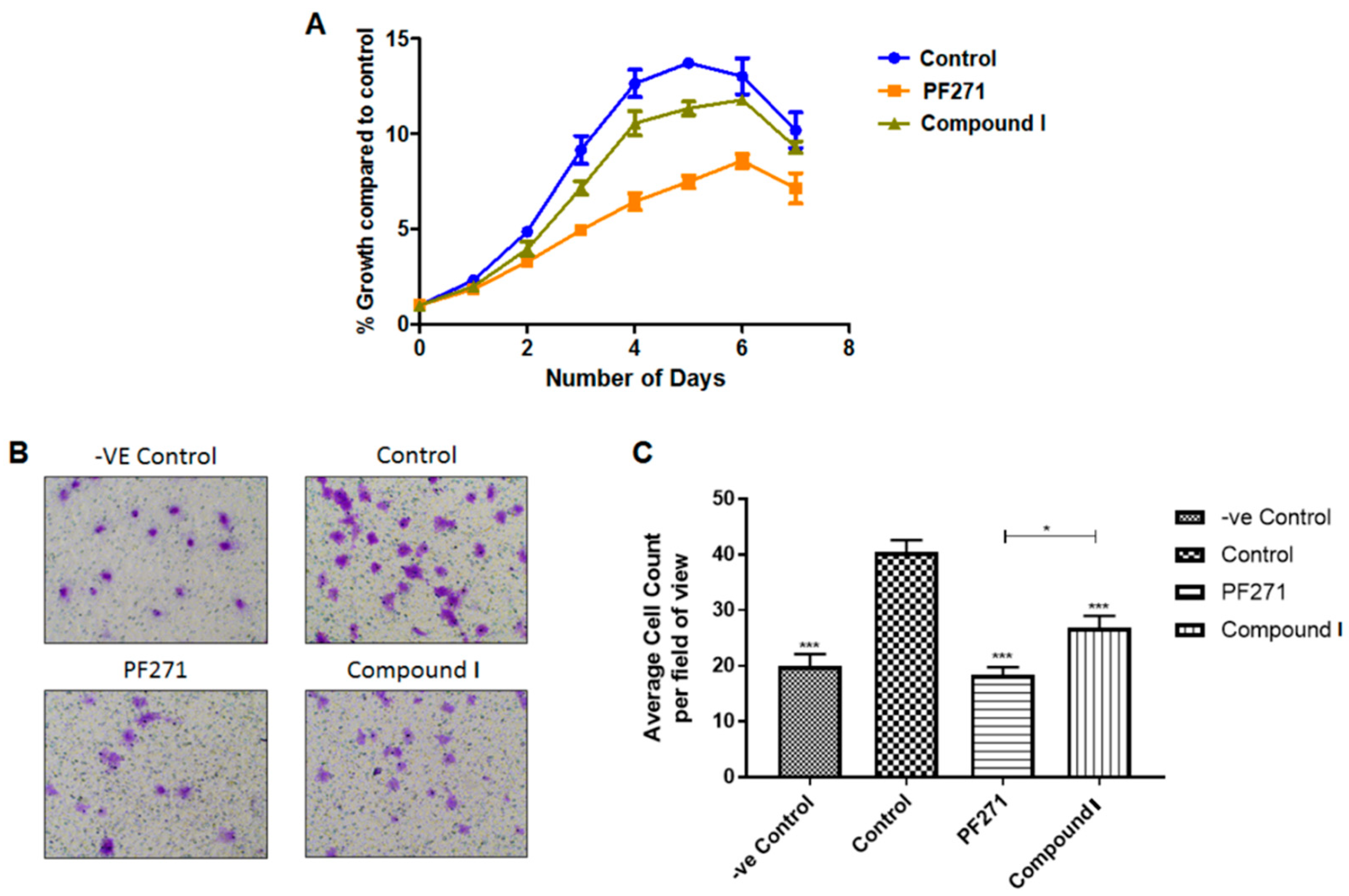
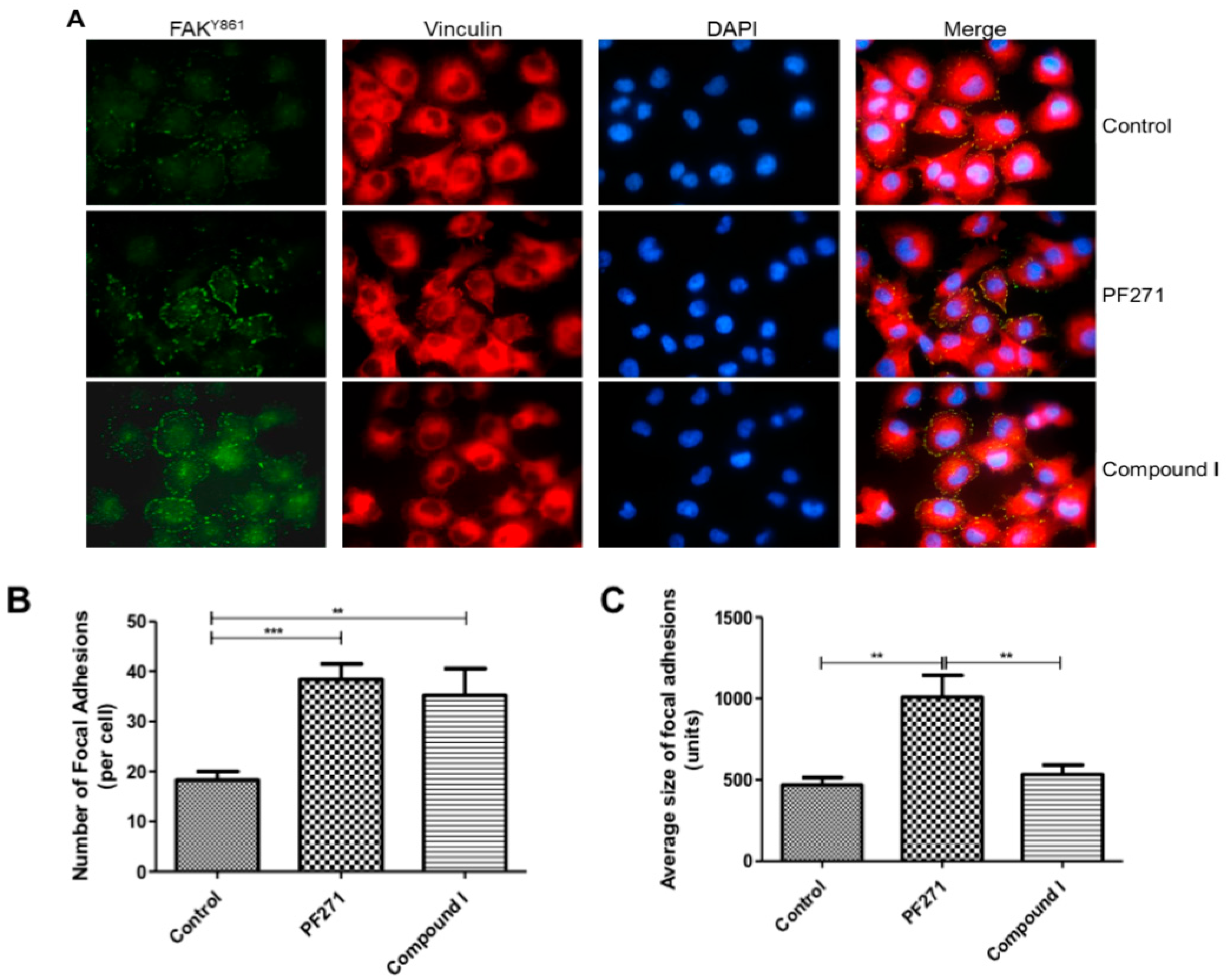
2.2. Characterisation of the Cellular Effects of Compound I
2.2.1. Compound I Inhibits TNBC Cell Proliferation and Migration
2.2.2. Compound I Causes Changes in Cellular Morphology and Cellular Localisation of Active FAK
2.3. Chemistry
2.4. Cell Viability Assay
2.5. Pharmacokinetic Studies
2.5.1. In Vitro Aqueous Solubility
2.5.2. Microsomal Metabolic Stability
2.5.3. Cardiotoxicity Assay
3. Materials and Methods
3.1. Molecular Modelling
3.2. Cell Proliferation Assay
3.3. Boyden Chamber Migration
3.4. Immunofluorescence
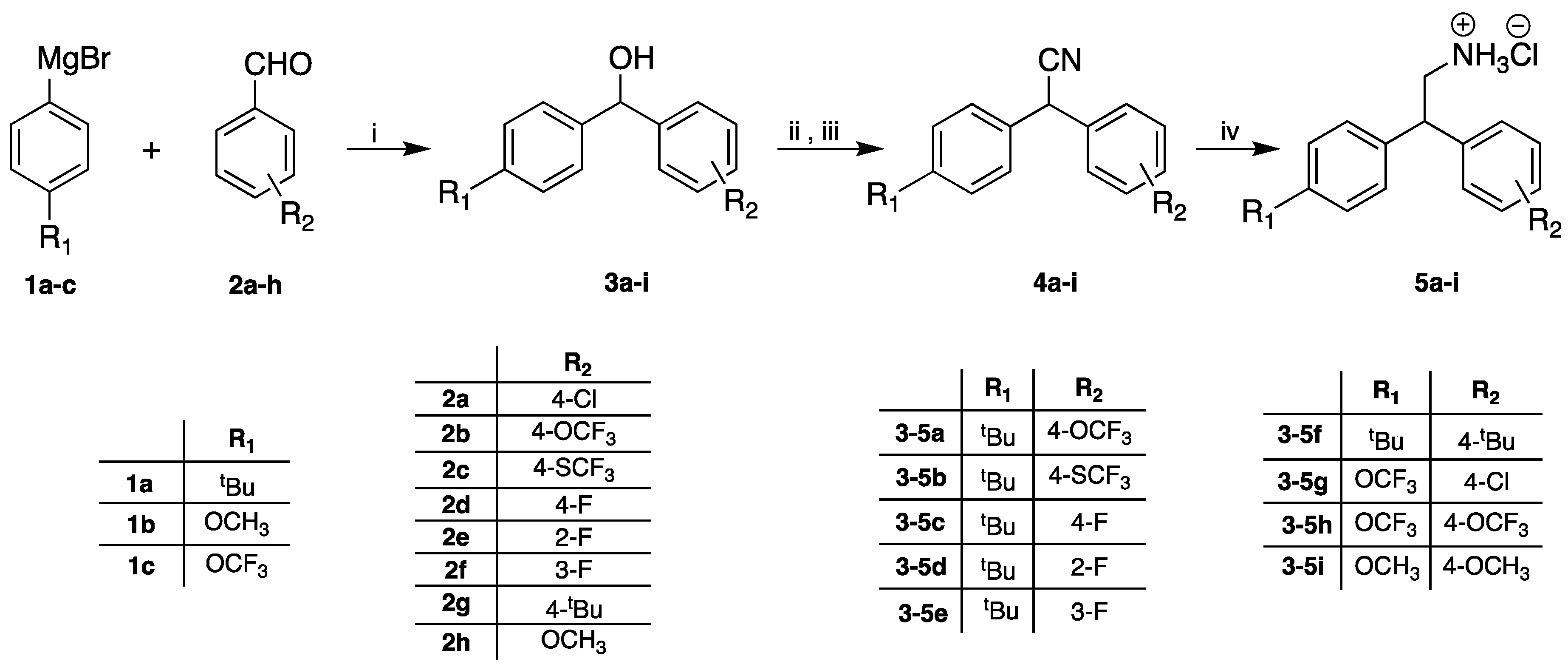
3.5. General Procedure for the Preparation of Diaryl Alcohols (3a–i)
3.6. General Procedure for Formation of Diaryl Nitrile (4a–i)
3.7. General Procedure for the Reduction of Nitrile to Amine Hydrochloride (5a–i)
3.8. In Vitro Aqueous Solubility, Metabolic Stability and Cardiotoxicity Studies
3.8.1. Turbidimetric Aqueous Solubility
Experimental Procedure
Data Analysis
3.8.2. Microsomal Metabolic Stability
Experimental Procedure
Quantitative Analysis
Data Analysis
3.8.3. In Vitro Cardiotoxicity; hERG Channel Inhibition (IC50 Determination)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sulzmaier, F.; Jean, C.; Schlaepfer, D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Oktay, M.H.; Oktay, K.; Hamele-Bena, D.; Buyuk, A.; Koss, L.G. Focal adhesion kinase as a marker of malignant phenotype in breast and cervical carcinomas. Hum. Pathol. 2003, 34, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Owens, L.V.; Xu, L.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumours. Cancer Res. 1995, 55, 2752–2755. [Google Scholar]
- Lark, A.L.; Livasy, C.A.; Calvo, B.; Caskey, L.; Moore, D.T.; Yang, X. Overexpression of focal adhesion kinase in primary colorectal carcinomas and colorectal liver metastases: Immuno- histochemistry and real-time PCR analyses. Clin. Cancer Res. 2003, 9, 215–222. [Google Scholar]
- Judson, P.L.; He, X.; Cance, W.G.; Van, L.L. Overexpression of focal adhesion kinase, a protein tyrosine kinase, in ovarian carcinoma. Cancer 1999, 86, 1551–1556. [Google Scholar] [CrossRef]
- Tremblay, L.; Hauck, W.; Aprikian, A.G.; Begin, L.R.; Chapdelaine, A.; Chevalier, S. Focal adhesion kinase (pp125FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int. J. Cancer 1996, 68, 164–171. [Google Scholar] [CrossRef]
- Weiner, T.M.; Liu, E.T.; Craven, R.J.; Cance, W.G. Expression of focal adhesion kinase gene and invasive cancer. Lancet 1993, 342, 1024–1025. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumour immunity. Cell. 2015, 163, 160–173. [Google Scholar] [CrossRef] [Green Version]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaller, M.D. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Guan, J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Mousson, A.; Sick, E.; Carl, P.; Dujardin, D.; De Mey, J.; Rondé, P. Targeting focal adhesion kinase using inhibitors of protein-protein interactions. Cancers 2018, 10, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Hjelmeland, A.B.; Keir, S.T.; Song, L.; Wickman, S.; Jackson, D.; Ohmori, O.; Bigner, D.D.; Friedman, H.S.; Rich, J.N. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol. Carcinog. 2007, 46, 488–496. [Google Scholar] [CrossRef]
- Liu, T.J.; LaFortune, T.; Honda, T.; Ohmori, O.; Hatakeyama, S.; Meyer, T.; Jackson, D.; de Groot, J.; Yung, W.K. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 1357–1367. [Google Scholar] [CrossRef] [Green Version]
- Halder, J.; Lin, Y.G.; Merritt, W.M.; Spannuth, W.A.; Nick, A.M.; Honda, T.; Kamat, A.A.; Han, L.Y.; Kim, T.J.; Lu, C. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007, 67, 10976–10983. [Google Scholar] [CrossRef] [Green Version]
- Lietha, D.; Eck, M.J. Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation. PLoS ONE 2008, 3, e3800. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Gao, C.; Gao, X.; Zhang, D.H.; Kuan, S.F.; Burns, T.F.; Hu, J. Wnt/beta-catenin pathway activation mediates adaptive resistance to BRAF inhibition in colorectal cancer. Mol. Cancer Ther. 2018, 17, 806–813. [Google Scholar] [CrossRef] [Green Version]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signalling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef]
- Kang, Y.; Hu, W.; Ivan, C.; Dalton, H.J.; Miyake, T.; Pecot, C.V.; Zand, B.; Liu, T.; Huang, J.; Jennings, N.B.; et al. Role of focal adhesion kinase in regulating YB1 mediated paclitaxel resistance in ovarian cancer. J. Natl Cancer Inst. 2013, 105, 1485–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.R.; Camidge, D.R.; Mileshkin, L.R.; Chen, E.X.; Hicks, R.J.; Rischin, D.; Fingert, H.; Pierce, K.J.; Xu, H.; Roberts, W.G.; et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF 00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J. Clin. Oncol. 2012, 30, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.-T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubovskaya, V.M.; Figel, S.; Ho, B.T.; Johnson, C.P.; Yemma, M.; Huang, G.; Zheng, M.; Nyberg, C.; Magis, A.; Ostrov, D.A.; et al. A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2 hydroxyethyl)-3, 5, 7 triaza-1 azoniatricyclo [3.3.1.1(3,7)]decane bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumour growth in vivo. Carcinogenesis 2012, 33, 1004–1013. [Google Scholar]
- Cance, W.G.; Kurenova, E.; Marlowe, T.; Golubovskaya, V. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. Sci. Signal 2013, 6, e10. [Google Scholar] [CrossRef] [Green Version]
- Golubovskaya, V.M.; Ho, B.; Zheng, M.; Magis, A.; Ostrov, D.; Morrison, C.; Cance, W.G. Disruption of focal adhesion kinase and p53 interaction with small molecule compound R2 reactivated p53 and blocked tumor growth. BMC Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [Green Version]
- Kurenova, E.V.; Hunt, D.L.; He, D.; Magis, A.T.; Ostrov, D.A.; Cance, W.G. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. J. Med. Chem. 2009, 52, 4716–4724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogate, P.N.; Ethirajan, M.; Kurenova, E.V.; Magis, A.T.; Pandey, R.K.; Cance, W.G. Design, synthesis and biological evaluation of novel FAK scaffold inhibitors targeting the FAK-VEGFR3 protein-protein interaction. Eur. J. Med. Chem. 2014, 80, 154–166. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Sood, A.K.; Armaiz-Pena, G.N.; Halder, J.; Nick, A.M.; Stone, R.L.; Hu, W.; Carroll, A.R.; Spannuth, W.A.; Deavers, M.T.; Allen, J.K.; et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J. Clin. Invest. 2010, 120, 1515–1523. [Google Scholar] [CrossRef] [Green Version]
- Ward, K.K.; Tancioni, I.; Lawson, C.; Miller, N.L.; Jean, C.; Chen, X.L.; Uryu, S.; Kim, J.; Tarin, D.; Stupack, D.G.; et al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumour progression. Clin. Exp. Metastasis 2013, 30, 579–594. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, K.K.; Sen, T.; Mandal, S.; Biswas, J.; Chatterjee, A. Studies on focal adhesion kinase in human breast cancer tissue. J. Cancer Ther. 2012, 3, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Rigiracciolo, D.C.; Santolla, M.F.; Lappano, R. Focal adhesion kinase (FAK) activation by estrogens involves GPER in triple-negative breast cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanteti, R.; Mirzapoiazova, T.; Riehm, J.J.; Dhanasingh, I.; Mambetsariev, B.; Wang, J.; Kulkarni, P.; Kaushik, G.; Seshacharyulu, P.; Ponnusamy, M.P.; et al. Focal adhesion kinase: A potential therapeutic target for pancreatic cancer and malignant pleural mesothelioma. Cancer Biol. Med. 2018, 19, 316–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, J.B.; Adair, S.J.; SlackDavis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of focal adhesion kinase by PF 562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumour microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Kiosses, W.; Sieg, D.; Otey, C.; Schlaepfer, D.; Schwartz, M. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 2000, 113, 3673–3678. [Google Scholar] [PubMed]
- Kandil, S.; Prencipe, F.; Jones, S.; Hiscox, S.; Westwell, A.D. The discovery of new and more potent chloropyramine (C4) analogues for the potential treatment of invasive breast cancer. Chem. Biol. Drug Des. 2018, 91, 314–321. [Google Scholar] [CrossRef]
- Arold, S.T.; Hoellerer, M.K.; Noble, M.E. The structural basis of localization and signalling by the focal adhesion targeting domain. Structure 2002, 10, 319–327. [Google Scholar] [CrossRef]
- Available online: http://cyprotex.com (accessed on 26 June 2020).
- Available online: www.specs.net (accessed on 26 June 2020).
Sample Availability: Samples of the compounds 5a–i are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
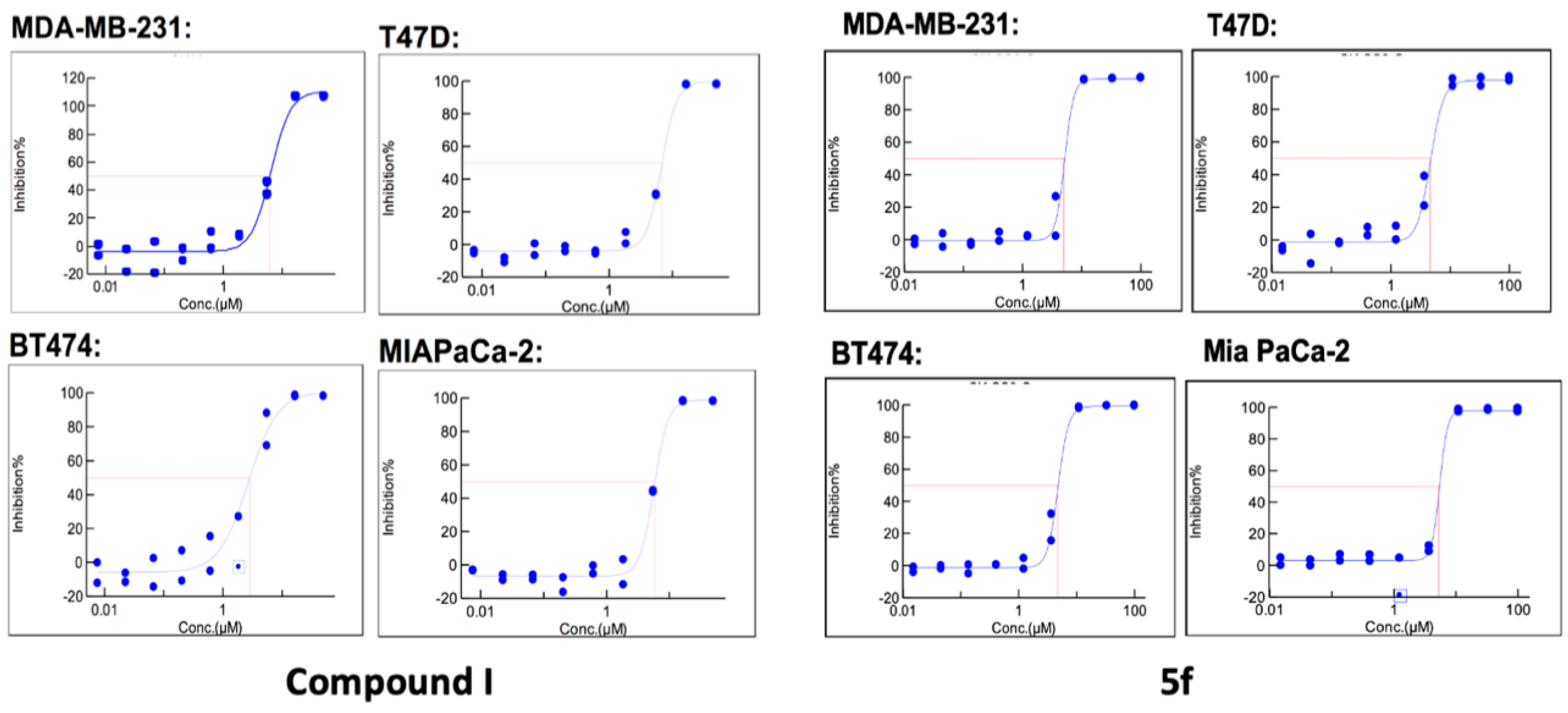
| ID | R1 | R2 | MDA-MB-231 ER− PR− HER− | T47D ER+ PR+/− HER− | BT474 ER+ PR+/− HER+ | MIAPaCa2 |
|---|---|---|---|---|---|---|
| I | 4-tBu | 4-Cl | 6.37 | 6.71 | 2.67 | 5.66 |
| 5a | 4-tBut | 4-OCF3 | 5.63 | 4.94 | 5.00 | 5.51 |
| 5b | 4-tBut | 4-SCF3 | 11.78 | 11.44 | 12.46 | 15.72 |
| 5c | 4-tBut | 4-F | 12.76 | 13.38 | 8.94 | 15.12 |
| 5d | 4-tBut | 2-F | 14.79 | 14.74 | 14.78 | 15.80 |
| 5e | 4-tBut | 3-F | 14.12 | 14.38 | 14.97 | 16.97 |
| 5f | 4-tBut | 4-tBut | 5.06 | 4.59 | 4.70 | 5.28 |
| 5g | 4-OCF3 | 4-Cl | 16.95 | 15.26 | 15.53 | 45.32 |
| 5h | 4-OCF3 | 4-OCF3 | 12.75 | 13.23 | 13.19 | 39.89 |
| 5i | 4-OMe | 4-OMe | 46.25 | 43.93 | 44.50 | 49.04 |
| C4 | - | - | 99.02 | >100 | >100 | >100 |
| ID | Estimated Precipitation Range (µM) | ||
|---|---|---|---|
| Lower Bound | Upper Bound | Calculated Mid-Range | |
| Compound I | 100 | >100 | >100 |
| 5a | 30 | 100 | 65 |
| 5b | 3 | 20 | 11.5 |
| 5c | 100 | >100 | >100 |
| 5f | 1 | 6.5 | 3.75 |
| 5h | 30 | 100 | 65 |
| nicardipine | 10 | 30 | 10.5 |
| pyrene | 3 | 10 | 6.5 |
| ID | Metabolic Stability (Human) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CLint (μL/min/mg protein) | SE CLint | t1/2 min | n | Compound Remaining (% of 0 min) | ||||||
| 0 min | 5 min | 15 min | 30 min | 45 min | Control | |||||
| Compound I | 9.10 | 3.35 | 152 | 5 | 100 | 89.4 | 92.3 | 77.9 | 81.3 | 95.7 |
| 5a | 8.91 | 0.532 | 156 | 5 | 100 | 95.8 | 92.3 | 86.0 | 81.4 | 91.0 |
| 5c | 8.26 | 1.15 | 168 | 5 | 100 | 94.7 | 95.0 | 86.1 | 82.5 | 98.0 |
| 5f | 9.34 | 0.699 | 148 | 5 | 100 | 97.2 | 92.2 | 88.0 | 80.7 | 88.0 |
| 5g | 13.9 | 2.14 | 99.9 | 5 | 100 | 99.0 | 96.2 | 86.8 | 72.8 | 86.6 |
| 5h | 2.35 | 2.31 | 589 | 5 | 100 | 102 | 108 | 100 | 95.8 | 97.8 |
| dextromethorphan | 28.2 | 3.73 | 49.2 | 5 | - | - | - | - | - | - |
| verapamil | 187 | 2.82 | 7.40 | 3 | - | - | - | - | - | - |
| ID | hERG Channel Inhibition | ||
|---|---|---|---|
| IC50 (µM) | SE IC50 (µM) | n | |
| Compound I | 5.47 | 0.616 | 15 |
| 5a | 3.59 | 0.833 | 11 |
| 5f | >25 | - | 12 |
| 5i | 6.01 | 1.22 | 11 |
| quinidine | 1.39 | 0.366 | 12 |
| dimethylsulfoxide | >25 | - | 15 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandil, S.B.; Jones, S.R.; Smith, S.; Hiscox, S.E.; Westwell, A.D. Structure-Based Virtual Screening, Synthesis and Biological Evaluation of Potential FAK-FAT Domain Inhibitors for Treatment of Metastatic Cancer. Molecules 2020, 25, 3488. https://doi.org/10.3390/molecules25153488
Kandil SB, Jones SR, Smith S, Hiscox SE, Westwell AD. Structure-Based Virtual Screening, Synthesis and Biological Evaluation of Potential FAK-FAT Domain Inhibitors for Treatment of Metastatic Cancer. Molecules. 2020; 25(15):3488. https://doi.org/10.3390/molecules25153488
Chicago/Turabian StyleKandil, Sahar B., Samuel R. Jones, Sonia Smith, Stephen E. Hiscox, and Andrew D. Westwell. 2020. "Structure-Based Virtual Screening, Synthesis and Biological Evaluation of Potential FAK-FAT Domain Inhibitors for Treatment of Metastatic Cancer" Molecules 25, no. 15: 3488. https://doi.org/10.3390/molecules25153488
APA StyleKandil, S. B., Jones, S. R., Smith, S., Hiscox, S. E., & Westwell, A. D. (2020). Structure-Based Virtual Screening, Synthesis and Biological Evaluation of Potential FAK-FAT Domain Inhibitors for Treatment of Metastatic Cancer. Molecules, 25(15), 3488. https://doi.org/10.3390/molecules25153488